

Reconstitution of the PMP22 protein into lipid bilayers results in membrane assemblies that share common features with myelin.
Keywords: Charcot-Marie-Tooth Disease, Electron Microscopy, Membrane, Myelin, Neuropathy, Peripheral Myelin Protein 22, Peripheral Nervous System, PNS, Reconstitution
Abstract
Peripheral myelin protein 22 (PMP22) is highly expressed in myelinating Schwann cells of the peripheral nervous system. PMP22 genetic alterations cause the most common forms of Charcot-Marie-Tooth disease (CMTD), which is characterized by severe dysmyelination in the peripheral nerves. However, the functions of PMP22 in Schwann cell membranes remain unclear. We demonstrate that reconstitution of purified PMP22 into lipid vesicles results in the formation of compressed and cylindrically wrapped protein-lipid vesicles that share common organizational traits with compact myelin of peripheral nerves in vivo. The formation of these myelin-like assemblies depends on the lipid-to-PMP22 ratio, as well as on the PMP22 extracellular loops. Formation of the myelin-like assemblies is disrupted by a CMTD-causing mutation. This study provides both a biochemical assay for PMP22 function and evidence that PMP22 directly contributes to membrane organization in compact myelin.
INTRODUCTION
Peripheral myelin protein 22 (PMP22) has important but poorly understood roles in peripheral nervous system (PNS) myelin. Myelin provides the insulating sheath to axons, serving to facilitate rapid conduction of electric impulses (1). In the PNS, compact myelin is formed when membrane tongues extend from myelinating Schwann cells to spirally wrap around an adjacent axon. The appropriate structural organization of myelin membranes is critical for proper nerve conduction.
PNS compact myelin membranes are composed of specific lipid and protein components that are essential to its function (1). The lipid bilayers of compact myelin contain high concentrations of cholesterol and sphingolipids. Proteins found in PNS compact myelin include myelin protein zero (MPZ), PMP22, and myelin basic protein (MBP) (2). Although the striking ultrastructure of myelin has been studied for many decades using various forms of imaging and scattering techniques (3–6), the molecular mechanism(s) responsible for the formation and maintenance of this complex membrane assembly is still being investigated.
PMP22 is a tetraspan helical integral membrane protein that is highly expressed (2 to 5% by weight of myelin proteins) in myelinating Schwann cells (7). Regulated expression and proper folding of PMP22 are essential for the development and maintenance of normal myelin in Schwann cells (7–9). A spectrum of heritable peripheral neuropathies is associated with aberrations in the PMP22 gene. These disorders include the most common (1:3500) inherited peripheral neuropathy Charcot-Marie-Tooth disease type 1A (CMT1A) that occurs with trisomy of PMP22 (10), resulting in overproduction of the protein. Hereditary neuropathy with liability to pressure palsies (HNPP), with a heterozygous deletion of PMP22, results in underexpression of the protein. CMT1E is caused by missense mutations of PMP22 that alter the protein amino acid sequence. Collectively, these defects affect 1 of 5000 individuals (11–18). Nerve biopsies from CMT1A patients show that the proliferative Schwann cells around axons resemble “onion bulbs” (19). Loss of one of the two PMP22 alleles results in HNPP, in which PNS myelin has characteristic “sausage-shape swellings that appear to be caused by abnormal membrane organization and/or myelin decompaction” (20, 21). Other disorders of widely varying severity are caused by dominant heterozygous PMP22 missense mutations (CMT1E), including the “trembler-J” (TrJ) L16P mutation of PMP22, which leads to abnormal interactions between the myelin sheath and the axon, abnormally thin myelin, and aberrant myelin wrapping (22). The connection between disease phenotypes and abnormal copy number or missense mutations in PMP22 highlights the importance of this membrane protein in myelin function.
Amino acid changes encoded by missense mutations disrupt the trafficking of PMP22 to the plasma membrane by increasing the propensity of the protein to misfold, resulting in targeting by the endoplasmic reticulum associated degradation (ERAD) system for disposal (23–30). However, degradation of misfolded PMP22 is likely not 100% efficient (26, 31–34). Thus, the severity of the phenotypes caused by PMP22 point mutations is postulated to result from a combined loss of functional PMP22 at the plasma membrane and cellular stress induced by misfolded protein. Additionally, some PMP22 disease mutants do reach the plasma membrane (28), suggesting that these mutants could directly disrupt or alter PMP22 functions in myelin membranes. All disease phenotypes resulting from PMP22 mutations lead to dysmyelination and axonal loss (7, 11).
Although PMP22 plays an essential role in myelinating Schwann cells, its biochemical function(s) is not well characterized. Here, we show that PMP22 reconstituted into model lipid bilayers causes the formation of protein-lipid superstructures that exhibit morphological similarities to compact myelin. These results reveal that PMP22 has an intrinsic capacity to promote the organization of membrane ultrastructure. This is the first biochemical evidence that isolated PMP22 can organize membranes, and provides mechanistic insight into the function of PMP22 in PNS myelin.
RESULTS
PMP22 forms myelin-like assemblies when reconstituted into lipid bilayers
In an attempt to determine the structure of PMP22, we began crystallization trials to generate two-dimensional (2D) crystals. We reconstituted human PMP22 into vesicles composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/egg sphingomyelin (ESM) (4:1 molar ratio) at a lipid-to-protein ratio (LPR) of 1.0 (w/w) via dialysis to remove the detergent β-n-decylmaltoside (DM) present in the starting PMP22-containing detergent/lipid mixed micelles [~4 mole percent (mol %) PMP22 relative to total moles of lipid]. Although this procedure did not result in the formation of single-layered 2D crystals, analysis by negative stain electron microscopy (EM) revealed that there were easily visualized lipid-protein assemblies that are distinctly multilayered (Fig. 1, A to C, and table S1). This indicates that the layer edges of these structures are exposed to bulk solvent, where they are accessible to stain. These assemblies appear to share several structural traits with PNS myelin (see Discussion) and are strikingly similar to intermediate assemblies observing the formation of myelin in calanoid copepods, which are invertebrate planktonic crustaceans [see Figs. 4, 6, and 9 in the study of Wilson and Hartline (35)]. We therefore refer to the PMP22-lipid superstructures as “myelin-like assemblies” (MLAs). MLAs were never observed in control reconstitutions carried out in the absence of PMP22 (Fig. 1, D and E, and table S2) or in reconstitutions using an unrelated tetraspan membrane protein, the voltage sensor domain of the human KCNQ1 potassium channel (Q1-VSD) (Fig. 1F and table S2). MLAs were not observed to form when a similar membrane reconstitution was applied to PMP22 using the lipid POPC, dipalmitoylphosphatidylcholine (DPPC), POPC/1-palmitoyl-2-oleoylphophatidylglycerol (POPG) (6:1 molar ratio), or DPPC/dipalmitoylphosphatidylglycerol (DPPG) (6:1 molar ratio). No MLAs were found in PMP22 reconstitutions using either total or polar brain lipids.
Fig. 1. PMP22 forms ordered assemblies upon reconstitution into lipid vesicles.
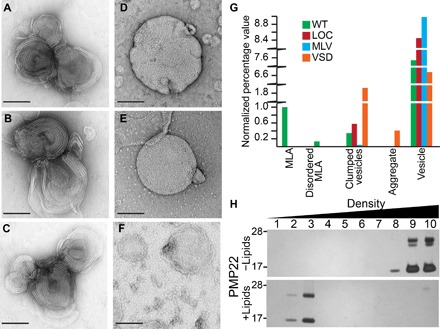
(A to C) Examples of protein-lipid MLAs created when PMP22 is reconstituted into 4:1 POPC/ESM vesicles via the dialysis method and visualized by negative stain EM. (D) Representative image of multilamellar vesicles (MLVs) prepared in the absence of protein via the dialysis method [lipid-only control (LOC)]. (E) MLVs prepared by spontaneous bilayer formation through hydration of lipids with water. (F) Control assemblies containing 4:1 POPC/ESM and the tetraspan VSD of KCNQ1 reconstituted via the dialysis method. Scale bars (all panels), 100 nm. (G) Quantification of the relative percentage of MLAs present in a series of negative stain EM images of wild-type (WT) PMP22, LOC, MLVs, and the tetraspan VSD domain of KCNQ1. All individual object counts were converted to percentage of total counts for a particular sample and were normalized to the percentage of total counts represented by MLAs in the WT PMP22 control, which was set to 1.0. Green, WT; red, LOC; blue, MLV; orange, VSD. (H) Sucrose gradient analysis of PMP22 reconstituted for 10 days without (−; top) or with (+; bottom) lipids. Fractions were collected from top (low density) to bottom (high density) and analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) with silver staining.
Although MLAs were seen only in 4:1 POPC/ESM lipid reconstitutions that contained PMP22 (tables S1 and S2), other types of assemblies, including vesicles, clustered vesicles, and protein aggregation, were also observed in MLA-containing samples (fig. S1). To more carefully characterize the protein-lipid reconstitutions, we quantified the objects seen on the negative stain EM grids. These were classified as MLAs, disordered MLAs (MLA-like assemblies composed of disorganized and/or “frayed” layers), vesicles, clumped vesicles (vesicles so close together they could not be individually counted), and aggregates that may be composed of protein, lipids, or a combination of protein and lipids (Fig. 1G). In all reconstitution experiments carried out with PMP22, vesicles were always the most commonly observed objects, followed by MLAs. Several other less common morphological classes of assemblies, including disordered MLAs, clumped vesicles, and aggregates, were also sometimes observed. The vesicles formed in PMP22-lipid reconstitutions and lipid-only reconstitutions were of similar appearance and size in negative stain EM.
To ensure that PMP22 is found in the lipid assemblies, we conducted a membrane flotation assay. PMP22 was subjected to the usual reconstitution method, both with 4:1 POPC/ESM and, in parallel, in the complete absence of lipids. The resulting mixtures were then loaded at the bottom of a stepwise sucrose gradient and centrifuged (Fig. 1H). PMP22 “reconstituted” without lipids remains pelleted in the high-density sucrose fractions, most likely as aggregated protein (Fig. 1H, top), whereas PMP22 reconstituted with lipids floats into the lower-density sucrose fractions, indicating that PMP22 is both stable during the reconstitution assay and incorporated into the membrane assemblies (Fig. 1H, bottom).
Cryo–electron microscopy of PMP22-containing MLAs
To confirm that MLAs are not an artifact of the negative staining protocol and to gain further insight into MLA ultrastructure, we vitrified PMP22 and control lipid reconstitutions and examined them using cryo–electron microscopy (cryo-EM). Visualizing the protein-free lipid reconstitutions in vitrified ice showed that multilamellar vesicles (MLVs) were the predominant assemblies formed by these reconstitutions (Fig. 2, A and B), whereas MLAs were never observed. However, when the product mixtures of PMP22-lipid reconstitutions were visualized by cryo-EM, we observed the presence of both MLAs and MLVs. The MLAs had similar architectures as observed in negative stain (Fig. 2, C and D). These appear to be compressed vesicles that stack and wrap around each other. By analyzing three independent MLA preparations frozen in vitrified ice, we measured an average interperiod line distance of 153 ± 7 Å (fig. S2). The average interperiod line distance for MLVs was 181 ± 7 Å. Figure S2 shows images of how these measurements were made. The average interperiod line distance of the MLAs is smaller than the repeat pattern of ~177 to 185 Å measured in neutron and x-ray diffraction studies of intact mammalian peripheral nerves, which would be expected because of the presence of other membrane proteins in vivo (5, 36–38); however, our measured repeat is similar to the repeat pattern (160 to 166 Å) reported for mammalian central nervous system (CNS) compact myelin (38). Cryo-EM analysis thereby confirms that PMP22 can drive the formation of lipid-protein structures with complex architectures that are morphologically different from the nested MLVs seen in lipid-only reconstitutions.
Fig. 2. Differences between MLVs and MLAs are visible by cryo-EM.
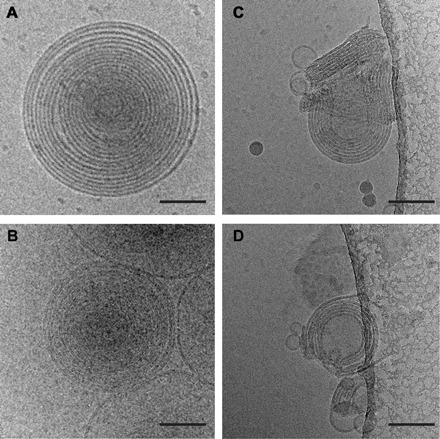
(A and B) Representative images of vitrified MLVs prepared in the absence of protein via the dialysis method (A) or by spontaneous bilayer formation through hydration of lipids only with water (B). (C and D) Examples of MLAs created when PMP22 is reconstituted into 4:1 POPC/ESM vesicles via the dialysis method and visualized using cryo-EM. Scale bars (all panels), 100 nm.
Tomography shows that MLAs are flattened, stacked, and wrapped vesicles
Although the negative stain and cryo-EM experiments indicate that MLAs are distinct from MLVs, a number of nonmutually exclusive morphological possibilities remain for how MLAs are organized. For example, MLAs could be a single, compressed, spiraled vesicle similar to myelin, a series of compressed and wrapped single unilamellar vesicles, and/or spiraling sheets of lipid bilayers. To more closely examine the morphology of MLAs, we used cryo–electron tomography (cryo-ET). We generated tomograms (Fig. 3, A to C) of PMP22-induced MLAs that allowed for the construction of a 3D model by segmenting the density in each z-slice (Fig. 3, D and F, and movies S1 and S2). We also generated a tomogram of an MLV as a comparison (fig. S3 and movie S3). These 3D reconstructions indicate that the examined MLAs are distinct from the architecture of MLVs and are composed of compressed, stacked, and wrapped unilamellar vesicles (Fig. 3F) and not nested vesicles (Fig. 3G). The 3D morphology of the MLAs, although not an exact recapitulation of the compressed and spiraling membranes found in Schwann cell–generated myelin, demonstrates that PMP22 can induce the flattening and wrapping of the vesicles into horseshoe-shaped stacks. These results also confirm that MLAs closely resemble superstructures observed as intermediates in the process of myelin formation in calanoid copepods (35). The above results indicate that PMP22 can directly alter membrane organization in vitro.
Fig. 3. MLAs examined by cryo-ET.
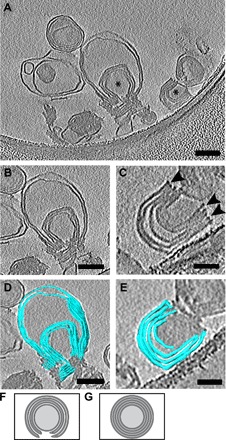
(A) Representative tomographic slices (1.47 nm) of two MLAs. *, MLAs in image. (B and C) Two MLAs from (A). Arrowheads indicate the ends of MLA. (D and E) Segmentation view of the corresponding MLA from (B) and (C). Scale bars, 100 nm (A, B, and D) and 50 nm (C and E). (F) Model demonstrating the compressed wrapped membranes of an MLA. (G) Model demonstrating the nesting vesicles of MLVs.
Formation of MLAs depends on the ratio of PMP22 to lipids
The most common type of CMTD, CMT1A, is caused by the presence of a third wild-type (WT) allele of PMP22 (10), resulting in a higher level of PMP22 than in healthy conditions. Nerve biopsies from CMT1A patients show that Schwann cells proliferate around axons without properly generating myelin, resembling onion bulbs (19). On the other hand, the presence of only a single PMP22 allele results in a different phenotype, called HNPP, in which PNS myelin has abnormal thickening and swelling of the myelin sheath; the myelin defects observed in HNPP appear to be caused by increased lamellae due to abnormal membrane organization around the lateral segment of the internode (20, 21). Together, these heritable conditions demonstrate that proper levels of PMP22 in Schwann cells are critical for proper myelin formation in vivo (1, 8, 11, 39, 40).
To determine whether MLA formation in vitro is also sensitive to PMP22 concentration, we performed a series of PMP22 reconstitution assays using a range of concentrations. LPRs ranged from 0.5 to 10 (w/w), spanning ~13 (8 mol % at LPR = 0.5) to ~256 (0.4 mol % at LPR = 10) lipid molecules per PMP22 in each reconstitution mixture. The highest percentage of MLAs, 17 ± 5%, was found at an LPR of 1.0, where there are ~26 lipid molecules per PMP22 (4 mol % protein) in the reconstitution assay (Fig. 4, C, D, and G, and table S3). At the lowest LPR of 0.5, where there are only ~13 lipid molecules per PMP22 in the reconstitution mixture (that is, the highest concentration of PMP22), well-organized MLAs were difficult to find. Instead, the sample contained a significant increase in the number of disordered MLAs, where the lipid bilayers are not tightly adhered and have a more frayed appearance (Fig. 4, A, B, and G, and table S3). These results demonstrate that higher concentrations of PMP22 in the membrane (that is, lower LPRs) do not lead to the formation of more MLAs but rather to a higher incidence of disordered MLAs.
Fig. 4. Altered PMP22 LPRs disrupt MLA formation.
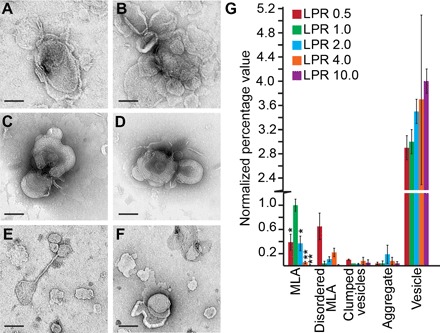
Representative negative stain images of PMP22 reconstitution assays carried out at LPRs (w/w) of 0.5 (A and B), 1.0 (C and D), and 10.0 (E and F). Scale bars (all panels), 100 nm. (G) Quantification of the relative percentage of MLAs present in a series of negative stain EM images of WT PMP22 reconstitutions at LPRs (w/w) of 0.5, 1.0. 2.0, 4.0, and 10.0. All individual object counts were converted to the percentage of total counts for a particular sample and were normalized to the percentage of counts represented by MLAs in the LPR 1.0 sample, which was set to 1.0. Red, LPR 0.5; green, LPR 1.0; blue, LPR 2.0; orange, LPR 4.0; purple, LPR 10.0. Error bars represent SEM between biological replicates. *P < 0.05, **P < 0.01. Statistical significance is only indicated for MLAs.
Successively lowering the amount of PMP22 in the reconstitutions from an LPR of 1.0 to higher LPRs led to progressively fewer MLAs. At an LPR of 2.0 (~51 lipid molecules per PMP22), we found an MLA prevalence of only 0.6 relative to reconstitutions at an LPR of 1.0 (table S3). At an LPR of 4.0 (~102 lipid molecules per PMP22 in the reconstitution assay), the MLA prevalence was further reduced to 0.1 relative to reconstitutions done at an LPR of 1.0 (table S3). At an LPR of 10, no MLAs were observed, although a small number of disordered MLAs that do not contain tightly condensed layers were still seen in the images (Fig. 4, E to G, and table S3). These studies confirm that PMP22 is responsible for MLA formation. Too little PMP22 leads to fewer MLAs, whereas too much PMP22 in the reconstitution assay increases the formation of disordered MLAs. Although the size and shape of the MLAs did not change as a function of the LPR, we did notice more clumped vesicles at LPRs of 0.5 and 4.0 (table S3). This suggests that PMP22 found in vesicles that do not form MLAs may be able to participate in trans-homophilic interactions that can cause vesicles to stick together. These results confirm a role for PMP22 in MLA formation and are reminiscent of in vivo findings that the proper level of PMP22 is important for the correct initiation and formation of myelin (8, 9, 40, 41). However, note that the LPR of individual MLAs may deviate from the bulk LPR used in the reconstitution process.
Removal of cysteine residues does not abrogate MLA formation
To explore which regions of PMP22 are required for MLA formation, we performed structure-function studies. We first tested whether disulfide bond formation in PMP22 might be related to MLA formation. PMP22 contains four native cysteine residues (fig. S4). Two of these residues are found in the extracellular loop and, based on homology between PMP22 and claudin-15, are predicted to form an intramolecular disulfide cross-link (42). We simultaneously mutated all four cysteine residues, creating a “Cys-less” PMP22 (C42S, C53S, C85A, and C109A) (Fig. 5, A and B), and purified it alongside WT PMP22. No significant differences were found between the ability of Cys-less and WT PMP22 to form MLAs (Fig. 5, A to C, and table S4). From these data, we conclude that the formation of disulfide bonds is not required for MLA formation.
Fig. 5. MLA formation is not dependent on intermolecular disulfide linkage.
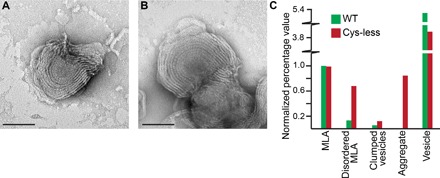
(A and B) Representative negative stain images of MLAs formed in a reconstitution assay using a Cys-less PMP22 mutant (C42S, C53S, C85A, and C109A). Scale bars (A and B), 100 nm. (C) Quantification of the percentage of MLAs present in a series of negative stain EM images of WT and Cys-less PMP22 reconstitutions. All individual object counts were converted to the percentage of total counts for a particular sample and were normalized to the percentage of total counts represented by MLAs in the WT PMP22 control, which was set to 1.0. Green, WT control; red, Cys-less PMP22.
PMP22 extracellular loops are important for MLA formation
A previous study using glutathione S-transferase (GST) fusion oligopeptides of PMP22 showed that its extracellular loops 1 and 2 (ECL1 and ECL2) could support both trans-homophilic interactions and trans-heterophilic interactions with MPZ (43). To test the importance of ECL1 and ECL2 for MLA formation, we expressed and purified GST-ECL1 and GST-ECL2 fusion proteins to use as a source of competitive binding to weaken any potential trans-homophilic loop interactions that form within the MLAs. We then also verified that GST alone did not reduce MLA formation by including GST at the same molar ratios in the PMP22-lipid reconstitution assay (fig. S5A and table S6). When GST-ECL1 was included in PMP22 reconstitution assays at a 1:1 or a 4:1 molar ratio (GST-ECL1/PMP22), MLA formation was reduced by 50 and 40%, respectively, compared to reconstitutions containing PMP22 alone (Fig. 6A and table S5). Addition of GST-ECL2 to the PMP22-lipid reconstitutions at a ratio of 1:1 (GST-ECL2/PMP22) reduced MLA formation by 60%, whereas a GST-ECL2–to–PMP22 ratio of 4:1 led to a 98% reduction (table S5). Reconstitution of GST, GST-ECL1, and GST-ECL2 alone (that is, without PMP22) did not lead to the formation of MLAs (table S7). The fact that high concentrations of both GST-ECL1 and GST-ECL2 reduced MLA formation suggests that both loops are important for the in vitro function of PMP22.
Fig. 6. ECL1 and ECL2 are important for MLA formation.
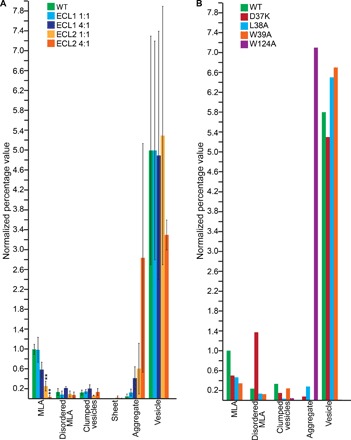
(A) Quantification of the relative percentage of MLAs present in a series of negative stain EM images of PMP22 reconstitutions of WT PMP22 only, WT PMP22 incubated with GST-ECL1, and WT PMP22 incubated with GST-ECL2. Green, WT control; light blue, GST-ECL1 + WT PMP22 (1:1 molar ratio); dark blue, GST-ECL1 + WT PMP22 (4:1 molar ratio); light orange, GST-ECL2 + WT PMP22 (1:1 molar ratio); dark orange, GST-ECL2 + WT PMP22 (4:1 molar ratio). Error bars represent SEM between biological replicates. **P < 0.01. Statistical significance is only indicated for MLAs. (B) Quantification of the relative percentage of MLAs present in a series of negative stain EM images of PMP22 reconstitutions of WT PMP22; ECL1 loop-mutants PMP22 D37K, L38A, or W39A; and ECL2 loop-mutant PMP22 W124A. Green, WT control; red, D37K; blue, L38A; orange, W39A; purple, W124A. For both panels, all individual object counts were converted to the percentage of total counts for a particular sample and were normalized to the percentage of total counts represented by MLAs in the WT PMP22 control. All values were normalized to the percentage of WT control MLAs, which was set to 1.0.
As another way to assess the roles of ECL1 and ECL2 in MLA formation, we prepared and purified PMP22 with mutations in either ECL1 or ECL2. PMP22 shares ~25% sequence identity with the claudins, including a claudin motif in ECL1 (42). In ECL1, individual residues from this motif were mutated (D37K, L38A, and W39A). Also, in ECL2, the highly conserved tryptophan residue (W124) was changed to alanine (see topology diagram in fig. S4). The three PMP22 ECL1 mutants disrupted MLA formation relative to a PMP22 WT control by 50, 50, and 70%, respectively (Fig. 6B and table S7). However, the PMP22 W124A mutation in ECL2 was especially deleterious, completely disrupting MLA formation (Fig. 6B and table S8). These results confirm that PMP22’s extracellular loops contribute to MLA formation, with ECL2 possibly having a more dominant role. However, it must be acknowledged that these experiments may not be the final word on the roles of ECL1 and ECL2 in MLA formation because they used recombinant protein in which PMP22 was not natively N-glycosylated at Asn41 located in ECL1 (44). Nevertheless, the above results make it very clear that the extracellular loops of PMP22 play a critical role in MLA formation.
The L16P Trembler-J (TrJ) disease mutation of PMP22 disrupts MLA formation
The L16P TrJ mutation located in the first transmembrane domain (TM1) of PMP22 causes severe dysmyelinating neuropathy in both humans and mice (fig. S4) (7, 45, 46). This mutation leads to abnormal differentiation of Schwann cells, which are arrested at the immature premyelination stage. As a consequence, interactions between the myelin sheath and the axon are disrupted, resulting in thin myelin. Additionally, the excessive immature Schwann cells surrounding axons fail to form compact myelin (22). Biophysical studies have shown that the L16P mutation disrupts the structural organization of the TM1 domain, reducing the thermodynamic stability of PMP22 (47), with most of the mutant protein adopting a partially folded state (24, 42, 47). To test the effect of the L16P mutation on MLA formation, L16P PMP22 was purified and used in lipid reconstitution assays. Reconstitutions using this mutant yielded fewer MLAs compared to WT control (Fig. 7A and table S9). The MLAs observed in the L16P PMP22 reconstitutions were composed of loosely packed layered assemblies that appear more disorganized than the MLAs created by WT PMP22 (Fig. 7, B and C, and table S9).
Fig. 7. The L16P PMP22 (TrJ) mutation disrupts MLA formation.
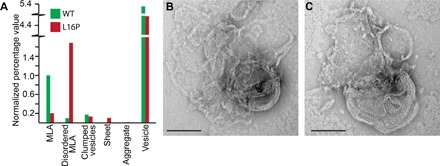
(A) Quantification of the relative percentage of MLAs present in a series of negative stain EM images in both WT and L16P PMP22 reconstituted on the same day. All individual object counts were converted to the percentage of total counts for a particular sample and were normalized to the percentage of total counts represented by MLAs in the WT PMP22 control, which was set to 1.0. Green, WT control; red, L16P. (B and C) Representative negative stain EM images of the disordered MLAs found in L16P PMP22 reconstitutions. Scale bars (B and C), 100 nm.
DISCUSSION
Similarity of MLAs to PNS myelin and related membrane assemblies
PMP22 has one or more essential roles in myelinating Schwann cells, as reflected by its central role in common forms of Charcot-Marie-Tooth and related dysmyelinating disorders (7, 11). However, the biochemical functions of PMP22 are not understood. Using in vitro reconstitution assays, we showed that PMP22 drives the formation of complex lipid-protein superstructures composed of compressed and stacked membranes that wrap around a central vesicle. The formation of these structures is absolutely dependent on the presence of PMP22 and can be disrupted by varying PMP22 concentration, introducing a disease mutation, including GST-ECL1 or GST-ECL2 fusion peptides during membrane reconstitution, or mutating residues in ECL1 and ECL2.
The organization of MLAs shares common traits with PNS myelin. Both MLAs and myelin include membrane compaction. For MLAs, the vesicles are flattened, whereas for PNS myelin, the bilayers that enclose the cytosol of a tongue-like membrane extension of a Schwann cell are drawn together to render the extension similar to double-layered tape. The flattened double bilayers of both MLAs and developing myelin are then multilayered—by stacking the flattened vesicles (in the case of MLAs) and by spirally wrapping the flattened double bilayers around a cylindrical segment of an axon similar to wrapping tape around a spool (in the case of myelin). MLAs seem also to usually wrap around a central cylindrical vesicle, although the stacked and flattened vesicles wrap collectively and only a single time—no spiraling. Although differences between the supramolecular organization of MLAs and myelin remain, it is notable that the inclusion of a single myelin protein in reconstituted lipid bilayers is capable of altering the organization of resulting membrane assemblies so as to confer several traits that resemble key features of the organization of PNS myelin. There is a previous report showing that MBP in aqueous n-dodecylphosphocholine (DPC) micelles can alter the organization of detergent into regions of parallel arrays (48). Thus, it seems likely that multiple myelin proteins have important structural roles in establishing myelin architecture. However, because MBP is not a transmembrane protein, its potential effect on myelin membrane organization is likely via a distinct mechanism than PMP22. Considering the importance of myelin ultrastructure in nerve conductance, it is not surprising that cells have developed multiple robust, and perhaps independent, ways to ensure the proper organization of myelin membranes.
MLAs are even more strikingly similar to membrane assemblies thought to represent intermediate structures on the pathway to myelin development in certain marine copepods. Myelin in copepods can be regarded as an early evolutionary alternative form of vertebrate myelin and is generated directly by axons rather than by glial cells. As seen in Figs. 4, 6, and 9 in the study of Wilson and Hartline (35), developing copepod myelin includes assemblies that are composed of flattened vesicles/cisternae that are stacked and nonspirally wrapped, which closely resemble MLAs. It is known that copepods [like all invertebrates (49)] lack MPZ, the major adhesive protein of vertebrate PNS myelin (50). Whether myelin-forming copepods have a PMP22 homolog is not yet established but seems feasible given that even Caenorhabditis elegans has a distant relative of PMP22 (51). Whether or not a PMP22 homolog is involved in copepod myelin formation, the fact that purified PMP22 can induce the formation of membrane assemblies that closely resemble a known intermediate structure in copepod myelin supports the likely mechanistic linkage between PMP22’s ability to modify membrane structure and its roles in PNS myelin.
PMP22-containing MLAs also resemble assemblies termed “intracellular myelin–like figures” (IMLFs) that form in Schwann and human embryonic kidney (HEK)–293 cells upon expression of PMP22 (23, 52). IMLFs are composed of multilayered “whorls” of spiraled membrane that form when overexpressed PMP22 fails to traffic to the cell surface and accumulates in the ER. The similarity of MLAs and IMLFs further supports the notion that PMP22 has an intrinsic ability to both flatten membrane double layers and promote stacking of these layers.
Adhesive function of PMP22 in MLA formation and stabilization
Our results indicate that the extracellular ECL1 and ECL2 loops of PMP22 have an important role in MLA formation. This result is reminiscent of studies involving PMP22 expressed in HeLa cell lines that showed that the protein can participate in trans homophilic interactions to form an adhesive PMP22 bridge between cells (43) and that these interactions can be disrupted by adding ECL1 peptide as a GST fusion protein to the milieu surrounding the cells. Additional evidence for PMP22 playing a role in adhesion between lipid bilayers includes studies showing expression of PMP22 in certain epithelia, where it appears to be involved in forming and/or stabilizing membrane junctions (53–56). Both the present and past studies support a direct role for PMP22 in bilayer-bilayer adhesion.
Although we have not yet directly observed the spatial distribution of PMP22 in MLAs, the properties of MLAs are consistent with stabilization of trans homophilic interactions by the extracellular loops of PMP22, both inside the flattened vesicles and between the stacked outer leaflets of the flattened and stacked vesicles. Trans homophilic interactions between PMP22 molecules on the outer surface of a cylindrical vesicle with the outermost leaflet of a stack of flattened vesicles are also likely responsible for the wrapping of the stacked multilayers around a central cylindrical vesicle in MLAs. It should be acknowledged that PMP22 likely assumes mixed topologies in MLAs, where half of the protein has its extracellular loops facing out of the vesicles and half with the loops inside. This is distinct from the environment within myelinating Schwann cells, where the loops are always extracellular and could only be involved in adhesion between juxtaposed outer leaflets of the spiraling Schwann cell–extended myelin double membrane.
Similarity of PMP22 function in MLA formation to other tetraspan proteins
Some other tetraspan proteins have the capacity to alter membrane architecture through trans homophilic interactions. The proteolipid protein (PLP) is the major protein of the CNS myelin. When this tetraspan membrane protein was purified and reconstituted into lipid bilayers, it led to the formation of structures that the authors suggested represent layers of stacked and compressed vesicles (57), assemblies very similar to MLAs. Furthermore, the two extracellular loops of PLP have been proposed to participate in trans homophilic interactions between adjacent membrane surfaces at the extracellular intraperiod line of CNS myelin (58). Analogous to PMP22, PLP is prone to mutation-induced misfolding that leads to a dysmyelinating disorder in the CNS, Pelizaeus-Merzbacher disease (59). Intriguingly, the average interperiod distance measured for MLAs in vitrified ice is 153 ± 7 Å (fig. S2). This is similar to the interperiod distance measured for mammalian CNS, which is 160 to 166 Å (38). Although it now seems clear that MPZ is the major adhesive protein of stable compact myelin under normal conditions (9, 39, 60, 61), it is still tempting to speculate that PMP22 may also play a structural role in PNS myelin that, to some degree, resembles the central role of PLP in the structure of CNS myelin.
Although the capacity of PMP22 to undergo trans homophilic adhesive interactions may not be critical to the stability of mature compact myelin, this does not preclude a possibly transient but important role for such interactions in Schwann cell differentiation and myelin formation. It has long been known that PMP22 appears to play an important role in these processes (7, 9, 11). Additionally, the trans adhesive biochemical properties of PMP22 may also help to stabilize other (noncompact) domains or junctions within mature PNS myelin. Another family of tetraspan membrane proteins that alter membrane organization through trans homophilic protein-protein interactions is the claudins, which are integral to the organization and stabilization of tight junctions (62, 63). Claudins facilitate adhesion by establishing trans homophilic interactions between its extracellular loops to form paracellular barriers and channels between adjacent epithelial cells (64, 65). PMP22 shares ~25% sequence identity with the claudins, including a claudin motif in ECL1, suggesting that they likely share similar folds (42). PMP22 has been found to be a component of tight junctions in some epithelial cells (55, 66). PMP22 has also been shown to be important for the integrity of some of the specialized junctions that control permeability in PNS myelin (56).
Thus, a common theme between the tetraspan membrane proteins PLP, claudins, and PMP22 and the single-span MPZ is that all these proteins can form networks of trans homophilic interactions across juxtaposed lipid bilayers that influence membrane ultrastructure. Given that the water-soluble but membrane-interactive MBP has long been known to cause aggregation of vesicles and even of micelles (48, 67, 68), trans adhesive properties seem to be intrinsic to multiple myelin proteins.
Possible additional (nonadhesive) roles for PMP22 in MLA formation
PMP22 may also promote MLA formation through additional mechanisms. For example, PMP22 could stabilize the high membrane curvature seen at the end of each pancaked vesicle, perhaps through cis-homophilic interactions, as has been suggested for ER membrane proteins that shape ER membrane organization (69). Additionally, it is possible that PMP22, if oligomerized within the same membrane, could promote membrane curvature through a protein-protein crowding mechanism (70). To generate a more detailed molecular understanding of PMP22’s role in organizing membranes, additional studies are needed to examine whether PMP22 forms cis-homophilic interactions in membranes and to map the spatial distribution of PMP22 in MLAs.
CONCLUSIONS
Here, we demonstrate that reconstitution of PMP22 into lipid bilayers results in the formation of MLA assemblies that share some similarity to myelin, albeit only to an extent. It was further demonstrated that MLA formation depends on the level of PMP22 relative to lipid, is dependent on both PMP22 ECL1 and ECL2 loops, and is disrupted by a misfolding-prone disease mutant form of PMP22. The intrinsic ability of PMP22 to alter the morphology of lipid bilayer assemblies supports the notion that PMP22 may play one or more direct structural roles in myelin organization, which includes contributing to adhesion across the extracellular space between apposed bilayers. Some of the roles may well be transient, occurring during Schwann cell differentiation and myelin formation. Also, although it is clear that MPZ is the major adhesive protein in compact PNS myelin, we speculate that PMP22 likely plays niche roles in myelin adhesion and/or myelin junctions that complement the more generally prevalent role of MPZ. This likely helps explain why myelin compaction is not completely eliminated even when MPZ is completely knocked out (71). It should be added that MPZ and PMP22 located on the surface of apposed cell membranes have been proposed to form glycosylation-independent trans-heterophilic complexes (43, 72), suggesting that the adhesive roles of these proteins may sometimes be directly cooperative.
Although PMP22 is thought to play multiple roles in Schwann cell biology and myelin homeostasis, the capacity of PMP22 to promote MLA formation appears to provide a convenient biochemical assay for PMP22 function in organizing membrane ultrastructure. Moreover, because the capacity of PMP22 to promote MLA formation is critically dependent on the correct folding of this protein, MLA formation might even be exploited as the basis for an assay for use in high-throughput screening of small molecules in a search for molecules that rescue misfolding-prone disease mutant forms of PMP22.
MATERIALS AND METHODS
Expression and purification of PMP22
PMP22 was expressed and purified as previously described, with minor adjustments (47, 73–75). Briefly, PMP22 was expressed in One Shot BL21 Star (DE3) Escherichia coli (Life Technologies) as a fusion protein construct consisting of an N-terminal 76–amino acid segment of the lambda repressor (which serves to drive the protein into inclusion bodies) (73), followed by a His10-tag, a 7–amino acid linker, a thrombin cleavage site, an 11–amino acid strep-tag, and, finally, the human PMP22 sequence. The fusion protein was solubilized from inclusion bodies using the zwitterionic detergent Empigen BB (Sigma-Aldrich) and purified using Ni(II)-NTA Superflow resin (Qiagen) (0.5 ml/1 g of original cell pellet) packed into a gravity column. Once bound to the Ni-NTA resin, the detergent was exchanged for DM (Anatrace), a mild, uncharged detergent, by repetitive pulsed washing of the column [20 column volumes (CVs) in one-half CV pulses] with 0.5% DM in 25 mM sodium phosphate buffer (pH 7.2). The fusion construct was cleaved by incubation with Recothrom Thrombin (The Medicines Company) overnight. This cleavage reaction was followed by a second purification over Ni-NTA resin. Cleaved PMP22 (no His-tag) has a modest affinity for divalent metal cations, including Ni(II) (74). Cleaved PMP22 was eluted from the Ni(II) resin in a stepwise fashion using a 10 to 30 mM imidazole (Sigma-Aldrich) in a 50 mM tris (pH 8.0) buffer. The uncleaved PMP22 and the His10-tag–containing fusion cleavage product remained bound to the resin at these imidazole concentrations. Protein purity was assessed via SDS-PAGE, and the pure fractions were pooled (28). In some cases, PMP22 was purified using a method in which the second Ni-NTA column used to purify the cleaved protein was replaced by an ion exchange chromatography column eluted with a salt gradient, essentially as described by Sakakura et al. (47).
Cleaved and purified PMP22 in DM micelles and a pH 8.0 buffer containing approximately 20 mM imidazole, 50 mM tris, and 0.5 mM dithiothreitol (DTT) were concentrated to 1.0 mg/ml, as determined by A280 (absorbance at 280 nm) using a molecular weight of 19.2 kDa and an extinction coefficient of 44,900 OD/M·cm before reconstitution into vesicles.
Reconstitution of PMP22 into vesicles
WT PMP22 (1.0 mg/ml) and the various PMP22 mutants used in these studies were combined with mixed micelles containing 53 mM DM plus 5.3 mM POPC and 1.3 mM ESM in water at the LPRs described in Results. Each PMP22-mixed micelle solution was pipetted into dialysis buttons (Hampton Research), which were covered by hydrated dialysis membrane with a molecular weight cutoff of 20 kDa (Spectra/Por Biotech RC Tubing) that had been pretreated by boiling in 1 mM EDTA for 5 min to remove metal ions. Buttons were dialyzed for 10 days at room temperature against a buffer containing 10 mM tris (pH 8.0), 150 mM NaCl, and 0.5 mM fresh DTT, which was changed daily. Protein-free vesicle controls were prepared by mixing mixed micelles and PMP22 elution buffer together at the same volume-to-volume ratio as the experimental conditions and dialyzed in the same buffer. Alternatively, MLV controls were prepared by mixing dry lipids with water and agitating.
Liposome flotation assay
After dialysis, a 750-μl sample of 20-μg PMP22 reconstituted into liposomes and dialysis buffer containing 50% (w/v) sucrose was prepared. This sample was placed at the bottom of a 5-ml polypropylene ultracentrifuge tube. A 750-μl sample of 20-μg PMP22 in DM micelles and dialysis buffer containing 50% sucrose was placed in another ultracentrifuge tube as a control. Dialysis buffer (3.38 ml) containing 40% sucrose was layered on top of the liposome or micelle layer, followed by 750 μl of dialysis buffer containing no sucrose. This created a stepwise sucrose gradient. All layers in the micelle sample contained 0.05% DM so as to not dilute the DM below the critical micelle concentration. Samples were then centrifuged at 160,000g at 4°C for 16 hours using a Beckman L-90K Ultracentrifuge equipped with an SW 55 Ti rotor. After ultracentrifugation, samples were collected from the sucrose gradient in 500-μl fractions from the top down using a glass Hamilton syringe; the fractions were analyzed by SDS-PAGE, followed by silver staining (GE Healthcare PlusOne Silver Staining Kit).
Expression and purification of KCNQ1-VSD
The human KCNQ1 potassium channel voltage sensor domain (Q1-VSD, residues 100 to 249) was purified into DPC (Anatrace) micelles, as previously described (76). The Q1-VSD in micelles was then combined with mixed micelles containing 174 mM DPC and 4:1 POPC/ESM and dialyzed alongside PMP22. DPC was used because the VSD has previously been successfully refolded from DPC into other micelle conditions (76).
Expression, purification, and addition of GST, GST-ECL1, and GST-ECL2 to MLA preparations
The following methods were adapted from Hasse et al. (43). DNA encoding the first extracellular loop (ECL1, residues 32 to 64) and second extracellular loop (ECL2, residues 120 to 133) of PMP22 were polymerase chain reaction (PCR)–amplified using the following primers: ECL1, 5′-TCCGCGTGGATCCCCAGGAATTCCCAATGGACACGCAACTGATCTC-3′ (forward) and 5′-GCCGCTCGAGTCGACTCAAGACTGCAGCCATTCGTTTGG-3′ (reverse); ECL2, 5′-TCCGCGTGGATCCCCAGGAATTCCCAGGCACCCGGAGTGGCATCTC-3′ (forward) and 5′-GCCGCTCGAGTCGACTCAACCGTAGGAGTAATCCGAGTT-3′ (reverse). PCR products were restriction-digested using Bam HI–HF and Sal I–HF enzymes (New England Biolabs) and ligated into a similarly digested pGEX-4T-1 vector (GE Healthcare Life Sciences). The final product of the ligation reaction was transformed into XL1-Blue E. coli cells, and plasmid DNA was purified with QIAprep Spin Miniprep Kit (Qiagen). The final constructs were verified by sequencing and were then transformed into BL21(DE3) E. coli cells. A single colony was used to inoculate an overnight starter culture in LB medium containing ampicillin (0.1 mg/ml). The starter culture was then added to 1 liter of LB medium and grown for 3 hours at 37°C with shaking at 230 rpm to an OD600 (optical density at 600 nm) of 0.9. Cultures were transferred to 25°C and induced over 5 hours with 1 mM isopropyl-β-d-thiogalactopyranoside. Cells were harvested by centrifugation, flash-frozen, and stored at −80°C until use.
To purify GST and the GST fusion peptides, the cell pellet from 1 liter of growth was thawed and resuspended in 30-ml buffer NETN [50 mM tris (pH 7.5), 150 mM NaCl, and 0.5% NP-40 detergent] plus 2 mM DTT, 1 mM phenylmethylsulfonyl fluoride, and lysozyme (1 mg/ml). This mixture was tumbled for 30 min at 4°C. Cells were sonicated at a 20% duty cycle (30 s on, 2 min off) for 1.5 min of total process time on ice at 4°C. The lysate was centrifuged in a JA-25.50 rotor at 20,000 rpm for 20 min, and the supernatant was collected and filtered (0.2 μm). Glutathione Sepharose resin (GE Healthcare Life Sciences) was prepared by washing with 2 CVs of water and 10 CVs of buffer NETN. For the supernatant from cells derived from a liter of culture, a 2-ml volume of glutathione resin was added and allowed to equilibrate for 2 hours at 4°C with tumbling. The resin was then transferred to a column and washed with 24 CVs of NETN containing 0.2 mM DTT, followed by 24 CVs of 50 mM tris (pH 8.0) containing 0.2 mM DTT. Protein was eluted with 50 mM tris (pH 8.0) containing 0.2 mM DTT and 10 mM glutathione. Elution was monitored at 280 nm, and the elution volume was collected in fractions. Protein purity was assessed by SDS-PAGE, and fractions containing pure fusion protein were combined. To prepare these purified protein solutions, we added 10% glycerol, followed by flash-freezing and storage at −80°C until use. Fusion peptides were added to the PMP22 dialysis conditions at final concentrations of 2.9 and 2.7 μM for GST-ECL1 and GST-ECL2, respectively (1:1 peptide/PMP22 molar ratio), 5.6 and 5.2 μM for GST-ECL1 and GST-ECL2, respectively (2:1 peptide/PMP22 molar ratio), or 11.2 and 10.4 μM for GST-ECL1 and GST-ECL2, respectively (4:1 peptide/PMP22 molar ratio). GST control protein was added to the PMP22 dialysis conditions at final concentrations of 2.8 μM (1:1 peptide/PMP22 molar ratio) and 11.2 μM (4:1 peptide/PMP22 molar ratio). As controls, GST-ECL1, GST-ECL-2, and GST alone were combined with mixed micelles containing 53 mM DM plus 5.3 mM POPC and 1.3 mM ESM and dialyzed as described for PMP22 reconstitutions. The final concentrations of GST-ECL1, GST-ECL-2, and GST in the reconstitutions were 2.9, 2.7, and 2.8 μM, respectively.
EM of PMP22 in vitro lipid reconstitutions
For negative stain EM grids, 2 μl of reconstituted PMP22-lipid assemblies was adsorbed to a glow-discharged 200-mesh copper grid covered with a carbon-coated collodion film (Electron Microscopy Sciences). Grids were washed in two drops of water and stained with two drops of uranyl formate (0.75%) (Electron Microscopy Sciences), as described (77). Samples were imaged using an FEI Morgagni equipped with a 1000 × 1000 charge-coupled device (CCD) camera [Advanced Microscopy Techniques (AMT)]. Measurements to determine the “interperiod distance” were carried out using AMT software. Approximately 200 measurements were made to determine the interperiod distance for multiple MLAs from different images. Images for presentation were contrast-adjusted in Photoshop, and a high-pass filter was applied to enhance contrast between layers. No imaging adjustments were made to images before any type of quantification.
For cryo-EM, 3 μl of PMP22/lipid assemblies was pipetted directly onto glow-discharged Quantifoil R2/2 Holey Carbon (200-mesh copper) grids (Electron Microscopy Sciences) and plunged into liquid ethane using a Vitrobot (2007, FEI) set to 60% humidity at 22°C. Vitrobot settings included a blot time of 3.5 s, an offset of −1, and a drain time of 1 s. Images shown in Fig. 2 (C and D) were collected using a Tecnai F20 electron microscope (FEI) equipped with a field emission gun at an acceleration voltage of 200 kV under low-dose conditions at a magnification of ×68,661 (2.18 Å per pixel) using a defocus value of −2.0 to 4.0 μm. Images were recorded on a 4000 × 4000 Gatan CCD camera. Images shown in Fig. 2 (A and B) were collected using a Tecnai F30 Polara electron microscope (FEI) equipped with a field emission gun at an acceleration voltage of 300 kV under low-dose conditions at a magnification of ×38,204 using a defocus value of 5.0 to 8.0 μm and recorded on a 4000 × 4000 UltraScan CCD camera in images with a pixel size of 3.5 Å per pixel (Gatan).
For cryo-ET, bovine serum albumin–conjugated 10-nm gold nanoparticles were mixed with the MLA sample to achieve an appropriate density for fiducial markers. Four microliters of the mixed solution was applied onto the glow-discharged 200-mesh copper R1.2/R1.3 Quantifoil transmission electron microscopy (TEM) grid (Quantifoil) before cryoplunging in liquid ethane using a Vitrobot Mark III system (FEI). Cryo-grids were stored in liquid nitrogen before imaging. Cryo-ET data were collected using a JEOL JEM-2200FS field emission TEM (FEG-TEM) at 200 kV (JEOL Ltd.), which is equipped with an in-column Omega energy filter with a slit width of 20 eV. Single-axis tilt series were recorded semiautomatically using the SerialEM package from −65° to 65° at 2° increment step (bidirectional), −6-μm defocus, with an accumulative dose under 130 e−/Å−2 (78). Images were recorded on a Direct Electron DE-20 camera (Direct Electron LP) at 24 frames per second at a nominal magnification of ×20,000, resulting in a pixel size of 0.294 nm.
Tilt series frames were motion-corrected before tomographic reconstruction using python scripts provided by the manufacturer (Direct Electron LP). Motion-corrected frames were used for tomographic reconstruction in the IMOD software package using the weighted back-projection algorithm, and the 10-nm gold nanoparticles were used as fiducial markers to align frames at the different tilt angles (79, 80). Images were contrast transfer function–corrected by phase-flipping before reconstruction of the 3D volumes (binned by a factor of 2). Volume-rendered segmentations were performed manually using the Amira package (FEI Visualization Sciences Group) (78). Visualization and movie making were carried out in IMOD and Amira software packages.
Quantification of MLAs
For experiments testing the effects of varying the lipid-to-PMP22 ratio, adding ECL1 and ECL2 peptides, or PMP22 mutations on MLA formation, PMP22 purifications were carried out for each group of experiments on the same day, and subsequent reconstitution experiments were run in parallel. For each set of experiments, WT PMP22 was purified and reconstituted with 4:1 POPC/ESM at an LPR of 1.0 as a positive control that was used to normalize quantification. Multiple negative stain grids were prepared for each reconstitution condition and imaged using an FEI Morgagni equipped with a 1000 × 1000 CCD (AMT) camera at ×14,000 magnification. Nikon Elements software was used to count and categorize objects viewed on the grid as MLA, disordered MLA, clumped vesicles, protein aggregate, lipid sheets, or vesicles. One person quantified all data sets under blind conditions (that is, the person counting did not know the experimental variables of each data set). Values for each experimental condition are found in tables S1 to S6. For presentation in figures, all percentage values for each category were normalized to the percentage of MLAs present in the WT control for each experimental grouping. The percentage of a counted object in an individual category from one experiment was divided by the percentage of counted MLAs in the corresponding control experiment. For statistical analysis in Figs. 4 and 6 and fig. S5, GraphPad Prism was used to perform a Welch’s t test to look for statistical significance between MLA formation in the WT control and different conditions.
Supplementary Material
Acknowledgments
We acknowledge cryo-EM facilities management support from Collier. We also thank members of the Ohi Lab—L. Lapointe, B. Carter, and T. Iverson—for critical reading of the manuscript. We thank the Vanderbilt Center for Structural Biology for support of the Structural Electron Microscopy Facility. Funding: This work was supported by grants R01 NS058815, R01 NS095989 (to C.R.S. and M.D.O.), R01 NS066927 (to C.R.S.), R01GM104540 (to E.R.W.), R01GM1145661 (E.R.W), and NSF-0923395 (to E.R.W.), as well as grants from the Emory University, the Children’s Healthcare of Atlanta, the Georgia Research Alliance, the Center for AIDS Research at Emory University (P30 AI050409), and the James B. Pendleton Charitable Trust (to E.R.W.). K.F.M. was supported by NIH T32 GM08320 and NSF Predoctoral Fellowship DGE090966. J.P.S. was supported by NIH F32 GM110929. J.T.M. was supported by T32 GM008320. Author contributions: K.F.M., J.P.S., J.T.M., A.H., C.L.L., C.M.H., Z.K., and M.D.O. as authors that contributed to experiments and M.D.O. conducted the experiments of this work. All authors participated in data analysis. K.F.M., J.T.M., J.L., C.R.S., and M.D.O. wrote the paper with input from all authors. K.F.M., J.T.M., C.R.S., E.R.W., and M.D.O. conceived this work and directed the approaches used. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Correspondence and requests for materials should be addressed to C.R.S. (chuck.sanders@vanderbilt.edu) or M.D.O. (melanie.ohi@vanderbilt.edu).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700220/DC1
movie S1. Cryo-ET of an MLA showing that it is composed of compressed wrapped vesicles (corresponding to Fig. 3B).
movie S2. Cryo-ET of an MLA showing that it is composed of compressed wrapped vesicles (corresponding to Fig. 3C).
movie S3. Cryo-ET of an MLV showing that it is composed of nested vesicles rather than compressed wrapped vesicles seen in MLAs (corresponding to fig. S3B).
fig. S1. Representative examples of objects observed in reconstitution experiments by negative stain.
fig. S2. Examples of interperiod repeat distance measurements taken for MLAs and MLVs imaged with cryo-EM.
fig. S3. MLV vesicles examined by cryo-ET.
fig. S4. Schematic showing the organization of PMP22.
fig. S5. Addition of GST does not reduce MLA formation.
table S1. Total counts, percentages, and SDs of WT PMP22 reconstitutions at the LPR of 1.0.
table S2. Total counts and normalized values from images of PMP22 reconstitutions compared to protein-free and KCNQ1 potassium channel voltage sensor domain (Q1-VSD) controls.
table S3. Total counts and normalized values from images of WT PMP22 reconstitutions at different LPRs.
table S4. Total counts and normalized values from images of reconstitutions of WT and Cys-less PMP22.
table S5. Total counts and normalized values from images of PMP22 reconstitutions containing GST-ECL1 and GST-ECL2.
table S6. Total counts and normalized values from images of PMP22 reconstitutions containing WT or GST.
table S7. Total counts and normalized values from images of reconstitutions containing only lipids and GST, GST-ECL1, or GST-ECL2.
table S8. Total counts and normalized values from images of reconstitutions of WT PMP22 compared to PMP22 constructs with mutations in conserved residues of ECL1 and ECL2.
table S9. Total counts and normalized values from images of reconstitutions of WT and the TrJ mutant PMP22 construct.
REFERENCES AND NOTES
- 1.Nave K. A., Werner H. B., Myelination of the nervous system: Mechanisms and functions. Annu. Rev. Cell Dev. Biol. 30, 503–533 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Scherer S. S., Arroyo E. J., Recent progress on the molecular organization of myelinated axons. J. Peripher. Nerv. Syst. 7, 1–12 (2002). [DOI] [PubMed] [Google Scholar]
- 3.S. Ramon y Cajal, Degeneration and Regeneration of the Nervous System (Oxford Univ. Press, 1928). [Google Scholar]
- 4.Revel J.-P., Hamilton D. W., The double nature of the intermediate dense line in peripheral nerve myelin. Anat. Rec. 163, 7–15 (1969). [DOI] [PubMed] [Google Scholar]
- 5.Hollingshead C. J., Caspar D. L. D., Melchior V., Kirschner D. A., Compaction and particle segregation in myelin membrane arrays. J. Cell Biol. 89, 631–644 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inouye H., Kirschner D. A., Evolution of myelin ultrastructure and the major structural myelin proteins. Brain Res. 1641, 43–63 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Jetten A. M., Suter U., The peripheral myelin protein 22 and epithelial membrane protein family. Prog. Nucleic Acid Res. Mol. Biol. 64, 97–129 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Adlkofer K., Martini R., Aguzzi A., Zielasek J., Toyka K. V., Suter U., Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat. Genet. 11, 274–280 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Carenini S., Neuberg D., Schachner M., Suter U., Martini R., Localization and functional roles of PMP22 in peripheral nerves of P0-deficient mice. Glia 28, 256–264 (1999). [PubMed] [Google Scholar]
- 10.Lupski J. R., An inherited DNA rearrangement and gene dosage effect are responsible for the most common autosomal dominant peripheral neuropathy: Charcot-Marie-Tooth disease type 1A. Clin. Res. 40, 645–652 (1992). [PubMed] [Google Scholar]
- 11.Li J., Parker B., Martyn C., Natarajan C., Guo J., The PMP22 gene and its related diseases. Mol. Neurobiol. 47, 673–698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger P., Niemann A., Suter U., Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease). Glia 54, 243–257 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Bertorini T., Narayanaswami P., Rashed H., Charcot-Marie-Tooth disease (hereditary motor sensory neuropathies) and hereditary sensory and autonomic neuropathies. Neurologist 10, 327–337 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Nicholson G. A., The dominantly inherited motor and sensory neuropathies: Clinical and molecular advances. Muscle Nerve 33, 589–597 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Pareek S., Notterpek L., Snipes G. J., Naef R., Sossin W., Laliberté J., Iacampo S., Suter U., Shooter E. M., Murphy R. A., Neurons promote the translocation of peripheral myelin protein 22 into myelin. J. Neurosci. 17, 7754–7762 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suter U., Scherer S. S., Disease mechanisms in inherited neuropathies. Nat. Rev. Neurosci. 4, 714–726 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Young P., Suter U., Disease mechanisms and potential therapeutic strategies in Charcot–Marie–Tooth disease. Brain Res. Brain Res. Rev. 36, 213–221 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Nelis E., Haites N., Van Broeckhoven C., Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum. Mutat. 13, 11–28 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Gabreels-Festen A. A., Bolhuis P. A., Hoogendijk J. E., Valentijn L. J., Eshuis E. J., Gabreëls F. J., Charcot-Marie-Tooth disease type 1A: Morphological phenotype of the 17p duplication versus PMP22 point mutations. Acta Neuropathol. 90, 645–649 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Madrid R., Bradley G., The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy): Studies on the formation of the abnormal myelin sheath. J. Neurol. Sci. 25, 415–448 (1975). [Google Scholar]
- 21.Behse F., Buchthal F., Carlsen F., Knappeis G. G., Hereditary neuropathy with liability to pressure palsies. Electrophysiological and histopathological aspects. Brain 95, 777–794 (1972). [DOI] [PubMed] [Google Scholar]
- 22.Robertson A. M., King R. H. M., Muddle J. R., Thomas P. K., Abnormal Schwann cell/axon interactions in the Trembler-J mouse. J. Anat. 190, 423–432 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dickson K. M., Bergeron J. J. M., Shames I., Colby J., Nguyen D. T., Chevet E., Thomas D. Y., Snipes G. J., Association of calnexin with mutant peripheral myelin protein-22 ex vivo: A basis for “gain-of-function” ER diseases. Proc. Natl. Acad. Sci. U.S.A. 99, 9852–9857 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fontanini A., Chies R., Snapp E. L., Ferrarini M., Fabrizi G. M., Brancolini C., Glycan-independent role of calnexin in the intracellular retention of Charcot-Marie-Tooth 1A Gas3/PMP22 mutants. J. Biol. Chem. 280, 2378–2387 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Naef R., Adlkofer K., Lescher B., Suter U., Aberrant protein trafficking in Trembler suggests a disease mechanism for hereditary human peripheral neuropathies. Mol. Cell. Neurosci. 9, 13–25 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Colby J., Nicholson R., Dickson K. M., Orfali W., Naef R., Suter U., Snipes G. J., PMP22 carrying the trembler or trembler-J mutation is intracellularly retained in myelinating Schwann cells. Neurobiol. Dis. 7, 561–573 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Naef R., Suter U., Impaired intracellular trafficking is a common disease mechanism of PMP22 point mutations in peripheral neuropathies. Neurobiol. Dis. 6, 1–14 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Schlebach J. P., Narayan M., Alford C., Mittendorf K. F., Carter B. D., Li J., Sanders C. R., Conformational stability and pathogenic misfolding of the integral membrane protein PMP22. J. Am. Chem. Soc. 137, 8758–8768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan M. C., Shooter E. M., Notterpek L., Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol. Dis. 10, 109–118 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Tobler A. R., Liu N., Mueller L., Shooter E. M., Differential aggregation of the Trembler and Trembler J mutants of peripheral myelin protein 22. Proc. Natl. Acad. Sci. U.S.A. 99, 483–488 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Notterpek L., Ryan M. C., Tobler A. R., Shooter E. M., PMP22 accumulation in aggresomes: Implications for CMT1A pathology. Neurobiol. Dis. 6, 450–460 (1999). [DOI] [PubMed] [Google Scholar]
- 32.D’Urso D., Prior R., Greiner-Petter R., Gabreëls-Festen A. A., Müller H. W., Overloaded endoplasmic reticulum–Golgi compartments, a possible pathomechanism of peripheral neuropathies caused by mutations of the peripheral myelin protein PMP22. J. Neurosci. 18, 731–740 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fortun J., Dunn W. A. Jr., Joy S., Li J., Notterpek L., Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 23, 10672–10680 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fortun J., Li J., Go J., Fenstermaker A., Fletcher B. S., Notterpek L., Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J. Neurochem. 92, 1531–1541 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Wilson C. H., Hartline D. K., Novel organization and development of copepod myelin. ii. nonglial origin. J. Comp. Neurol. 519, 3281–3305 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Hoglund G., Ringertz H., X-ray diffraction studies on peripheral nerve myelin. Acta Physiol. Scand. 51, 290–295 (1961). [DOI] [PubMed] [Google Scholar]
- 37.Parsons D. F., Akers C. K., Neutron diffraction of cell membranes (myelin). Science 165, 1016–1018 (1969). [DOI] [PubMed] [Google Scholar]
- 38.Chandross R. J., Bear R. S., Montgomery R. L., An X-ray diffraction comparison of myelins from the human nervous system. J. Comp. Neurol. 177, 1–9 (1978). [DOI] [PubMed] [Google Scholar]
- 39.Shapiro L., Doyle J. P., Hensley P., Colman D. R., Hendrickson W. A., Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron 17, 435–449 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Huxley C., Passage E., Robertson A. M., Youl B., Huston S., Manson A., Sabéran-Djoniedi D., Figarella-Branger D., Pellissier J. F., Thomas P. K., Fontés M., Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum. Mol. Genet. 7, 449–458 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Huxley C., Passage E., Robertson A. M., Youl B., Huston S., Manson A., Sabéran-Djoniedi D., Figarella-Branger D., Pellissier J. F., Thomas P. K., Fontés M., Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum. Mol. Genet. 5, 563–569 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Mittendorf K. F., Kroncke B. M., Meiler J., Sanders C. R., The homology model of PMP22 suggests mutations resulting in peripheral neuropathy disrupt transmembrane helix packing. Biochemistry 53, 6139–6141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasse B., Bosse F., Hanenberg H., Müller H. W., Peripheral myelin protein 22 kDa and protein zero: Domain specific trans-interactions. Mol. Cell. Neurosci. 27, 370–378 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Kitamura K., Uyemura K., Shibuya K., Sakamoto Y., Yoshimura K., Nomura M., Structure of a major oligosaccharide of PASII/PMP22 glycoprotein in bovine peripheral nerve myelin. J. Neurochem. 75, 853–860 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Suter U., Welcher A. A., Özcelik T., Snipes G. J., Kosaras B., Francke U., Billings-Gagliardi S., Sidman R. L., Shooter E. M., Trembler mouse carries a point mutation in a myelin gene. Nature 356, 241–244 (1992). [DOI] [PubMed] [Google Scholar]
- 46.Valentijn L. J., Baas F., Wolterman R. A., Hoogendijk J. E., van den Bosch N. H. A., Zorn I., Gabreëls-Festen A. A. W. M., de Visser M., Bolhuis P. A., Identical point mutations of PMP–22 in Trembler–J mouse and Charcot–Marie–Tooth disease type 1A. Nat. Genet. 2, 288–291 (1992). [DOI] [PubMed] [Google Scholar]
- 47.Sakakura M., Hadziselimovic A., Wang Z., Schey K. L., Sanders C. R., Structural basis for the Trembler-J phenotype of Charcot-Marie-Tooth disease. Structure 19, 1160–1169 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mendz G. L., Moore W. J., Kaplin I. J., Cornell B. A., Separovic F., Miller D. J., Brown L. R., Characterization of dodecylphosphocholine/myelin basic protein complexes. Biochemistry 27, 379–386 (1988). [DOI] [PubMed] [Google Scholar]
- 49.Gould R. M., Oakley T., Goldstone J. V., Dugas J. C., Brady S. T., Gow A., Myelin sheaths are formed with proteins that originated in vertebrate lineages. Neuron Glia Biol. 4, 137–152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mobius W., Patzig J., Nave K.-A., Werner H. B., Phylogeny of proteolipid proteins: Divergence, constraints, and the evolution of novel functions in myelination and neuroprotection. Neuron Glia Biol. 4, 111–127 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Simske J. S., Köppen M., Sims P., Hodgkin J., Yonkof A., Hardin J., The cell junction protein VAB-9 regulates adhesion and epidermal morphology in C. elegans. Nat. Cell Biol. 5, 619–625 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Niemann S., Sereda M. W., Suter U., Griffiths I. R., Nave K.-A., Uncoupling of myelin assembly and Schwann cell differentiation by transgenic overexpression of peripheral myelin protein 22. J. Neurosci. 20, 4120–4128 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohsawa Y., Murakami T., Miyazaki Y., Shirabe T., Sunada Y., Peripheral myelin protein 22 is expressed in human central nervous system. J. Neurol. Sci. 247, 11–15 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Roux K. J., Amici S. A., Notterpek L., The temporospatial expression of peripheral myelin protein 22 at the developing blood-nerve and blood-brain barriers. J. Comp. Neurol. 474, 578–588 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Roux K. J., Amici S. A., Fletcher B. S., Notterpek L., Modulation of epithelial morphology, monolayer permeability, and cell migration by growth arrest specific 3/peripheral myelin protein 22. Mol. Biol. Cell 16, 1142–1151 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo J., Wang L., Zhang Y., Wu J., Arpag S., Hu B., Imhof B. A., Tian X., Carter B. D., Suter U., Li J., Abnormal junctions and permeability of myelin in PMP22-deficient nerves. Ann. Neurol. 75, 255–265 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palaniyar N., Semotok J. L., Wood D. D., Moscarello M. A., Harauz G., Human proteolipid protein (PLP) mediates winding and adhesion of phospholipid membranes but prevents their fusion. Biochim. Biophys. Acta 1415, 85–100 (1998). [DOI] [PubMed] [Google Scholar]
- 58.Bakhti M., Aggarwal S., Simons M., Myelin architecture: Zippering membranes tightly together. Cell. Mol. Life Sci. 71, 1265–1277 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yool D. A., Edgar J. M., Montague P., Malcolm S., The proteolipid protein gene and myelin disorders in man and animal models. Hum. Mol. Genet. 9, 987–992 (2000). [DOI] [PubMed] [Google Scholar]
- 60.Shy M. E., Peripheral neuropathies caused by mutations in the myelin protein zero. J. Neurol. Sci. 242, 55–66 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Arroyo E. J., Scherer S. S., On the molecular architecture of myelinated fibers. Histochem. Cell Biol. 113, 1–18 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Furuse M., Fujita K., Hiiragi T., Fujimoto K., Tsukita S., Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 141, 1539–1550 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Furuse M., Sasaki H., Fujimoto K., Tsukita S., A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 143, 391–401 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Furuse M., Tsukita S., Claudins in occluding junctions of humans and flies. Trends Cell Biol. 16, 181–188 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Angelow S., Ahlstrom R., Yu A. S. L., Biology of claudins. Am. J. Physiol. Renal Physiol. 295, F867–F876 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Notterpek L., Roux K. J., Amici S. A., Yazdanpour A., Rahner C., Fletcher B. S., Peripheral myelin protein 22 is a constituent of intercellular junctions in epithelia. Proc. Natl. Acad. Sci. U.S.A. 98, 14404–14409 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jo E., Boggs J. M., Aggregation of acidic lipid vesicles by myelin basic protein: Dependence on potassium concentration. Biochemistry 34, 13705–13716 (1995). [DOI] [PubMed] [Google Scholar]
- 68.ter Beest M. B. A., Hoekstra D., Interaction of myelin basic protein with artificial membranes. Parameters governing binding, aggregation and dissociation. Eur. J. Biochem. 211, 689–696 (1993). [DOI] [PubMed] [Google Scholar]
- 69.Hu J., Prinz W. A., Rapoport T. A., Weaving the web of ER tubules. Cell 147, 1226–1231 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stachowiak J. C., Schmid E. M., Ryan C. J., Ann H. S., Sasaki D. Y., Sherman M. B., Geissler P. L., Fletcher D. A., Hayden C. C., Membrane bending by protein–protein crowding. Nat. Cell Biol. 14, 944–949 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Patzig J., Kusch K., Fledrich R., Eichel M. A., Lüders K. A., Möbius W., Sereda M. W., Nave K.-A., Martini R., Werner H. B., Proteolipid protein modulates preservation of peripheral axons and premature death when myelin protein zero is lacking. Glia 64, 155–174 (2016). [DOI] [PubMed] [Google Scholar]
- 72.D’Urso D., Ehrhardt P., Muller H. W., Peripheral myelin protein 22 and protein zero: A novel association in peripheral nervous system myelin. J. Neurosci. 19, 3396–3403 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mobley C. K., Myers J. K., Hadziselimovic A., Ellis C. D., Sanders C. R., Purification and initiation of structural characterization of human peripheral myelin protein 22, an integral membrane protein linked to peripheral neuropathies. Biochemistry 46, 11185–11195 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Myers J. K., Mobley C. K., Sanders C. R., The peripheral neuropathy-linked Trembler and Trembler-J mutant forms of peripheral myelin protein 22 are folding-destabilized. Biochemistry 47, 10620–10629 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schlebach J. P., Peng D., Kroncke B. M., Mittendorf K. F., Narayan M., Carter B. D., Sanders C. R., Reversible folding of human peripheral myelin protein 22, a tetraspan membrane protein. Biochemistry 52, 3229–3241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peng D., Kim J.-H., Kroncke B. M., Law C. L., Xia Y., Droege K. D., Van Horn W. D., Vanoye C. G., Sanders C. R., Purification and structural study of the voltage-sensor domain of the human KCNQ1 potassium ion channel. Biochemistry 53, 2032–2042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ohi M., Li Y., Cheng Y., Walz T., Negative staining and image classification—Powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hampton C. M., Strauss J. D., Ke Z., Dillard R. S., Hammonds J. E., Alonas E., Desai T. M., Marin M., Storms R. E., Leon F., Melikyan G. B., Santangelo P. J., Spearman P. W., Wright E. R., Correlated fluorescence microscopy and cryo-electron tomography of virus-infected or transfected mammalian cells. Nat. Protoc. 12, 150–167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kremer J. R., Mastronarde D. N., McIntosh J. R., Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 80.Mastronarde D. N., Dual-axis tomography: An approach with alignment methods that preserve resolution. J. Struct. Biol. 120, 343–352 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700220/DC1
movie S1. Cryo-ET of an MLA showing that it is composed of compressed wrapped vesicles (corresponding to Fig. 3B).
movie S2. Cryo-ET of an MLA showing that it is composed of compressed wrapped vesicles (corresponding to Fig. 3C).
movie S3. Cryo-ET of an MLV showing that it is composed of nested vesicles rather than compressed wrapped vesicles seen in MLAs (corresponding to fig. S3B).
fig. S1. Representative examples of objects observed in reconstitution experiments by negative stain.
fig. S2. Examples of interperiod repeat distance measurements taken for MLAs and MLVs imaged with cryo-EM.
fig. S3. MLV vesicles examined by cryo-ET.
fig. S4. Schematic showing the organization of PMP22.
fig. S5. Addition of GST does not reduce MLA formation.
table S1. Total counts, percentages, and SDs of WT PMP22 reconstitutions at the LPR of 1.0.
table S2. Total counts and normalized values from images of PMP22 reconstitutions compared to protein-free and KCNQ1 potassium channel voltage sensor domain (Q1-VSD) controls.
table S3. Total counts and normalized values from images of WT PMP22 reconstitutions at different LPRs.
table S4. Total counts and normalized values from images of reconstitutions of WT and Cys-less PMP22.
table S5. Total counts and normalized values from images of PMP22 reconstitutions containing GST-ECL1 and GST-ECL2.
table S6. Total counts and normalized values from images of PMP22 reconstitutions containing WT or GST.
table S7. Total counts and normalized values from images of reconstitutions containing only lipids and GST, GST-ECL1, or GST-ECL2.
table S8. Total counts and normalized values from images of reconstitutions of WT PMP22 compared to PMP22 constructs with mutations in conserved residues of ECL1 and ECL2.
table S9. Total counts and normalized values from images of reconstitutions of WT and the TrJ mutant PMP22 construct.