

Abstract
Bone is a major organ in the skeletal system that supports and protects muscle and other organs, facilitates movement and hematopoiesis, and forms a reservoir of minerals including calcium. The cells in the bone, such as osteoblasts, osteoclasts, and osteocytes, orchestrate sequential and balanced regulatory mechanisms to maintain bone and are capable of differentiating in bones. Bone development and remodeling require a precise regulation of gene expressions in bone cells, a process governed by epigenetic mechanisms such as histone modification, DNA methylation, and chromatin structure. Importantly, lineage-specific transcription factors can determine the epigenetic regulation of bone cells. Emerging data suggest that perturbation of epigenetic programs can affect function and activity of bone cells and contributes to pathogenesis of bone diseases, including osteoporosis. Thus, understanding epigenetic regulations in bone cells would be important for early diagnosis and future therapeutic approaches.
Keywords: epigenetic regulation, osteoclasts, osteoblasts, Histone modification, DNA methylation
Introduction
Epigenetics refers to the changes in gene expression or phenotype without alterations in DNA sequence [1]. Epigenetic regulation controls gene expression by chromatin regulators that bind to DNA, by modifications of chromatin such as histone modification and DNA methylation, or by non-coding RNAs. Increasing evidence suggests that epigenetic regulation plays an important role in physiological and pathological conditions. It has become clear that epigenetic regulation is a key molecular mechanism by which environmental influences and cues are imprinted on DNA/chromatin and determine patterns of gene expression, responses to environmental challenges, and disease causation and pathogenesis [2].
Bone is a dynamic tissue that undergoes constant remodeling and controls skeletal development and regeneration. Bone remodeling is a complex process whereby old, mature bone is removed by bone resorption and is replaced by new bone formation. Bone remodeling is required for bone homeostasis including maintaining appropriate bone cell physiology and bone microarchitecture. It is tightly regulated by a balance between bone resorption by osteoclasts and bone formation by osteoblasts (Figure 1). Osteoblasts are bone-forming cells and are derived from mesenchymal stem cells (MSCs) [3]. Various extrinsic regulators and corresponding transcription factors important for osteogenic differentiation of MSCs have been well characterized. Osteoblasts not only play a central role in bone formation by synthesizing multiple bone matrix proteins, but they also regulate osteoclast maturation and bone resorption by secreting soluble factors and by direct cell-cell interactions. Osteoclasts are bone-resorbing cells important for bone homeostasis and pathological bone resorption [4–8]. Osteoclasts are differentiated from hematopoietic stem cells (HSCs) of the myeloid lineage under influence of M-CSF and RANKL. Various autocrine and paracrine factors modulate bone cell differentiation and function. One such factor is RANKL which is produced by several cell types, including osteoblasts, activated T cells, and osteocytes. RANKL binds to its receptor, RANK that is expressed in osteoclast-lineage cells and the RANKL-RANK interaction activates signaling pathways that induce NFATc1, a master transcription factor of osteoclast differentiation. OPG is secreted by osteoblasts and other cell types and functions as a decoy receptor by binding to RANKL, subsequently preventing the activation of RANK. Thus, OPG is a key negative regulator of osteoclastogenesis, a property that has already been therapeutically exploited. In addition to RANK/RANKL/OPG, cytokines from immune cells also influence osteoclast differentiation and function; and various proteins have been shown to control the bone integrity.
Figure 1. The process of bone remodeling and epigenetic factors.
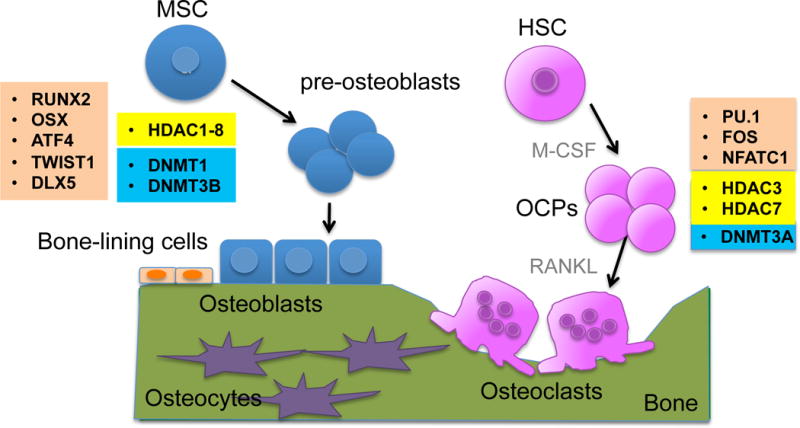
Bone is maintained by bone remodeling which is a continuous cycle of bone resorption and bone formation. Bone remodeling is a coupled and balanced process and takes place in basic multicellular units. In a resting stage, the non-remodeling bone surface is covered by bone lining cells. Osteocytes sense bone damages and recruit osteoclast precursor cells (OCPs), which further differentiate into mature osteoclasts. Osteoclasts resorb a damaged or old bone matrix and subsequently osteoblasts are recruited into resorbed sites and form new bone. Osteoclasts are differentiated from hematopoietic stem cells (HSCs). M-CSF and RANKL are key drivers of osteoclastogenesis. M-CSF enables HSCs to differentiate into OCPs which express RANK, a receptor for RANKL. RANKL further promotes OCPs into mature osteoclasts. Mesenchymal stem cells (MSCs) differentiate into osteoblasts and classical coupling factors, which are released from resorbed bone matrix or produced by osteoclasts, promote osteogenic differentiation through the osteoblast lineage. Key transcription factors (orange boxes), epigenetic enzymes that participate in DNA methylation (blue boxes), and histone deacetylases (yellow boxes) are indicated and described in the text.
Over the past years, substantive progress in epigenetic research on osteoclasts, osteoblasts, and their differentiation from hematopoietic stem cells and mesenchymal stem cells (MSCs) respectively has been made, highlighting the importance of epigenetic regulation in bone remodeling. The impairment of bone remodeling is associated with many skeletal diseases such as osteoporosis and, to date, anti-resorptive or osteoanabolic treatments have been developed but all have significant side effects limiting their prolonged use. Investigating epigenetic regulations may provide novel treatment targets for promoting bone formation and suppressing excessive bone resorption. Understanding the epigenetic regulation during physiological bone remodeling further provides insight into clinical and mechanistic characterizations of bone metabolism and into pathophysiology of bone diseases. Although osteocytes are recognized as important regulators of bone turnover, this review summarizes current knowledge of epigenetic mechanisms in relation to the mechanisms regulating the differentiation of osteoclasts and osteoblasts, and how these regulations can be applied to therapies for bone diseases.
Epigenetic regulation
A notable feature of multicellular organisms is their capacity to create functionally unique cell types from the same genome sequence that is shared by all cells in the organism. This diversity results from the capacity of individual cell types to initiate and then maintain specific gene expression patterns during development. To achieve this, cellular signaling events are thought to regulate the activity of cell type-specific DNA-binding transcription factors that function as master regulators of gene expression networks. Recently, epigenetic regulation has also been identified as an important factor that is involved in the regulation of gene expression and cell fate decision.
Due to their unique properties of self-renewal and their ability to undergo multilineage differentiation in response to appropriate signaling cues, pluripotent embryonic stem cells (ESCs) are widely used to study epigenetic mechanisms and show various characteristic epigenetic phenotypes based on prevalence of different histone markers [9]. A public research consortium named the Encyclopedia Of DNA Elements (ENCODE) [10] and the Epigenomics Roadmap [11] carried out a project to identify all functional elements in the human genome sequence and generated an epigenomic map. This data allows researchers to understand molecular and cellular processes of particular cell types and to elucidate fundamental processes of human diseases at a genomic level. Extensive epigenomic data sets of human osteoblasts are currently available in ENCODE database and the chromatin landscape of human osteoblasts has been shown by studies of ENCODE consortium.
Beyond the sequences of the genome, a large regulatory network controls cell specific identity and function of cells. Each cell type has its own unique chromatin structure and organization. Whereas mRNA expression profile indicates the current state of a cell, epigenomic regulation can give perspectives of history of cell states. Advanced high throughput sequencing makes the genome-wide analysis of chromatin profiling feasible, resulting in generating a profiling of the epigenome [12]. As chromatin patterns are related to underlying regulatory processes, chromatin profiling enables identification of the regulatory network in bone cells. Increasing knowledge of regulatory network in bone cells will reveal a cell-type specific chromatin structure and organization leading to affecting cell-specific gene regulation.
Transcription factor network
Transcription factors bind to specific DNA sequences (called binding motif) in promoter or enhancer regions to regulate gene expression. Transcription factor networks and transcriptional processes help shape epigenetic regulation as well as chromatin structure [13]. However, transcription factor binding to the promoter is insufficient to fully regulate transcription and the interaction between the promoter and the regulatory elements, called enhancers, provide another level of dynamic regulation of transcription [14]. Unlike other cells in mammals, interaction with bone and bone marrow environment would be important factors to control the gene expression profile in bone cells [15, 16].
Molecular and biochemical studies reveal principal regulators of transcription in bone cells, while ChIP-based assays have mapped the binding sites of many important transcription factors. During osteoblastogenesis, both the transcriptome and the epigenome of osteoblast lineage cells undergo dynamic changes [17–21]. Several transcription factors are known to control bone development and osteoblast differentiation [22]. Runt-related transcription factor 2 (RUNX2, Cbfa1) is a master transcription factor of osteoblasts and is indispensible in all stages of osteoblast differentiation [23]. RUNX2 ablation causes bone defects in human and mouse [24–26]. RUNX2 regulates gene expression during osteoblastogenesis and detailed epigenetic regulation has been identified for RUNX2-mediated gene expression [27–29]. In MC3T3-E1 osteoblast-like cells, ChIP-sequencing demonstrated that Runx2 occupancy is actively regulated during osteoblast differentiation [19]. Other transcription factors have been identified to control osteoblast differentiation, including Osterix (OSX)[30], ATF4[31], twist homolog 1 (TWIST1)[32], and distal-less homeobox 5 (DLX5)[33].
Various transcription factors coordinate a broad spectrum of gene expression programs in osteoclastogenesis [34]. NFATc1 is a master transcriptional factor of osteoclastogenesis and drives the early stages of osteoclast differentiation [35–37]. NFATc1-deficient mice develop severe osteopetrosis in vivo [38], and NFATc1-deficient cells are incapable of differentiating into osteoclasts in vitro [36, 37, 39]. However, it has been shown that NFATc1 forms a complex with osterix and both are required for osteoblastic bone formation [40]. This data suggests that NFATc1 cooperatively regulates osteoclastogenesis by interacting with other transcription factors. c-Fos is another essential transcription factor for osteoclastogenesis. c-Fos belongs to AP-1 family and c-Fos-deficient cells can differentiate into macrophages but not into osteoclasts, suggesting the role of c-Fos in osteoclastogenesis [41, 42]. The cis-regulatory elements enriched with binding sites of transcription factor and the lineage determining transcription factors play an important role in pioneering activity of transcription. Following transcriptional program sets cell-specific gene expression program and establishes dynamic epigenetic states. Therefore, the complicated interaction between transcription factors and epigenetic regulation endow cell identify to bone cells.
Histone modification
The basic unit of chromatin is the nucleosome[43]. Each nucleosome consists of a short (around 147 base pair) DNA segment wrapped around a histone octamer that comprises two copies each of core histones: H2A, H2B, H3 and H4, as shown figure 2A. The N-terminal tails of core histones are subject to various post-translational modifications by chromatin remodelers [44, 45]. More than eight different histone modifications are identified, including acetylation, methylation, phosphorylation, ubiquitination, and sumolylation. A large-scale mapping of histone modifications and related chromatin structure enables the characterization and determination of functional consequences of changes in chromatin structure [46, 47]. Currently more than 14,000 data sets are deposited in public domain such as Gene Expression Omnibus (GEO), allowing computational integration of different data sets. These integrated approaches can identify novel interactions between histone modification and genomic elements and supports the idea that a different combination of histone modifications serves as a “switch” to fine-tune genomic elements [48, 49]. Unique patterns of histone modifications are found in different genomic elements such as promoters, gene bodies, enhancers, and chromatin insulators (boundary elements) and reflect the status of transcription (Figure 2B). For example, active promoters are enriched in histone H3 lysine 4 dimethylation (H3K4me2), H3 lysine trimethylation (H3K4me3), histone acetylation (H3Ac and H4 Ac), and H2A.Z.
Figure 2.
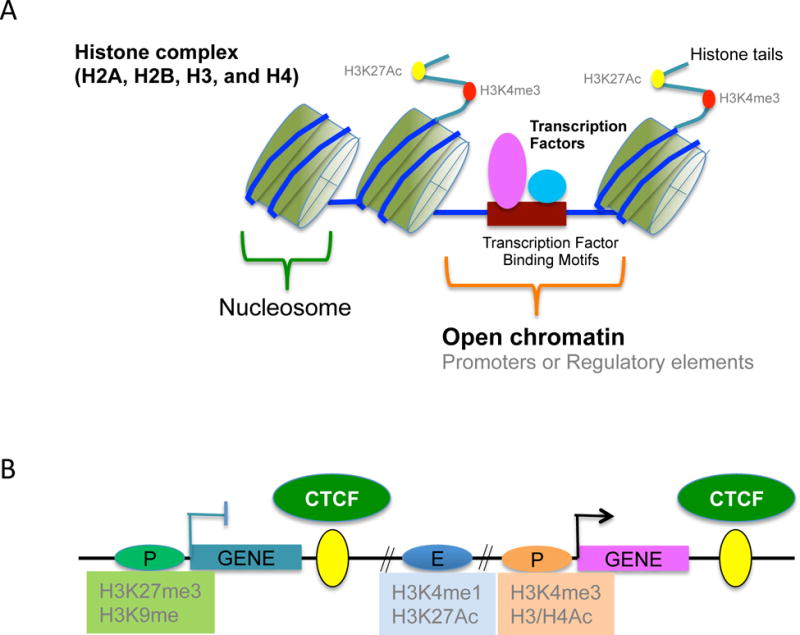
A. The core proteins of nucleosomes are two copies of H2A, H2B, H3, and H4 and 146 base pairs of DNA wraps around these core histone octamer. Core histones are highly conserved and have amino-terminal tails which are subject to various post-translational modifications. Histone modifications, such as methylation and acetylation, play an important role in gene expression and active promoter regions are distinguished by specific histone modifications including H3K4me3 and H3K27Ac. Nucleosomes are depleted in highly active regions, called “open chromatin”. Transcription factors and lineage determining factors such as NFAT and RUNX2 bind to a specific binding motif in promoters or enhancers. Ac, acetylation; Me, methylation.
B. Histone modifications of functional elements including promoters, enhancers, and insulators. Insulator is a genetic boundary element that blocks the interaction between enhancers and promoters and is defined by CCCTC-binding factor (CTCF) binding although CTCF has dual effects on enhancers; either blocking or activating. Active promoters are enriched for H3K4me3 and H3/H4 acetylation. H3K9 me2/3 and H3K27me3 are associated with repressed promoter regions. Active enhancers are marked by H3Kme1 and H3K27Ac.
Histone modifications are dynamically regulated and have an influence on chromatin structure and function [45]. Charting different combination of histone modifications in the chromatin can predict the stage of transcription at a specific gene [49]. Histone modification is mediated by three components: writers, erasers, and readers. Histone acetylation is an important determinant in multiple chromatin-dependent processes, including gene expression, DNA replication, and DNA damage repair. Acetylation is generally associated with elevated gene expression and open chromatin by reducing a positive charge of histone. The N-ε-lysine acetylation and deacetylation of histones are also controlled by three groups of enzymes: histone acetyltransferase (HAT, reviewed [50]), histone deacetylase (HDAC, reviewed [51]), and histone acetylation readers (reviewed [52]). Histone acetylation by HATs plays a key role in transcriptional activation, whereas deacetylation of histones promotes transcriptional repression and silencing of genes. However, dysregulated acetylation of histone is often associated with the development of a wide variety of human cancers. An excessive level of histone acetylation induces apoptotic cell death, whereas extensive histone deacetylation has been linked to the repression of tumor regulatory genes promoting cancer pathologies. Recruitment of HATs and subsequent histone acetylation are dynamically regulated during the differentiation of bone cells [53, 54] and intervening histone acetylation by small molecule inhibitors results in altering the differentiation of bone cells.
Due to the importance of histone acetylation, various small molecule inhibitors which target HATs or HDACs have been developed and contain therapeutic potential for various diseases. HDACs(Histone deacetylases) function as “erasers” which remove an acetyl group from histones and also have activity on non-histone proteins [55]. HDACs are comprised of three classical classes (I, II, and IV) that have 11 HDACs and class III (sirtuins) that containing NAD-dependent catalytic sties. HDAC inhibitors (HDACi) cause an increase in the acetylated level of histones, which in turn promotes the re-expression of the silenced regulatory genes in cancer cells and reverse the malignant phenotype and thus HDAC inhibitors have recently emerged as potential cancer therapeutic agents. Two HDAC inhibitors, vorinostat and romidepsin, have received an approval from the US FDA for the treatment of cancer and certain neurological conditions.
Many studies testing the effect of HDAC inhibitors (HDACi; TSA, SAHA, MS-275, sodium butyrate, and valproic acid) on osteoblasts and osteoclasts have been documented, implicating that downregulation or deletion of HDACs may play a role in the differentiation of osteoclasts and osteoblasts. Histone acetylation of RUNX2 is one of key regulatory mechanisms for RUNX2-mediated gene programs [56]. HDAC inhibitors show positive effects on in vitro osteoblast differentiation and maturation, in part, mediated by promoting RUNX2 function. The treatment with a HDAC inhibitor promotes osteoblastogenesis [57–63] and also increases genome-wide distribution of H4 acetylation [64]. A conditional deletion of HDAC3 in osteochondroprogenitor cells results in osteopenia and increases marrow adiposity [65]. Resveratrol or isonicotinamide is an agonist of Sirtuin 1, class III HDAC, and blocks spontaneous adipocyte differentiation and thereby promotes osteoblast differentiation and bone mineralization [66].
Although HDAC inhibitors suppress osteoclastogenesis [67], the deletion of individual HDAC shows differential effects on osteoclast differentiation depending on the class. The downregulation of HDAC3 by shRNA suppresses osteoclastogenesis [68]. However, Stemig et al shows that a conditional deletion of HDAC7 in osteoclasts causes osteopenia [69], suggesting that HDAC7 functions as an inhibitor of osteoclastogenesis. Although 24% of 41 patients with epilepsy who received HDACi, Valproate, experienced osteopenia, there was no significant correlation between HDACi treatment and bone density, suggesting bone metabolism may not be significantly affected by valproate monotherapy [70]. Thus, a clinical association between HDACi and bone metabolism is not clear yet.
The readers of histone acetylation contain bromo domain [71], a tandom PHD domain [72] that recognizs acetylated lysines [52]. The readers of histone modification are regulated by non-coding RNAs, binding partners and conformational changes of readers. After recognizing modified histones, the readers function as chromatin architectural proteins, chromatin remodelers, and chromatin modifiers and recruit other components to regulate transcription, repair, recombination, replication, and RNA processing [52]. Bromodomain and extra terminal domain (BET) family proteins are readers of acetylated histones as well as other acetylated proteins such as NF-kB and regulate gene expression [73]. BET proteins are often deregulated in cancers leading to an aberrant expression of proinflammatory cytokines and growth-promoting genes. Small molecule inhibitors of BET family proteins have demonstrated efficacy in treatment of cancer [73–79]. We and others reported that BET inhibitors, I-BET151 and JQ1, effectively suppress osteoclastogenesis, attenuate inflammatory bone loss, and protect mice from ovariectomy-induced bone loss as well as tumor metastasis induced osteolysis [54, 80]. Interestingly, BET inhibitors also suppress osteoblast differentiation but sensitivity to inhibitors of osteoblasts is lower than in osteoclasts [54, 80]. These results indicated that proper dosing of BET inhibitors can show beneficial effects on pathologic bone diseases, despite ubiquitous expression of BET proteins.
Histone methylation occurs on basic residues such as lysine and arginine and plays an important role in controlling gene expression. Histone methylation has been shown to display differential effects on transcription [81]. For example, trimethylation of H3K4, H3K36, and H3K79 positively control gene expression. In contrast, trimethylation of H3K27 and H3K9 and H4K20 is associated with repression. Protein methylation process was thought to be irreversible because the half-life of methylation is similar to that of histone. However, the discovery of histone demethylases had changed this paradigm to indicate that histone methylation is dynamically regulated. Two families of lysine demethylases are identified: the amine oxidases (LSD1[82]) and jumonji C (JmjC)-domain-containing family[83]. JMJD3 is a H3K27 demethylase [84]. Yasui et al has shown that RANKL stimulation removes trimethylation of H3K27, a repressive mark from the promoter of NFATC1 by, in part, inducing JMJD3, demonstrating the dynamic regulation of histone methylation during osteoclast differentiation [85]. JMJD3 expression is also induced during osteoblast differentiation and regulates the expression of bone-related genes including Runx2, osterix, osteopontin, bone sialoprotein (BSP), and osteocalcin (OCN) [86]. Consistently, Jmjd3−/− embryos exhibited open fontanells, less-mineralized cranial bones and hypoplastic clavicles compared to wild type littermates at E18.5, suggesting that JMJD3 is required for osteoblast maturation during both intramembraneous and endochondral bone formation[87].
The recruitment of histone demethylases is mediated by specific DNA sequences such as PRE (polycomb responsive element), cofactors, and DNA-binding factors. Polycomb proteins are transcriptional repressors essential for maintaining tissue specific gene expression program and consist of two groups, polycomb repressive complex 1 (PRC1) and polycomb repressive complex 2 (PRC2) [88]. PRC1 and PRC2 target promoters and are responsible for methylation of H3K27 that is known as a repressive epigenetic mark and mediates transcriptional repression in cancer cells [89]. PRC1-type complexes also contain an ubiquitin ligase activity for monoubiquitination of histone H2A to promote H3K27 trimethylation [90]. The enhancer of zeste 2 (EZH2) is a methyltransferase and the catalytic subunit of the PRC2 and trimethylates H3K27 and plays an essential role in the epigenetic repression [89, 91]. Wei et al report that EZH2 suppresses mesenchymal stem cells (MSCs) from differentiating into osteoblasts[92]. Recently, Dudakovic et al demonstrate the expression profiles of epigenetic regulators during osteogenic differentiation of human mesenchymal cells derived from the stromal vascular fraction of adipose tissue (AMSCs) and show that EZH2 is downregulated during osteoblastogenesis [64]. Whereas EZH2 inhibition suppresses adipogenic differentiation, EZH2 functions as a repressor of osteoblastogenesis, supported by experiments using EZH2 inhibitor, siRNAs and knock-out mice. WD repeat domain 5 (WDR5) is another histone methyltransferase and is a component of the mixed lineage leukemia (MLL) complex, which methylates lysine 4 of histone H3. In contrast to EZH2, WDR5 accelerates osteoblast differentiation [93–95]. Osteoblasts with reduced expression of WDR5 exhibit persistent acceleration of osteoblast differentiation through the activation of the canonical Wnt pathway. Altogether, dynamic regulation of histone methylases and demethylases is required for skeletal development and histone lysine methylation plays an important role in bone remodeling by controlling the differentiation of both osteoclasts and osteoblasts although their specific function needs to be further clarified.
Open Chromatin
In order for transcripton factors to access and interact with genomic regulatory elements, local chromatin needs to undergo rearrangement. Nucleosomes are able to change their location and are depleted in highly active regulatory regions, called “open chromatin”[96]. DNA in open chromatin regions has higher accessibility to proteins including transcription factors and open chromatin has a signature of active histone modification. Combined with next-generation sequencing, methods for searching genome-wide DNA accessibility have proven extremely effective in identifying regulatory elements across a variety of cell types [96, 97] and quantifying changes that lead to both activation or repression of gene expression. Open chromatin has been first identified by its sensitivity to cleavage by non-specific nucleases such as deoxyribonuclease I (DNase I) and micrococcal nuclease (MNase) at a genome-wide level [98]. Several methods to map open chromatin are developed including formaldehyde-assisted isolation of regulatory elements (FAIRE)[99], and sonication of cross-linked chromatin sequencing (Sono-seq) [100]. Assay for transposase-accessible chromatin using sequencing (ATAC-seq) is a newly developed method to detect open chromatin faster and with a higher sensitivity and applying the preferential integration of transposons into nucleosome-depleted region to interrogate chromatin structure using Tn5 transposase[101]. Measuring open chromatin is a powerful tool for identifying regulatory elements including transcription factors and enhancers. Binding of proteins, such as transcription factors to genomic DNA, protects the underlying sequences from cleavage by DNase I. DNase I footprinting assay allows identification of transcription factor-DNA interaction at single base pair resolution and provide information on regulatory elements [102–104]. Identification of open chromatin regions further allows predicting cell-type specific function by patterning of chromatin accessibility [96].
Open chromatin regions have been identified in osteoblasts and osteoclasts. Tai et al performed DNase seq analysis during osteoblast differentiation including growth phase, matrix deposition stage, and mineralization stage (GEO55046) and identified a role of a long-range interaction between bone specific RUNX2 P1 promoter-Supt3h (suppressor of Ty 3 homolog) promoters in murine pre-osteoblastic MC3T3-E1 cells using DNase seq in combination with the ENCODE data set [105, 106]. Although these interactions are detected in cells in which RUNX2 is silent, the interaction frequency between RUNX2-Supt3h is significantly increased during osteoblastogenesis as shown by chromosome conformation capture (3C) analysis, suggesting Supt3h as a potential regulator of bone specific RUNX2 P1 promoter. DNase-seq analysis also detected that RANKL facilitates the dynamic changes of chromatin accessibility in RAW 264.7 cells, a murine monocytic cell line that is capable of differentiating into osteoclasts upon RANKL stimulation [107]. Using motif discovery analysis, Inoue et al further profiled the transcription motifs in open chromatin regions and identified novel players for osteoclastogenesis in addition to other well-defined osteoclastogenic transcription factors. Therefore, linking DNase seq footprinting analysis with cell-specific gene expression profiles will expand our knowledge of a complicated transcriptomic network of bone cells.
DNA methylation
DNA methylation is a reversible and covalent modification of the 5′-carbon of cytosine residue with addition of methyl group from SAM and occurs mostly within CpG dinucleotides that cluster as a region called CpG islands in mammal [108, 109]. DNA methylation can be mapped at base-pair resolution with bisulfite sequencing [110]. In general, DNA methylation establishes the pattern of a repressive state of gene expression by maintaining a stable condensed chromatin configuration. DNA methylation is preserved during DNA replication and mitosis and thus a repressive state of genome can be inherited into the next generation. Local deposition and removal of DNA methylation is tightly coupled with occupancy of transcription factors [111]. It has been suggested that DNA methylation-mediated silencing plays a role in the expression of important osteogenic transcriptional factors. During osteoblast differentiation, osteocalcin expression is increased but methylation in the promoter of osteocalcin is significantly decreased. However, CpG methylation of osteocalcin promoter does not alter binding of RUNX2, basal transcriptional activity, or vitamin-D mediated enhancement of OC genes, supporting an indirect association between OC promoter and CpG methylation [112]. Hagh et al has shown that the methylation status of promoters of RUNX2, DLX5 and BSP are not changed during osteoblastic differentiation. In contrast, OSX promoter showed a dynamic change in its methylation pattern [113]. DNA methylation is also not overlapped with active and open chromatin architecture, supporting a negative correlation between H3K4me3 and DNA methylation [114].
Enzymes responsible for DNA methylation are DNA methyltransferases (DNMTs), which consist of DNMT1, DNMT3a, and DNMT3b. DNMT3a and 3b are de novo DNA methyltransferases and transfer methyl groups to unmethylated sites. Mice with DNMT deficiencies are embryonic lethal with reduced DNA methylation [115, 116]. These studies reveal that DNA methylation is essential for mammalian development, suggesting methylation patterns are pre-deposited during development. Genome-wide sequencing in wild-type mouse ESCs reveals that most methylated genes are differentiation-associated genes, which are repressed in mouse ESCs. The cell-specific roles of DNMT3a and 3b have been investigated, showing that DNMTs have a large impact on bone cells and the importance of DNMTs in skeletal biology. Recently, Nishikawa et al has demonstrated that DNMT3a is needed for osteoclast differentiation and bone resorption by suppressing IRF8, a negative regulator of osteoclast differentiation. DNMT3a suppresses IRF8 via increased methylation on distal regulatory elements of IRF8; methylation was promoted by increased S-Adenosyl methionine (SAM) concentration. Both conditional osteoclast-specific DNMT3a deletion and treatment of mice with TF-3, a DNMT3 inhibitor, protected mice from ovariectomy-induced bone loss, supporting a potential role of epigenetic therapy in bone diseases. 5-aza-cytidine, an another DNMT inhibitor, inhibits differentiation of MSCs into osteoblasts, adipocytes, and chondrocytes [117], suggesting DNMT regulates the differentiation potential of MSCs. Consistently, oxidized adenosine (ADOX), an inhibitor of SAM-dependent methyltransferases suppresses osteoblast differentiation[118]. Bone mineral density and body weight decrease with aging by reduced DNMT1 activity [119] and differentially methylated regions are enriched in genes associated with cell differentiation and skeletal embryogenesis in bone samples from patients with osteoporotic hip fracture [120]. These data suggest that DNA methylation plays an important role in aging [117] and changes of DNA methylation can be linked to disease pathogenesis of age-associated bone diseases. In addition to DNMTs, the Ten-Eleven Translocation (TET) proteins are recently discovered 5-methylcytosine hydroxylases that convert 5-methylcytosine (5mc) to (5hmc). It has been suggested that 5-hydroxymethylcytosine (5hmC) plays a role as an early intermediate in active DNA demethylation [121]. Although there is no direct evidence of association of TET proteins with bone cells, the increase in AP activity and matrix mineralization in Ad-MSCs has been shown to be associated with an increased presence of 5hmC as well as with an increased TET2/3 gene expression [122]. DNA methylation changes during osteoclast differentiation are also highly associated with TET2-mediated demethylation and DNMT3b-mediated methylation [123]. Although DNA methylation is important for the differentiation of bone cells, the role of DNA methylation in bone cells remains elusive and needs further investigation.
Non-coding RNAs
Only 1–2% of the human genome is translated into protein [124, 125] and the remaining transcribed RNAs are not annotated into protein-coding genes. The transcripts from non-coding regions of genomes are called non-coding RNAs (ncRNAs)[126, 127]. Non-coding RNAs have essential roles in regulating gene expression as well as other ncRNAs both in cis and in trans. It has been shown that non-coding RNAs affect various biological processes during development and in some pathological conditions. Importantly, ncRNAs are expressed in a stage specific and cell specific manner and provide another view of transcriptional circuit. The major classes of ncRNAs are micro RNAs (miRNAs) and long-non-coding RNAs (lncRNAs). lncRNAs are non-coding transcripts length over 200 nucleotides long [127] and are involved in many biological functions [128]. More than 28,000 distinct lncRNA transcripts are currently identified and lncRNAs regulate gene expression by various mechanisms [129]. Large intergenic non-coding RNAs (lincRNAs) are the largest class of lncRNAs and other classes of lncRNAs are intronic lncRNA, sense lncRNA, and antisense lncRNA. Tong et al recently reported that lncRNA-DANCR expression is unregulated in circulating monocytes in patients with low-BMD and DANCR promotes the expression of inflammatory cytokines such as IL-6 and TNF [130]. LncRNA-DANCR directly interacts with EZH2 and suppresses RUNX2 expression and osteoblast differentiation [131]. Although lncRNAs have been shown to engage in various biological processes, the identity of lncRNAs in skeletal biology is not well known [132]. However, many studies about the function and regulation of miRNAs in skeletal biology have been reported (reviewed in [133, 134]). Thus, this review briefly discusses the recent prospective view of miRNAs in bone diseases.
MicroRNAs (miRNAs) are a class of short non-coding RNAs that mediate post-transcriptional silencing and are important for cell differentiation, embryonic organ development, and apoptosis [135–138]. miRNAs control the expression of more than 30% of genes and have profound effects on physiological and pathological conditions. miRNAs are transcribed as primary miRNAs (pri-miRNAs) containing a secondary loop structure, a 5-cap structure and a poly(A) tail [139]. Pri-mRNAs are processed to pre-miRNA by Drosha (nuclear RNase III) and cofactor, DGCR8. Dicer (cytosolic RNase III) sequentially processes pre-miRNAs to a mature form. miRNAs are regulated during bone development [140] and Dicer inactivation, which results in defects on miRNAs cleavage, regulates bone mass, suggesting that Dicer-generated miRNAs play an important role in osteoblastogenesis and osteoclastogenesis [141–144]. Whereas DICER deficiency inhibits both in vitro and in vivo osteoclastogenesis, CD11b-specific DICER null mice exhibit mild osteopetrosis [144]. Mizoguchi et al has shown that osteoclast-specific DICER deletion by cathepsin K cre also increases bone mass and, surprisingly, decreased osteoblastogenesis in addition to inhibition of osteoclastogenesis [143]. These studies point to the potential functions of miRNAs from osteoclasts on osteoblast differentiation. Indeed, miRNAs secreted from osteoclasts are delivered into osteoblasts and regulate osteoblast differentiation. miRNAs are regulated by important signaling pathways such as WNT, RANKL, and BMP in osteoclasts and osteoblasts and inversely control and target multiple signaling pathways in osteoclasts and osteoblasts. In addition to the in vitro studies that identified the regulation and function of miRNAs in osteoblast and osteoclast differentiation [145–148], several studies have examined the expression of miRNAs during bone development in vivo and skeletal diseases. Extracellular miRNAs have recently been found in a variety of body fluids including synovial fluids, plasma, serum, saliva, and urine. Emerging evidence shows that miRNAs can be used a diagnostic tool to detect skeletal diseases such as osteoporosis. Osteoporosis is characterized by low bone mass that results from imbalanced bone remodeling: high osteoclast activity and low osteoblast activity [149]. Bone fractures associated with osteoporosis can be predicted by measuring bone mineral density (BMD) using dual-energy X-ray absorptiometry (DXA) and clinical history [150, 151]. However, current methods have a low detection rate and thus improved fracture risk assessment methods are required. miRNAs can be extruded into extracellular space, supporting the idea that miRNAs may function as biomarkers. Several miRNAs are detected in the blood of osteoporosis patients, although the function of individual miRNAs is not well characterized in the pathogenesis of osteoporosis. A recent study shows that nine cell free miRNAs in the serum are significantly upregulated in patients with osteoporotic fractures, opening up a possibility of using miRNAs as a biomarker for diagnosing osteoporotic fracture [152]. In addition, abnormal miRNA expressions are often found in several diseases such as cancer, leading to the first miRNA mimics reaching the clinical phase I trial in patients with primary liver cancer in 2013 [153]. Since one miRNA can interact with many genes simultaneously, miRNA replacement therapy using synthetic miRNA mimics would be an option to treat diseases, if it can avoid off-target effects.
Conclusions
The crucial role of bone cells in bone homeostasis and in skeletal diseases makes them potential therapeutic targets. For example, understanding the osteogenic potential of MSCs can be used to treat bone defect repair and bone diseases[154]. Suppressing osteoclast activity or differentiation allows one to block pathological bone resorption. Epigenetics regulations were implicated in skeletal diseases by a few studies [120, 152], suggesting that epigenetic changes can be predisposited or used as a signature of skeletal diseases. Epigenetic regulation affects differentiation and activity of bone cells and the response of bone cells to exogenous stimulus. Understanding bone cells-specific epigenetic regulation using genome-wide sequencing technology will provide the foundation for cell-specific therapeutic approaches. Recent drug development targeting epigenetic mechanisms shows a promising prospect for the treatment of cancers and inflammatory conditions. The development of epigenetic drugs or therapies that changes epigenetic states will contribute to such treatments. In this review, we provide a brief overview of epigenetic regulations in bone cells but the understanding of chromatin landscapes in bone cell is still incomplete.
Over the past years, the study of histone post-translational modifications (PTMs) has made progress to show the association of epigenetic modifications with fundamental biological processes and pathological conditions. During the differentiation of bone cells, histone PTMs are dynamically regulated. Recent advances in developing small molecules to modulate pharmacologically epigenetic enzymes and interfere with these biochemical mechanisms offer great promise for therapy of bone diseases. In this article, we provide an overview of modulating histone PTMs by small molecule inhibitors including HDAC inhibitor and BET inhibitor which provides elucidation of the basic epigenetic regulation of bone cells and devising future epigenetic therapy for bone diseases.
Recent progress in genome-wide technologies using small cell numbers have allowed researchers to capture transcriptional and epigenetic snapshots of cells from small or rare biological samples. These new methods allow identifying single-cell transcriptional and epigenomic variability across several cell types and suggests that controlling single-cell variance is a fundamental characteristic of different biological states [155]. Heterogeneity within cellular populations has been obvious since the first microscopic observation of individual cells. Dissecting single-cell based epigenetic heterogeneity and linked cis- and trans-effectors to variability in chromatin accessibility provides new opportunities for studying multiple different layers of regulation and advances our understanding of physiological development and the changes in pathological conditions and aging. Much effort has been made to identify osteoclast-specific or osteoblast-specific precursors from stem cells. Specific populations have been sorted using flow cytometry techniques. However, single cell ChIP sequencing or RNA sequencing would provide an advantage over isolating cells by flow cytometry and would allow investigation of a larger number of genes or epigenetic regulation in a single cell which has the potential for differentiation into bone cells. Thus, single cell sequencing techniques will allow for identifying bone-cell specific epigenetic mechanisms and for detailed new information about cellular differentiation or disease in different biological contexts.
Acknowledgments
I thank Drs. Lionel Ivashkiv, Mary-Beth Humphrey, Sungho Park for discussions and critical review of the manuscript. The research was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS, R00AR061430).
Footnotes
Declaration of interest
The author reports no conflicts of interest. The author alone is responsible for the content and writing of the article.
References
- 1.Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nature reviews Molecular cell biology. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- 2.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 3.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 4.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 5.Lorenzo J, Horowitz M, Choi Y. Osteoimmunology: interactions of the bone and immune system. Endocr Rev. 2008;29:403–40. doi: 10.1210/er.2007-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 7.Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. 2008;3:457–84. doi: 10.1146/annurev.pathmechdis.3.121806.151431. [DOI] [PubMed] [Google Scholar]
- 8.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411–25. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng HH, Surani MA. The transcriptional and signalling networks of pluripotency. Nature cell biology. 2011;13:490–6. doi: 10.1038/ncb0511-490. [DOI] [PubMed] [Google Scholar]
- 10.Stamatoyannopoulos JA. What does our genome encode? Genome research. 2012;22:1602–11. doi: 10.1101/gr.146506.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nature biotechnology. 2010;28:1045–8. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zentner GE, Henikoff S. High-resolution digital profiling of the epigenome. Nature reviews Genetics. 2014;15:814–27. doi: 10.1038/nrg3798. [DOI] [PubMed] [Google Scholar]
- 13.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 14.Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nature reviews Genetics. 2014;15:69–81. doi: 10.1038/nrg3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purdue PE, Crotti TN, Shen Z, Swantek J, Li J, Hill J, et al. Comprehensive profiling analysis of actively resorbing osteoclasts identifies critical signaling pathways regulated by bone substrate. Scientific reports. 2014;4:7595. doi: 10.1038/srep07595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF-beta signaling in bone remodeling. The Journal of clinical investigation. 2014;124:466–72. doi: 10.1172/JCI70050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer MB, Benkusky NA, Lee CH, Pike JW. Genomic determinants of gene regulation by 1,25-dihydroxyvitamin D3 during osteoblast-lineage cell differentiation. The Journal of biological chemistry. 2014;289:19539–54. doi: 10.1074/jbc.M114.578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer MB, Benkusky NA, Pike JW. The RUNX2 cistrome in osteoblasts: characterization, down-regulation following differentiation, and relationship to gene expression. The Journal of biological chemistry. 2014;289:16016–31. doi: 10.1074/jbc.M114.552216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu H, Whitfield TW, Gordon JA, Dobson JR, Tai PW, van Wijnen AJ, et al. Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome biology. 2014;15:R52. doi: 10.1186/gb-2014-15-3-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.St John HC, Bishop KA, Meyer MB, Benkusky NA, Leng N, Kendziorski C, et al. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Molecular endocrinology. 2014;28:1150–65. doi: 10.1210/me.2014-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome research. 2010;20:1352–60. doi: 10.1101/gr.107920.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–55. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 23.Komori T. Regulation of osteoblast differentiation by transcription factors. Journal of cellular biochemistry. 2006;99:1233–9. doi: 10.1002/jcb.20958. [DOI] [PubMed] [Google Scholar]
- 24.Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Human mutation. 2002;19:209–16. doi: 10.1002/humu.10043. [DOI] [PubMed] [Google Scholar]
- 25.Hecht J, Seitz V, Urban M, Wagner F, Robinson PN, Stiege A, et al. Detection of novel skeletogenesis target genes by comprehensive analysis of a Runx2(−/−) mouse model. Gene expression patterns: GEP. 2007;7:102–12. doi: 10.1016/j.modgep.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 26.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 27.Bradley EW, McGee-Lawrence ME, Westendorf JJ. Hdac-mediated control of endochondral and intramembranous ossification. Critical reviews in eukaryotic gene expression. 2011;21:101–13. doi: 10.1615/critreveukargeneexpr.v21.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelletier N, Champagne N, Stifani S, Yang XJ. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene. 2002;21:2729–40. doi: 10.1038/sj.onc.1205367. [DOI] [PubMed] [Google Scholar]
- 29.Villagra A, Cruzat F, Carvallo L, Paredes R, Olate J, van Wijnen AJ, et al. Chromatin remodeling and transcriptional activity of the bone-specific osteocalcin gene require CCAAT/enhancer-binding protein beta-dependent recruitment of SWI/SNF activity. The Journal of biological chemistry. 2006;281:22695–706. doi: 10.1074/jbc.M511640200. [DOI] [PubMed] [Google Scholar]
- 30.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 31.Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 2004;117:387–98. doi: 10.1016/s0092-8674(04)00344-7. [DOI] [PubMed] [Google Scholar]
- 32.Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, et al. A twist code determines the onset of osteoblast differentiation. Developmental cell. 2004;6:423–35. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- 33.Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S, et al. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development. 1999;126:3795–809. doi: 10.1242/dev.126.17.3795. [DOI] [PubMed] [Google Scholar]
- 34.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–9. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 35.Negishi-Koga T, Takayanagi H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev. 2009;231:241–56. doi: 10.1111/j.1600-065X.2009.00821.x. [DOI] [PubMed] [Google Scholar]
- 36.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 37.Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM, et al. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118:3775–89. doi: 10.1172/JCI35711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Endo-Munoz L, Cumming A, Rickwood D, Wilson D, Cueva C, Ng C, et al. Loss of osteoclasts contributes to development of osteosarcoma pulmonary metastases. Cancer research. 2010;70:7063–72. doi: 10.1158/0008-5472.CAN-09-4291. [DOI] [PubMed] [Google Scholar]
- 39.Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–9. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koga T, Matsui Y, Asagiri M, Kodama T, de Crombrugghe B, Nakashima K, et al. NFAT and Osterix cooperatively regulate bone formation. Nature medicine. 2005;11:880–5. doi: 10.1038/nm1270. [DOI] [PubMed] [Google Scholar]
- 41.Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, et al. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–8. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- 42.Wang ZQ, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360:741–5. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- 43.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–50. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 44.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–53. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 45.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 46.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature genetics. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nature reviews Genetics. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 50.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nature reviews Molecular cell biology. 2007;8:284–95. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 51.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nature reviews Drug discovery. 2014;13:673–91. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 52.Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell research. 2011;21:564–78. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim S, Shevde NK, Pike JW. 1,25-Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2005;20:305–17. doi: 10.1359/JBMR.041112. [DOI] [PubMed] [Google Scholar]
- 54.Park-Min KH, Lim E, Lee MJ, Park SH, Giannopoulou E, Yarilina A, et al. Inhibition of osteoclastogenesis and inflammatory bone resorption by targeting BET proteins and epigenetic regulation. Nature communications. 2014;5:5418. doi: 10.1038/ncomms6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 56.Westendorf JJ. Histone deacetylases in control of skeletogenesis. Journal of cellular biochemistry. 2007;102:332–40. doi: 10.1002/jcb.21486. [DOI] [PubMed] [Google Scholar]
- 57.Jensen ED, Schroeder TM, Bailey J, Gopalakrishnan R, Westendorf JJ. Histone deacetylase 7 associates with Runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2008;23:361–72. doi: 10.1359/JBMR.071104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schroeder TM, Nair AK, Staggs R, Lamblin AF, Westendorf JJ. Gene profile analysis of osteoblast genes differentially regulated by histone deacetylase inhibitors. BMC genomics. 2007;8:362. doi: 10.1186/1471-2164-8-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iwami K, Moriyama T. Effects of short chain fatty acid, sodium butyrate, on osteoblastic cells and osteoclastic cells. The International journal of biochemistry. 1993;25:1631–5. doi: 10.1016/0020-711x(93)90522-g. [DOI] [PubMed] [Google Scholar]
- 60.Schroeder TM, Westendorf JJ. Histone deacetylase inhibitors promote osteoblast maturation. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2005;20:2254–63. doi: 10.1359/JBMR.050813. [DOI] [PubMed] [Google Scholar]
- 61.Di Bernardo G, Squillaro T, Dell’Aversana C, Miceli M, Cipollaro M, Cascino A, et al. Histone deacetylase inhibitors promote apoptosis and senescence in human mesenchymal stem cells. Stem cells and development. 2009;18:573–81. doi: 10.1089/scd.2008.0172. [DOI] [PubMed] [Google Scholar]
- 62.Lee HW, Suh JH, Kim AY, Lee YS, Park SY, Kim JB. Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Molecular endocrinology. 2006;20:2432–43. doi: 10.1210/me.2006-0061. [DOI] [PubMed] [Google Scholar]
- 63.Schroeder TM, Kahler RA, Li X, Westendorf JJ. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. The Journal of biological chemistry. 2004;279:41998–2007. doi: 10.1074/jbc.M403702200. [DOI] [PubMed] [Google Scholar]
- 64.Dudakovic A, Evans JM, Li Y, Middha S, McGee-Lawrence ME, van Wijnen AJ, et al. Histone deacetylase inhibition promotes osteoblast maturation by altering the histone H4 epigenome and reduces Akt phosphorylation. The Journal of biological chemistry. 2013;288:28783–91. doi: 10.1074/jbc.M113.489732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McGee-Lawrence ME, Carpio LR, Schulze RJ, Pierce JL, McNiven MA, Farr JN, et al. Hdac3 Deficiency Increases Marrow Adiposity and Induces Lipid Storage and Glucocorticoid Metabolism in Osteochondroprogenitor Cells. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2015 doi: 10.1002/jbmr.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Backesjo CM, Li Y, Lindgren U, Haldosen LA. Activation of Sirt1 decreases adipocyte formation during osteoblast differentiation of mesenchymal stem cells. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2006;21:993–1002. doi: 10.1359/jbmr.060415. [DOI] [PubMed] [Google Scholar]
- 67.Rahman MM, Kukita A, Kukita T, Shobuike T, Nakamura T, Kohashi O. Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood. 2003;101:3451–9. doi: 10.1182/blood-2002-08-2622. [DOI] [PubMed] [Google Scholar]
- 68.Pham L, Kaiser B, Romsa A, Schwarz T, Gopalakrishnan R, Jensen ED, et al. HDAC3 and HDAC7 have opposite effects on osteoclast differentiation. The Journal of biological chemistry. 2011;286:12056–65. doi: 10.1074/jbc.M110.216853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stemig M, Astelford K, Emery A, Cho JJ, Allen B, Huang TH, et al. Deletion of histone deacetylase 7 in osteoclasts decreases bone mass in mice by interactions with MITF. PloS one. 2015;10:e0123843. doi: 10.1371/journal.pone.0123843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Triantafyllou N, Lambrinoudaki I, Armeni E, Evangelopoulos EM, Boufidou F, Antoniou A, et al. Effect of long-term valproate monotherapy on bone mineral density in adults with epilepsy. Journal of the neurological sciences. 2010;290:131–4. doi: 10.1016/j.jns.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 71.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–6. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 72.Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258–62. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–77. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–23. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2010;478:529–33. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33:146–53. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 77.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014 doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lamoureux F, Baud'huin M, Rodriguez Calleja L, Jacques C, Berreur M, Redini F, et al. Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nature communications. 2014;5:3511. doi: 10.1038/ncomms4511. [DOI] [PubMed] [Google Scholar]
- 81.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature reviews Genetics. 2012;13:343–57. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 83.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–6. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 84.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–94. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 85.Yasui T, Hirose J, Tsutsumi S, Nakamura K, Aburatani H, Tanaka S. Epigenetic regulation of osteoclast differentiation: possible involvement of Jmjd3 in the histone demethylation of Nfatc1. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2011;26:2665–71. doi: 10.1002/jbmr.464. [DOI] [PubMed] [Google Scholar]
- 86.Yang D, Okamura H, Nakashima Y, Haneji T. Histone demethylase Jmjd3 regulates osteoblast differentiation via transcription factors Runx2 and osterix. The Journal of biological chemistry. 2013;288:33530–41. doi: 10.1074/jbc.M113.497040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang F, Xu L, Xu L, Xu Q, Karsenty G, Chen CD. Histone demethylase JMJD3 is required for osteoblast differentiation in mice. Scientific reports. 2015;5:13418. doi: 10.1038/srep13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nature reviews Molecular cell biology. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 89.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 90.Kalb R, Latwiel S, Baymaz HI, Jansen PW, Muller CW, Vermeulen M, et al. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nature structural & molecular biology. 2014;21:569–71. doi: 10.1038/nsmb.2833. [DOI] [PubMed] [Google Scholar]
- 91.Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–4. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 92.Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nature cell biology. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gori F, Divieti P, Demay MB. Cloning and characterization of a novel WD-40 repeat protein that dramatically accelerates osteoblastic differentiation. The Journal of biological chemistry. 2001;276:46515–22. doi: 10.1074/jbc.M105757200. [DOI] [PubMed] [Google Scholar]
- 94.Gori F, Friedman LG, Demay MB. Wdr5, a WD-40 protein, regulates osteoblast differentiation during embryonic bone development. Developmental biology. 2006;295:498–506. doi: 10.1016/j.ydbio.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 95.Zhu ED, Demay MB, Gori F. Wdr5 is essential for osteoblast differentiation. The Journal of biological chemistry. 2008;283:7361–7. doi: 10.1074/jbc.M703304200. [DOI] [PubMed] [Google Scholar]
- 96.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome research. 2011;21:1757–67. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor protocols. 2010;2010 doi: 10.1101/pdb.prot5384. pdb prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome research. 2007;17:877–85. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Auerbach RK, Euskirchen G, Rozowsky J, Lamarre-Vincent N, Moqtaderi Z, Lefrancois P, et al. Mapping accessible chromatin regions using Sono-Seq. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14926–31. doi: 10.1073/pnas.0905443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods. 2013;10:1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.!!! INVALID CITATION !!!
- 103.Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boyle AP, Song L, Lee BK, London D, Keefe D, Birney E, et al. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome research. 2011;21:456–64. doi: 10.1101/gr.112656.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barutcu AR, Tai PW, Wu H, Gordon JA, Whitfield TW, Dobson JR, et al. The bone-specific Runx2-P1 promoter displays conserved three-dimensional chromatin structure with the syntenic Supt3h promoter. Nucleic acids research. 2014;42:10360–72. doi: 10.1093/nar/gku712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou X, Lowdon RF, Li D, Lawson HA, Madden PA, Costello JF, et al. Exploring long-range genome interactions using the WashU Epigenome Browser. Nature methods. 2013;10:375–6. doi: 10.1038/nmeth.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Inoue K, Imai Y. Identification of novel transcription factors in osteoclast differentiation using genome-wide analysis of open chromatin determined by DNase-seq. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2014;29:1823–32. doi: 10.1002/jbmr.2229. [DOI] [PubMed] [Google Scholar]
- 108.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1412–7. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature reviews Genetics. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 110.Krueger F, Kreck B, Franke A, Andrews SR. DNA methylome analysis using short bisulfite sequencing data. Nature methods. 2012;9:145–51. doi: 10.1038/nmeth.1828. [DOI] [PubMed] [Google Scholar]
- 111.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome research. 2010;20:320–31. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Villagra A, Gutierrez J, Paredes R, Sierra J, Puchi M, Imschenetzky M, et al. Reduced CpG methylation is associated with transcriptional activation of the bone-specific rat osteocalcin gene in osteoblasts. Journal of cellular biochemistry. 2002;85:112–22. [PubMed] [Google Scholar]
- 113.Farshdousti Hagh M, Noruzinia M, Mortazavi Y, Soleimani M, Kaviani S, Abroun S, et al. Different Methylation Patterns of RUNX2, OSX, DLX5 and BSP in Osteoblastic Differentiation of Mesenchymal Stem Cells. Cell journal. 2015;17:71–82. doi: 10.22074/cellj.2015.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guo X, Wang L, Li J, Ding Z, Xiao J, Yin X, et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature. 2015;517:640–4. doi: 10.1038/nature13899. [DOI] [PubMed] [Google Scholar]
- 115.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 116.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 117.Tsai CC, Su PF, Huang YF, Yew TL, Hung SC. Oct4 and Nanog directly regulate Dnmt1 to maintain self-renewal and undifferentiated state in mesenchymal stem cells. Molecular cell. 2012;47:169–82. doi: 10.1016/j.molcel.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 118.Vaes BL, Lute C, van der Woning SP, Piek E, Vermeer J, Blom HJ, et al. Inhibition of methylation decreases osteoblast differentiation via a non-DNA-dependent methylation mechanism. Bone. 2010;46:514–23. doi: 10.1016/j.bone.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 119.Liu L, van Groen T, Kadish I, Li Y, Wang D, James SR, et al. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clinical epigenetics. 2011;2:349–60. doi: 10.1007/s13148-011-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Delgado-Calle J, Fernandez AF, Sainz J, Zarrabeitia MT, Sanudo C, Garcia-Renedo R, et al. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis and rheumatism. 2013;65:197–205. doi: 10.1002/art.37753. [DOI] [PubMed] [Google Scholar]
- 121.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nature reviews Molecular cell biology. 2013;14:341–56. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yan X, Ehnert S, Culmes M, Bachmann A, Seeliger C, Schyschka L, et al. 5-azacytidine improves the osteogenic differentiation potential of aged human adipose-derived mesenchymal stem cells by DNA demethylation. PloS one. 2014;9:e90846. doi: 10.1371/journal.pone.0090846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.de la Rica L, Rodriguez-Ubreva J, Garcia M, Islam AB, Urquiza JM, Hernando H, et al. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome biology. 2013;14:R99. doi: 10.1186/gb-2013-14-9-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 125.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 126.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & development. 2011;25:1915–27. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome research. 2012;22:1775–89. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nature reviews Genetics. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 129.Huarte M. The emerging role of lncRNAs in cancer. Nature medicine. 2015;21:1253–61. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 130.Tong X, Gu PC, Xu SZ, Lin XJ. Long non-coding RNA-DANCR in human circulating monocytes: a potential biomarker associated with postmenopausal osteoporosis. Bioscience, biotechnology, and biochemistry. 2015;79:732–7. doi: 10.1080/09168451.2014.998617. [DOI] [PubMed] [Google Scholar]
- 131.Zhu L, Xu PC. Downregulated LncRNA-ANCR promotes osteoblast differentiation by targeting EZH2 and regulating Runx2 expression. Biochemical and biophysical research communications. 2013;432:612–7. doi: 10.1016/j.bbrc.2013.02.036. [DOI] [PubMed] [Google Scholar]
- 132.Hassan MQ, Tye CE, Stein GS, Lian JB. Non-coding RNAs: Epigenetic regulators of bone development and homeostasis. Bone. 2015;81:746–56. doi: 10.1016/j.bone.2015.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gamez B, Rodriguez-Carballo E, Ventura F. MicroRNAs and post-transcriptional regulation of skeletal development. Journal of molecular endocrinology. 2014;52:R179–97. doi: 10.1530/JME-13-0294. [DOI] [PubMed] [Google Scholar]
- 134.Lian JB, Stein GS, van Wijnen AJ, Stein JL, Hassan MQ, Gaur T, et al. MicroRNA control of bone formation and homeostasis. Nature reviews Endocrinology. 2012;8:212–27. doi: 10.1038/nrendo.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 136.Plasterk RH. Micro RNAs in animal development. Cell. 2006;124:877–81. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 137.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nature reviews Genetics. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 138.Hobert O. Gene regulation by transcription factors and microRNAs. Science. 2008;319:1785–6. doi: 10.1126/science.1151651. [DOI] [PubMed] [Google Scholar]
- 139.Starega-Roslan J, Koscianska E, Kozlowski P, Krzyzosiak WJ. The role of the precursor structure in the biogenesis of microRNA. Cellular and molecular life sciences: CMLS. 2011;68:2859–71. doi: 10.1007/s00018-011-0726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kapinas K, Delany AM. MicroRNA biogenesis and regulation of bone remodeling. Arthritis research & therapy. 2011;13:220. doi: 10.1186/ar3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10898–903. doi: 10.1073/pnas.0504834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gaur T, Hussain S, Mudhasani R, Parulkar I, Colby JL, Frederick D, et al. Dicer inactivation in osteoprogenitor cells compromises fetal survival and bone formation, while excision in differentiated osteoblasts increases bone mass in the adult mouse. Developmental biology. 2010;340:10–21. doi: 10.1016/j.ydbio.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mizoguchi F, Izu Y, Hayata T, Hemmi H, Nakashima K, Nakamura T, et al. Osteoclast-specific Dicer gene deficiency suppresses osteoclastic bone resorption. Journal of cellular biochemistry. 2010;109:866–75. doi: 10.1002/jcb.22228. [DOI] [PubMed] [Google Scholar]
- 144.Sugatani T, Hruska KA. Impaired micro-RNA pathways diminish osteoclast differentiation and function. The Journal of biological chemistry. 2009;284:4667–78. doi: 10.1074/jbc.M805777200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sugatani T, Vacher J, Hruska KA. A microRNA expression signature of osteoclastogenesis. Blood. 2011;117:3648–57. doi: 10.1182/blood-2010-10-311415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hassan MQ, Gordon JA, Beloti MM, Croce CM, van Wijnen AJ, Stein JL, et al. A network connecting Runx2, SATB2, and the miR-23a~27a~24-2 cluster regulates the osteoblast differentiation program. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19879–84. doi: 10.1073/pnas.1007698107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zeng Y, Qu X, Li H, Huang S, Wang S, Xu Q, et al. MicroRNA-100 regulates osteogenic differentiation of human adipose-derived mesenchymal stem cells by targeting BMPR2. FEBS letters. 2012;586:2375–81. doi: 10.1016/j.febslet.2012.05.049. [DOI] [PubMed] [Google Scholar]
- 148.Zhang Y, Xie RL, Croce CM, Stein JL, Lian JB, van Wijnen AJ, et al. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9863–8. doi: 10.1073/pnas.1018493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kanis JA. Diagnosis of osteoporosis and assessment of fracture risk. Lancet. 2002;359:1929–36. doi: 10.1016/S0140-6736(02)08761-5. [DOI] [PubMed] [Google Scholar]
- 150.Kanis JA, Johnell O, Oden A, De Laet C, Jonsson B, Dawson A. Ten-year risk of osteoporotic fracture and the effect of risk factors on screening strategies. Bone. 2002;30:251–8. doi: 10.1016/s8756-3282(01)00653-6. [DOI] [PubMed] [Google Scholar]
- 151.Berry SD, Samelson EJ, Pencina MJ, McLean RR, Cupples LA, Broe KE, et al. Repeat bone mineral density screening and prediction of hip and major osteoporotic fracture. Jama. 2013;310:1256–62. doi: 10.1001/jama.2013.277817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Seeliger C, Karpinski K, Haug AT, Vester H, Schmitt A, Bauer JS, et al. Five freely circulating miRNAs and bone tissue miRNAs are associated with osteoporotic fractures. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2014;29:1718–28. doi: 10.1002/jbmr.2175. [DOI] [PubMed] [Google Scholar]
- 153.Bouchie A. First microRNA mimic enters clinic. Nat Biotech. 2013;31:577. doi: 10.1038/nbt0713-577. [DOI] [PubMed] [Google Scholar]
- 154.Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nature medicine. 2013;19:35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–90. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]