

Abstract
The kinamycin family of aromatic polyketide natural products contains an atypical angucycline ring system substituted with a diazo group. The enzymatic chemistry involved in constructing both of these structural features has been largely unexplored. Here we report the in vivo and in vitro production of seongomycin, a shunt product from this pathway, and stealthin C, a proposed biosynthetic precursor to the kinamycins. We show that a single enzyme, the flavin-dependent monooxygenase AlpJ, can generate these metabolites from N-acetyl-L-cysteine and L-cysteine, respectively, and that the synthesis of stealthin C likely proceeds via a non-enzymatic S–N-type Smiles rearrangement. This unexpected route to stealthin C reveals a distinct approach to install aromatic amino groups in metabolites and raises questions about the intermediacy of this species in kinamycin production.
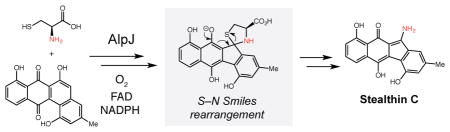
Graphical Abstract
The diazofluorenes are a group of bioactive natural products produced by both soil and marine Actinomycetes (Figure 1A).1 Family members, including the kinamycins and the lomaiviticins, share a common diazotetrahydrobenzo[b]fluorene core scaffold. Both metabolite classes exhibit cytotoxicity due to their ability to damage DNA, with the dimeric scaffold of lomaiviticin A greatly increasing potency.2 The unusual structures of these natural products have been a rich source of inspiration for synthetic chemists and chemical biologists who have pursued syntheses of the natural products as well as simplified analogs for biological evaluation.1
Figure 1.

Biosynthesis of the diazofluorene natural products. A) Representative diazofluorene natural products. B) Proposed biosynthetic pathway to the kinamycins and seongomycin.
Despite intense interest in these molecules, our understanding of diazofluorene biosynthesis is limited. Past efforts largely consisted of in vivo experiments involving the kinamycins, monomeric metabolites produced by Streptomyces murayamaensis and Streptomyces ambofaciens.3 Gould and co-workers identified additional metabolites from S. murayamaensis, including the aromatic polyketide dehydrorabelomycin (1). 1 is a key intermediate in the production of all angucycline polyketides, including the atypical angucyclines gilvocarcin and jadomycin.4 Feeding deuterated 1 to S. murayamaensis confirmed its connection to kinamycin D, suggesting a biosynthetic route involving C-ring contraction and nitrogen insertion events (Figure 1B).5
Seongomycin (2), stealthin C (3), and kinobscurinone (4) are kinamycin-related metabolites isolated from S. murayamaensis mutants generated via chemical mutagenesis.6 Feeding studies implicated both 3 and 4 as intermediates in kinamycin D biosynthesis (Figure 1B),6a,b while 2 and its counterpart from lomaiviticin biosynthesis (homoseongomycin) were proposed to be shunt products derived from the reaction of the diazo-containing metabolites prekinamycin and homoprekinamycin with cellular thiols.7 Notably, 2–4 were produced upon heterologous expression of a partial kinamycin biosynthetic gene cluster.8
These initial studies left many biosynthetic questions unanswered, including the identities of the enzymes involved in benzo[b]fluorene ring formation and diazo generation. The discovery of the complete kinamycin and lomaiviticin biosynthetic gene clusters has provided new opportunities to investigate these intriguing transformations.3c,9 Here we report the in vivo and in vitro characterization of enzymatic machinery capable of synthesizing 2 and 3. We demonstrate that a single enzyme, flavin-dependent monooxygenase AlpJ, can access these two products from 1 and either N-acetyl-L-cysteine or L-cysteine. Assays with labeled substrates and substrate analogs suggest that formation of 3 involves a non-enzymatic S–N-type Smiles rearrangement. This discovery, along with preliminary feeding experiments with S. murayamaensis, calls into question the proposed role of stealthin C as a biosynthetic intermediate.
We began by identifying the enzymes involved in generating the benzo[b]fluorene ring system. Yang and co-workers reported two oxidases from S. ambofaciens (AlpJ/K) that produce this scaffold from 1 in vivo and in vitro.10 In-frame deletion of alpJ and alpK individually in S. ambofaciens led to the accumulation of 1 and a trimeric benzo[b]fluorene, respectively. Generation of 2 in vitro in the presence of AlpJ, AlpK, and N-acetyl-L-cysteine and in vivo upon heterologous expression of these enzymes suggested that these enzymes mediate ring contraction.
Simultaneously, we pursued a similar line of inquiry that led us to identify these enzymes as not only being involved in the biosynthesis of 2, but also capable of producing proposed biosynthetic intermediate 3 in vivo and in vitro. We uncovered candidate benzo[b]fluorene ring-forming enzymes by comparing the kin and lom biosynthetic gene clusters to gene clusters that produce other angucycline polyketides.11 In the jadomycin and gilvocarcin pathways, flavin dependent monooxygenases JadG and GilOII perform a Bayer–Villiger-type oxidative C–C bond cleavage reaction.4 Hypothesizing that a similar transformation should be involved in kinamycin and lomaiviticin biosynthesis, we located homologous flavin-dependent monooxygenases encoded in the kin and lom clusters (AlpJ/KinG and Lom28). These JadG homologs were always co-localized with a second flavin-dependent oxidase (AlpK/KinO1 and Lom27) that was absent from the gil and jad gene clusters (Figure S1), suggesting that this enzyme might also play a role in benzo[b]fluorene formation.
To initiate our studies, we cloned alpJ and alpK from S. ambofaciens into the Streptomyces expression vector pUWL201PW in an operon under the control of the ermE* promoter. The resulting plasmid pAlpJK was transformed into Streptomyces lividans TK64 to afford recombinant strain pAlpJK/TK64. Feeding 1 to pAlpJK/TK64 led to production of 2 (m/z [M + H]+ = 454.0960) (Figures 2 and S2), confirming that the AlpJK enzyme pair catalyzed the C–C bond cleavage and ring contraction reactions.
Figure 2.

AlpJ converts dehydrorabelomycin to seongomycin and stealthin C in vivo. HPLC analysis of culture extracts.
Interestingly, we also detected a minor metabolite that had a similar UV-visible absorption spectrum to stealthin C (3) (m/z [M + H]+ = 308.0924) (Figures 2 and S3).6b This observation was unexpected as the formation of an aminobenzo[b]fluorene is proposed to require at least one additional enzyme for nitrogen transfer.6b Though this result suggested that AlpJK could generate 3 in vivo, the yield of this metabolite was extremely low compared to that of 2. This difference could arise from the availability of biosynthetic precursors to these metabolites. The N-acetyl-cysteine group of 2 is likely derived from mycothiol, which is present at high concentrations in Streptomyces lividans cells (~ 2.2 mM).12 The source of the amino group of 3 has not been identified; however, we wondered if it might come from L-cysteine (present at ~0.2 mM).12 We fed 1 and L-cysteine (~ 11 mM) to pAlpJK/TK64 and observed both a loss of 2 and an 11-fold increase in 3 (Figure 2). Feeding 1 15N-labeled L-cysteine shifted the mass of 3 by +1 mass unit, confirming that the nitrogen atom of this amino acid is incorporated into stealthin C (Figure S4).
To explore these processes in vitro, AlpJ and the AlpK homolog KinO1 were expressed in E. coli (Figure S5). While purified AlpJ was colorless, purified KinO1 was bright yellow. LC-MS analysis of the supernatants of both enzymes revealed FAD (Figures S6 and S7). To enable the production of reduced cofactor in in vitro assays, we also cloned and expressed the E. coli FAD reductase Fre (Figure S5).13
When 1 was incubated with AlpJ, Fre, FAD, and NADPH, we observed a new peak by HPLC that did not correspond to the hydroxyoxepinone intermediate noted previously in in vitro assays with 1 and GilOII or JadG.4d Subsequent LC-MS analysis indicated that this new compound had m/z [M – H]– = 581.1218 (Figure S8, trace ii and Figure S9), which corresponds to a dimeric benzofluorene (5). This product was also identified by Yang and co-workers.10 When we replaced Fre with KinO1, the consumption of 1 was slower and 5 was not detected (Figure S8, trace iii). Furthermore, AlpJ alone generated 5 with reduced efficiency (Figure S8, trace i). These results show that AlpJ can catalyze both the oxidative C–C bond cleavage reaction and the subsequent ring-closing reaction. The altered product distribution in the presence of KinO1 implies that in addition to reducing FAD, KinO1 might work as an ancillary oxygenase to enhance the production of hydroquinone-kinobscurinone and avoid 5, a function also proposed by Yang and co-workers.10
We next added N-acetyl-L-cysteine to the HPLC assays described above and observed efficient formation of a new product with a mass corresponding to 2 (Figures S10 and S11). AlpJ produced 2 both in the presence and absence of KinO1/Fre, although the qualitative rate of production was lower without a partner oxidoreductase. The dispensability of KinO1/Fre suggests a supportive but nonessential role in the formation of 2 in vitro. This contrasts with the results of Yang and co-workers.
The discovery that 2 can be accessed directly from 1 revises our thinking about its origins. Our work, and that of Yang and coworkers, demonstrates an alternate route to 2 that does not require a diazo-containing metabolite. We propose that AlpJ first catalyzes the ring opening reaction via a Baeyer–Villiger oxidation mechanism, giving rise to a previously posited aldehyde/acid intermediate (Scheme S1).4a,b This species may then undergo ring closure, decarboxylation, and dehydration to give the benzofluorene ring. The C-5 position of benzofluorene could be further oxidized to give hydroquinone-kinobscurinone, which could be attacked by the thiol of N-acetyl-cysteine, affording 2.
We then explored the reactivity of AlpJ and Fre toward 1 and L-cysteine in vitro. We observed the formation of two new peaks that displayed UV-vis absorbances similar to that of 3 (Figures S12 trace ii, S13, and S14). An assay time course suggested that one of these products (3a) was a reaction intermediate, as its depletion accompanied the formation of the second product (3) (Figure 3A). The two products were also generated by AlpJ and KinO1 (Figure S12, trace iii). Isolation and high-resolution mass spectrometry (HRMS) analysis of the final product revealed m/z [M + H]+ = 308.0929, which matches the molecular formula of 3. The complete characterization of this product was complicated by the fact that 3 is NMR-silent.14 We therefore derivatized 3 obtained from a large scale assay with methyl iodide.3b This provided a new product (6) that co-eluted with an authentic standard of dimethylstealthin C, thereby confirming the identity of 3 (Figures 3B and S15). Further HRMS analysis of intermediate 3a (Figure S14) showed a molecular weight m/z [M + H]+ = 531.0898 and fragmentation pattern consistent with appendage of a cysteine disulfide to the benzofluorene scaffold. Inclusion of other proteinogenic amino acids in AlpJ/Fre in vitro assays did not provide new products (data not shown).
Figure 3.

AlpJ converts 1 and L-cysteine to stealthin C (3). A) HPLC analysis of in vitro assays containing AlpJ and Fre. 5 = diazofluorene dimer. Additional control assays are shown in Figures S16 and S17. B) Derivatization of AlpJ assay product 3.
The production of 3 from 1 and L-cysteine by AlpJ and Ki-nO1/Fre is interesting from a mechanistic perspective. To verify the origin of the amino substituent of 3, we performed the in vitro assay with 15N-labeled L-cysteine (Figure S12, trace iv) and observed the masses of 3 and 3a increase by +1 and +2 mass units, respectively (Figures S18 and S19). As AlpJ could convert 1 to either 3 in the presence of L-cysteine or 2 in the presence of N-acetyl-L-cysteine, we deduced that the presence of a free amine might be a critical determinant of reaction outcome. We hypothesize that in a manner analogous to the formation of 2, generation of 3 involves attack by the L-cysteine thiol to form a thioether intermediate (N-deacetylseongomycin, 7). This species could undergo an intramolecular, S–N-type Smiles rearrangement (Scheme 1).15 This reactivity resembles the S- to N-acyl transfer mechanisms of native chemical ligation and protein splicing.16 The resulting N-linked intermediate (8) could form a disulfide bond with L-cysteine to afford 3a, which could be further oxidized to imine 9. Hydrolysis of this intermediate would provide 3.
Scheme 1.

Proposed mechanism for stealthin C formation.
To test this mechanistic proposal, we ran AlpJ/Fre in vitro assays with 1 and a panel of alternative amino acid co-substrates (Figure 4). Amino acids lacking a free thiol group, including L-serine and S-methyl-L-cysteine, failed to provide new products, confirming the essential role of a thiol nucleophile (Figure S20, traces i and ii). L-cysteine analogs containing non-nucleophilic amino groups, including the dipeptide γ-Glu-Cys and N-carbamoyl-L-cysteine, generated new S-linked products (10 and 11). This result shows that a nucleophilic amino group is necessary for C–N bond formation. (Figure S20, traces iii and iv, Figures S21 and S22). Assays with N-methyl-L-cysteine provided N-methyl-stealthin C (12) (Figure S20, trace v, Figure S23). We investigated the importance of the carboxylic acid of L-cysteine by examining cysteamine. HPLC and LC-HRMS analyses revealed a single new product (m/z [M – H]– = 441.0949) that likely corresponds to mixed disulfide 13 (Figure S20, trace vi, and Figure S24). This result suggests that the carboxylic acid is not needed for the Smiles rearrangement but is critical for facilitating the final oxidation step. Assays with L-homocysteine gave a single product with a mass corresponding to homocysteine adduct 14 (m/z [M – H]– = 424.0862) (Figure S20, trace vii and Figure S25). The failure to form 3 could reflect a reduced rate of formation for a six membered ring-containing intermediate. Finally, assays with D-cysteine generated 3 with efficiencies similar to that observed for L-cysteine (Figure S20, trace viii).
Figure 4.

Reactivity of AlpJ-derived intermediates toward alternate nucleophiles.
The broad reactivity observed with thiol nucleophiles and the lack of a preference for L- vs. D-cysteine led us to question whether AlpJ and/or Fre catalyzed the thiol addition and Smiles rearrangement reactions involved in forming 2 and 3. To examine this issue, we performed competition experiments in which AlpJ and Fre were incubated with 1 and equal concentrations of two thiols. Assays containing L-cysteine and N-acetyl cysteine, as well as L-cysteine and cysteamine, generated comparable amounts of both expected products (Figure S20, trace ix). A third product with a mass corresponding to a mixed disulfide was also produced in the assay containing L-cysteine and cysteamine (Figure S20, trace x and Figure S26). Overall, the failure to observe a strong preference for L-cysteine over other thiol substrates suggests this reactivity is non-enzymatic. Conversion of 3a to 3 was observed when purified 3a was incubated alone, as well as after assay mixtures containing 3a were boiled to denature enzymes (Figure S27), indicating that the oxidation of 3a and imine hydrolysis are likely also not enzyme catalyzed.
The non-enzymatic, in vitro generation of 3 from L-cysteine and an AlpJ-derived reaction partner has implications for the in vivo production of this metabolite. 3 has only been isolated from mutant strains,6b consistent with non-enzymatic production from an accumulating, reactive species. This general phenomenon has been identified in other biosynthetic pathways.17 The relevance of 3 to kinamycin biosynthesis was established using feeding experiments,6b and this result was the basis for the hypothesis that diazo group construction involves N–N bond formation on the benzo[b]fluorene scaffold. However, the 3 used for these feeding experiments was labeled on the carbon scaffold, not nitrogen, and only low levels of incorporation were observed. Though additional, unidentified enzymes could provide an alternate route to 3 in vivo, the observation that this metabolite is readily accessed via non-enzymatic reactions and the propensity of this benzo[b]fluorene ring system to undergo substitution chemistry leads us to question the stability of 3 in the original feeding experiments and its role as a biosynthetic intermediate.
To investigate this possibility, we performed preliminary feeding experiments with the kinamycin producer S. murayamaensis. Feeding 15N-labeled L-cysteine results in undetectable labeling of kinamycin D, whereas feeding 15N4-labeled L-arginine as a control provided low but detectable incorporation (Table S3). The lack of kinamyin D enrichment upon feeding 15N-labeled L-cysteine suggests that 3 arises from off-pathway reactions of a shunt product generated in mutants blocked in kinamycin synthesis. Significantly, this finding, along with recent investigations of hydrazide assembly in fosfazinomycin biosynthesis,18 raises serious doubts about the long-assumed intermediacy of stealthin C in this pathway and will reshape future efforts to decipher the enzymatic chemistry of diazo formation.
Discovering new approaches for aromatic C–N bond formation is important because of this structural motif’s predominance in pharmaceuticals and other bioactive compounds.18 In natural product biosynthesis, aromatic C–N bonds have been generated by cytochrome P450s.19a,b Aminotransferases are hypothesized to be involved in aromatic C–N bond formation in azamerone20a and streptonigrin20b biosynthesis, respectively. Additionally, structure-based mutagenesis of a type III polyketide synthase enabled C–N bond formation in a polyketide-alkaloid aromatic scaffold.21 Interfacing the non-enzymatic S–N-type Smiles rearrangement with a natural product biosynthetic pathway represents a distinct strategy for accessing aromatic C–N bond containing compounds. Our findings should therefore inspire related strategies in synthetic biology.
Supplementary Material
Acknowledgments
We thank Prof. Sean F. Brady (The Rockefeller University) for the gift of BAC AB649/1850, Prof. Wenjun Zhang (University of California, Berkeley) for the gifts of S. lividans TK64 and pUL201PW, and Bristol-Myers Squibb for the strain Streptomyces lividans K4-114. We thank Prof. Philip J. Proteau (Oregon State University) for providing the dimethyl-stealthin C and kinamycin D standards and Jennifer Wang (Harvard University) for assistance with LC-MS analyses. We acknowledge financial support from Harvard University, the Searle Scholars Program, and the NIH (DP2 GM105434).
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interests.
Full experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Herzon SB, Woo CM. Nat Prod Rep. 2012;29:87. doi: 10.1039/c1np00052g. [DOI] [PubMed] [Google Scholar]
- 2.Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. Nat Chem. 2014;6:504. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Seaton PJ, Gould SJ. J Antibiot (Tokyo) 1989;42:189. doi: 10.7164/antibiotics.42.189. [DOI] [PubMed] [Google Scholar]; (b) Cone MC, Seaton PJ, Halley KA, Gould SJ. J Antibiot (Tokyo) 1989;42:179. doi: 10.7164/antibiotics.42.179. [DOI] [PubMed] [Google Scholar]; (c) Bunet R, Song L, Mendes MV, Corre C, Hotel L, Rouhier N, Framboisier X, Leblond P, Challis GL, Aigle B. J Bacteriol. 2011;193:1142. doi: 10.1128/JB.01269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Rix U, Wang C, Chen Y, Lipata FM, Remsing Rix LL, Greenwell LM, Vining LC, Yang K, Rohr J. Chembiochem. 2005;6:838. doi: 10.1002/cbic.200400395. [DOI] [PubMed] [Google Scholar]; (b) Chen YH, Wang CC, Greenwell L, Rix U, Hoffmeister D, Vining LC, Rohr J, Yang KQ. J Biol Chem. 2005;280:22508. doi: 10.1074/jbc.M414229200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fan K, Pan G, Peng X, Zheng J, Gao W, Wang J, Wang W, Li Y, Yang K. Chem Biol. 2012;19:1381. doi: 10.1016/j.chembiol.2012.09.009. [DOI] [PubMed] [Google Scholar]; (d) Tibrewal N, Pahari P, Wang G, Kharel MK, Morris C, Downey T, Hou Y, Bugni TS, Rohr J. J Am Chem Soc. 2012;134:18181. doi: 10.1021/ja3081154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seaton PJ, Gould SJ. J Am Chem Soc. 1987;109:5282. [Google Scholar]
- 6.(a) Gould SJ, Melville CR. Bioorg Med Chem Lett. 1995;5:51. [Google Scholar]; (b) Gould SJ, Melville CR, Cone MC, Chen J, Carney JR. J Org Chem. 1997;62:320. doi: 10.1021/jo961486y. [DOI] [PubMed] [Google Scholar]; (c) Carney JR, Hong ST, Gould SJ. Tetrahedron Lett. 1997;38:3139. [Google Scholar]
- 7.Woo CM, Gholap SL, Herzon SB. J Nat Prod. 2013;76:1238. doi: 10.1021/np400355h. [DOI] [PubMed] [Google Scholar]
- 8.Gould SJ, Hong ST, Carney JR. J Antibiot (Tokyo) 1998;51:50. doi: 10.7164/antibiotics.51.50. [DOI] [PubMed] [Google Scholar]
- 9.(a) Janso JE, Haltli BA, Eustaquio AS, Kulowski K, Waldman AJ, Zha L, Nakamura H, Bernan VS, He H, Carter GT, Koehn FE, Balskus EP. Tetrahedron. 2014;70:4156. doi: 10.1016/j.tet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kersten RD, Lane AL, Nett M, Richter TK, Duggan BM, Dorrestein PC, Moore BS. Chembiochem. 2013;14:955. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang B, Ren JW, Li LY, Guo F, Pan GH, Ai GM, Aigle B, Fan KQ, Yang KQ. Chem Commun. 2015;51:8845. doi: 10.1039/c5cc01986a. [DOI] [PubMed] [Google Scholar]
- 11.Kharel MK, Pahari P, Shepherd MD, Tibrewal N, Nybo SE, Shaaban KA, Rohr J. Nat Prod Rep. 2012;29:264. doi: 10.1039/c1np00068c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. J Bacteriol. 1996;178:1990. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spyrou G, Haggard-Ljungquist E, Krook M, Jornvall H, Nilsson E, Reichard P. J Bacteriol. 1991;173:3673. doi: 10.1128/jb.173.12.3673-3679.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gould SJ, Melville CR. Tetrahedron Lett. 1997;38:1473. [Google Scholar]
- 15.Castagnolo D, Pagano M, Bernardini M, Botta M. Tetrahedron Lett. 2012;53:5008. [Google Scholar]
- 16.(a) Kane PM, Yamashiro CT, Wolczyk DF, Neff N, Goebl M, Stevens TH. Science. 1990;250:651. doi: 10.1126/science.2146742. [DOI] [PubMed] [Google Scholar]; (b) Hirata R, Ohsumk Y, Nakano A, Kawasaki H, Suzuki K, Anraku Y. J Biol Chem. 1990;265:6726. [PubMed] [Google Scholar]
- 17.(a) Rix U, Zheng J, Remsing Rix LL, Greenwell L, Yang K, Rohr J. J Am Chem Soc. 2004;126:4496. doi: 10.1021/ja031724o. [DOI] [PubMed] [Google Scholar]; (b) Yan Y, Yang J, Yu Z, Yu M, Ma Y, Wang L, Su C, Luo J, Horsman GP, Huang S. Nat Commun. 2016;7:13083. doi: 10.1038/ncomms13083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hu Y, Potts MB, Colosimo D, Herrera-Herrera ML, Legako AG, Yousufuddin M, White MA, MacMillan JB. J Am Chem Soc. 2013;135:13387. doi: 10.1021/ja403412y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Z, Wang KK, van der Donk WA. Chem Sci. 2016;7:5219. doi: 10.1039/c6sc01389a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hili R, Yudin AK. Nat Chem Biol. 2006;2:284. doi: 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- 19.(a) Barry SM, Kers JA, Johnson EG, Song L, Aston PR, Patel B, Krasnoff SB, Crane BR, Gibson DM, Loria R, Challis GL. Nat Chem Biol. 2012;8:814. doi: 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huynh MU, Elston MC, Hernandez NM, Ball DB, Kajiyama S, Irie K, Gerwick WH, Edwards DJ. J Nat Prod. 2010;73:71. doi: 10.1021/np900481a. [DOI] [PubMed] [Google Scholar]
- 20.(a) Winter JM, Jansma AL, Handel TM, Moore BS. Angew Chem Int Ed. 2009;48:767. doi: 10.1002/anie.200805140. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu F, Kong D, He X, Zhang Z, Han M, Xie X, Wang P, Cheng H, Tao M, Zhang L, Deng Z, Lin S. J Am Chem Soc. 2013;135:1739. doi: 10.1021/ja3069243. [DOI] [PubMed] [Google Scholar]
- 21.Morita H, Yamashita M, Shi SP, Wakimoto T, Kondo S, Kato R, Sugio S, Kohno T, Abe I. Proc Natl Acad Sci U S A. 2011;108:13504. doi: 10.1073/pnas.1107782108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.