

Abstract
Hypoparathyroidism, sensorineural deafness, and renal dysplasia (HDR) syndrome is a rare autosomal dominant disease caused by GATA3 mutations. Although several cases with variable renal features have been reported, the presence of histological changes within the glomeruli in adult patients is unclear. We herein report an adult case of HDR syndrome with a novel p.C288W (TGC>TGG) missense mutation in GATA3. His renal histology showed a membranoproliferative glomerulonephritis-like glomerular lesion. Additional renal histological analyses of HDR syndrome patients will be needed to clarify the role of GATA3 in both the developing and adult kidney.
Keywords: HDR syndrome, GATA3, membranoproliferative glomerulonephritis-like lesion
Introduction
Hypoparathyroidism, sensorineural deafness, and renal dysplasia (HDR) syndrome (OMIM 146255) is a hereditary disease with autosomal dominant transmission. This syndrome was first described in 1992 (1) and was subsequently named HDR syndrome by Hasegawa et al., who detected a deletion on chromosome 10p15 (2). Esch et al. went on to identify the causal genetic abnormality of this disease within this region: mutations in the GATA3 gene (3).
GATA3 is mainly expressed in the parathyroid, inner ear, and kidney during embryonic development. In the embryonic kidney, GATA3 is strongly expressed in the collecting ducts and weakly expressed in glomerular mesangial cells (4, 5). In the normal adult kidney, GATA3 expression is rarely found, but there are a few positive cells in the mesangial area (6). Experimental findings show that GATA3 is indispensable for kidney development, especially for ureteric bud formation (7), and is regulated by Pax2 and Pax8 (5).
The common renal phenotype of HDR syndrome is a structural anomaly introduced during development. A recent review of previous cases showed that renal dysplasia, hypoplasia, or aplasia was most common (41% of HDR patients), and a cyst or pelvicalyceal deformity was observed in 11% of patients (8). Of note, this mutation has variable expressivity, even for the same mutation in the same family (8, 9). Regarding the glomerular phenotype, there have been only a few reports on glomerular lesions in cases of HDR syndrome. In a 4-year-old patient who presented nephrotic range proteinuria and moderate hematuria, unusual proliferative glomerulonephritis was observed on light microscopy, but immunofluorescence and electron microscopy findings were not available (10). Other reports on adult biopsy cases with HDR syndrome complicated with vesicoureteral reflux revealed that 50% of glomeruli had focal segmental glomeruloscrelosis lesions (8). However, there have been no report of adult HDR patients with unusual proliferative glomerulonephritis.
We herein report an adult case of HDR syndrome with a novel GATA3 mutation and unusual proliferative glomerulonephritis and discuss the possible mechanisms underlying the development of glomerular lesions in HDR patients.
Case Report
A 52-year-old Japanese man was admitted to our hospital with moderate proteinuria and renal dysfunction. Proteinuria was initially detected at 18 years old. At 40 years old, he was diagnosed with sensorineural deafness. At 47 years old, he was diagnosed with hypoparathyroidism and alfacalcidol was initially administered. At the same time, he was diagnosed with viral hepatitis B infection based on positive blood tests for hepatitis B surface antigen (HBsAg) and hepatitis B virus (HBV)-DNA. Regarding the other risk factors for the progression of kidney diseases, he was diagnosed with mild hypertension and moderate hyperlipidemia several years prior to the admission, but these were appropriately controlled thereafter. His sister and nephew had deafness and renal disease, presenting mild to moderate renal insufficiency and mild proteinuria, and his son and father had deafness (Fig. 1).
Figure 1.
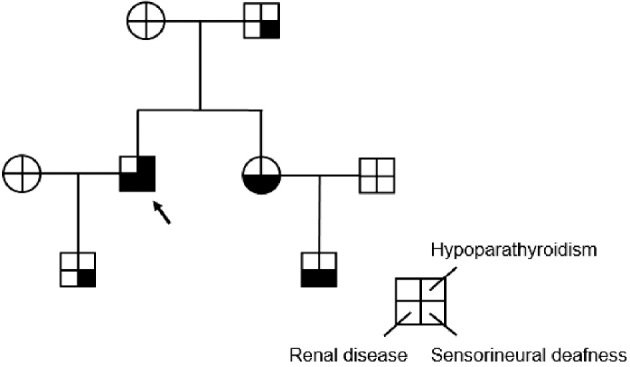
Family tree. The proband is indicated by an arrow. The presence of the hypoparathyroidism, sensorineural deafness, and renal disease triad is indicated by colors in each quartered area.
A laboratory examination on admission showed moderate renal insufficiency, and a urinalysis revealed moderate proteinuria and mild elevation of N-acetyl-β-D-glucosaminidase and β2-microglobulin; however, hematuria was not apparent. Serological findings showed no increase in autoantibodies, but serum samples tested positive for HBsAg and HBV-DNA (Table). On abdominal echography, the size of both kidneys was preserved, and some renal cysts were observed, but urinary tract deformity was not present (Fig. 2).
Table.
Laboratory Data Obtained at Admission.
| Hematology | Serology | ||||
| WBC | 7,100 | /μL | IgG | 1,551 | mg/dL |
| Hb | 13.9 | g/dL | IgA | 375 | mg/dL |
| Platelet | 22.9×104 | /μL | IgM | 131 | mg/dL |
| complement3 | 67.9 | mg/dL | |||
| Biochemistry | complement4 | 13.4 | mg/dL | ||
| Total protein | 6.8 | g/dL | CH50 | 35.9 | U/mL |
| Albumin | 3.6 | g/dL | C-reactive protein | 0.21 | mg/dL |
| Urea nitrogen | 30.2 | mg/dL | ANA | <×40 | |
| Creatinine | 1.86 | mg/dL | HBs-Ag | 60,618 | IU/mL |
| eGFR | 31.7 | mL/min/1.73m2 | HBV-DNA | 8.6 | log copies/mL |
| Sodium | 137 | mEq/L | HCV-Ab | - | |
| Potassium | 5.1 | mEq/L | Cryoglobulin | - | |
| Chloride | 105 | mEq/L | |||
| Calcium | 9.2 | mg/dL | Urinalysis | ||
| Phosphorus | 4.1 | mg/dL | sp gr | 1.014 | |
| Total bilirubin | 0.43 | mg/dL | pH | 8.5 | |
| AST | 27 | IU/L | Protein | 1.52 | g/gCreatinine |
| ALT | 33 | IU/L | Glucose | - | |
| LDH | 210 | IU/L | RBC sediment | 1-4 | /high power field |
| ALP | 202 | IU/L | beta-2-MG | 767 | µg/gCreatinine |
| γ-GTP | 20 | IU/L | NAG | 22 | U/gCreatinine |
| Hemoglobin A1c | 5.4 | % | BJP | - |
eGFR: estimated glomerular filtration rate, AST: aspartate aminotransferase, ALT: alanine aminotransferase, LDH: lactate dehydrogenase, ALP: alkaline phosphatase, γ-GTP: gamma-glutamyl transpeptidase, CH50: 50% hemolytic unit of complement, ANA: antinuclear antibody, HBs-Ag: hepatitis B surface antigen, HBV-DNA: hepatitis B virus-DNA, HCV-Ab: hepatitis C virus antibody, sp gr: specific gravity, RBC sediment: red blood cell sediment, beta-2-MG: beta-2-microglobulin, NAG: N-acetyl-beta-D-glucosaminidase, BJP: Bence Jones protein
Figure 2.

Echography of both kidneys. Some renal cysts were observed, but no atrophy or anomalies were present in either kidney.
To determine the pathogenesis of proteinuria in this patient, we performed a kidney biopsy. On light microscopy, 25% of glomeruli showed global sclerosis, and the major lesion in the rest of the glomeruli was segmental mesangial proliferation with a partial double contour of the glomerular basement membrane, indicating unusual proliferative glomerulonephritis (Fig. 3a and b). The other lesions were focal segmental sclerosis (5% of glomeruli), and 1 glomerulus showed fibro-cellular crescent formation. Regarding tubulo-interstitial lesions, mild to moderate fibrosis and tubular atrophy were found (Fig. 3b). The patient had had hypertension and hyperlipidemia, but there was no apparent arterio-arteriolosclerosis.
Figure 3.

Kidney biopsy findings. a) Light microscopy findings of periodic acid-methenamine-silver staining (magnification×400). Glomerular basement membrane duplication (arrowhead) and endocapillary proliferation (small square) were observed. b) Light microscopy findings of periodic acid-Schiff staining (magnification×100). Segmental mesangial proliferation (small square) and moderate interstitial injury (arrowheads) were observed. c) The findings of the immunofluorescence staining. IgG (-), IgM (+). d) The findings of immunohistochemistry for hepatitis B surface antigen (HBsAg) (magnification×400), which was negative in the glomeruli. e) The electron microscopy findings of the glomerulus (magnification×2,500 and ×15,000). A magnified view of the outlined region in the lower-magnification picture is shown as a separate picture. Ill-demarcated and low-electron-density deposits were observed within the glomerular basement membrane. Mesangial interposition and foot process effacement were also present.
Immunofluorescence examinations showed neither immunoglobulin nor complement deposition except for faint IgM staining along the glomerular basement membrane (Fig. 3c). On electron microscopy, ill-demarcated and low-electron-density deposits were localized at the subendothelial area and within the glomerular basement membrane (Fig. 3e). Mesangial interposition and foot process effacement were also observed (Fig. 3e). Given these results, we diagnosed his kidney disease as unusual proliferative glomerulonephritis type I. To determine whether or not the hepatitis B virus played a role in the glomerular lesion development, we performed immunostaining for HBsAg and obtained negative staining results (Fig. 3d).
We strongly suspected that the patient had HDR syndrome and performed a genetic analysis of the causal gene (i.e. GATA3) for confirmation. We detected a novel heterozygous p.C288W (TGC>TGG) missense mutation in GATA3 at the codon encoding a cysteine residue that forms ZnF1. Accordingly, based on the genetic analysis and the clinical parameters, we diagnosed the patient with HDR syndrome. These findings together with the lack of notable arterio-arteriolo sclerosis on light microscopy suggest that the glomerular lesion, which might have been introduced by a GATA3 mutation, eventually led to renal insufficiency. His renal function gradually deteriorated, and maintenance hemodialysis was initiated two years after the kidney biopsy.
Discussion
We herein describe a patient with HDR syndrome and hypoparathyroidism, sensorineural deafness, and glomerulonephritis. We also identified an unusual proliferation based on the renal histology. Genetic tests showed a novel mutation in the GATA3 gene.
Human GATA3 encodes 444 amino acids and contains 2 transactivating domains (TA1 and TA2) and 2 zinc finger DNA-binding domains (ZnF1 and ZnF2). Experimental findings have shown that ZnF2 is the actual DNA binding site of GATA3, and ZnF1 stabilizes this binding and interacts with other zinc finger proteins, such as Friends of GATA (FOG) (11-14). The mutation in our patient was found at a ZnF1-encoding cysteine residue, where zinc directly binds. Ali et al. analyzed 38 GATA3 mutations and classified HDR syndrome patients into 3 groups according to alterations in the binding between DNA and the mutant GATA3 (13). One group is characterized by a complete loss of DNA binding, which is mainly caused by a lack of ZnF2, where DNA directly binds. The second group exhibits a decrease in DNA-binding affinity; this mainly involves mutations in ZnF1, which strengthen the binding between DNA and GATA3. The last group is characterized by a loss of transcription owing to a conformational change in GATA3, despite normal DNA binding and normal affinity. Under this classification system, our patient fell into the second group.
Even though the GATA3 mutation in our patient caused HDR syndrome, it is still unclear whether or not this mutation was related to the membranoproliferative glomerulonephritis-like lesion observed in the renal histology. Many previous reports have examined the GATA3 expression in the developing kidney; however, very few studies have examined its expression in the adult kidney. Chan et al. have shown that GATA3 is weakly expressed in lupus nephritis kidneys (15). Sun et al. analyzed the GATA3 expression in allograft kidneys and detected intraglomerular GATA3 expression in 76.5% of the antibody-mediated rejection group, 93.5% of the T-cell-mediated rejection group, and 27.3% of recipients with a normal graft function. In their report, the GATA3 expression was scattered in the glomeruli and located mainly in the mesangial area (6).
Regarding the relationship between GATA3 mutations and the development of glomerular lesions in our patient, one possible mechanism is the imbalance of immunoreaction through the two subsets of T-helper cells: Th1 and Th2. GATA3 is indispensable for the differentiation of naïve T cells into Th2 cells, subsequently upregulating Th2 cytokines like IL-4 (16). IL-4 suppresses the differentiation of naïve T cells into Th1 cells, which further shifts the Th1/Th2 pendulum toward the Th2-related immunoresponse. Holdsworth et al. found that Th1/Th2 predominance affected the patterns and severity of glomerulonephritis and that membranoproliferative glomerulonephritis was an entity introduced by a Th1-dominant immunoresponse (17). Despite the lack of direct evidence, these recent observations suggest that a loss of the function of GATA3 due to a genome mutation might lead to a Th1-dominant immunoresponse, eventually contributing to the progression of membranoproloferative lesions observed in the present case.
In our patient, a serological examination revealed high levels of HBsAg and HBV-DNA with normal liver enzyme values, indicating that he is an HBV carrier. The common morphological forms of HBV-associated glomerulonephritis are membranous nephropathy, mesangial proliferative glomerulonephritis, and membranoproliferative glomerulonephritis (18), which was found in our patient. HBV-associated membranoproliferative glomerulonephritis is usually characterized by the deposition of circulating antigen-antibody complexes in the mesangial and subendothelial spaces, which are positive for IgG and C3 (18). However, immunostaining results for IgG and HBsAg were negative in the glomeruli, suggesting that HBV antigens and the immune responses against them might not have contributed to the glomerular lesion development in our patient. In addition, electron microscopy showed that the electron-dense deposits did not exhibit a fibrillary or band-like structure, which excludes the possibility that the glomerular lesions were caused by renal amyloidosis or Randall-type monoclonal immunoglobulin deposition diseases. These findings, together with those from a previous report on a similar membranoproliferative glomerulonephritis-like glomerular lesion in a 4-year-old HDR syndrome patient (10), suggest that the renal lesion in our patient might have been caused by his HDR syndrome.
In summary, we presented an HDR patient with a novel missense mutation in GATA3 affecting the zinc finger 1 domain. We also described the likely clinical impact of this disease-namely the development of unusual proliferation in the renal histology. Additional histological analyses of the kidneys of HDR syndrome patients are warranted, and these analyses may clarify the role of GATA3 not only in the developing kidney but also in the adult kidney, especially in the glomerulus.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Bilous RW, Murty G, Parkinson DB, et al. . Brief report: autosomal dominant familial hypoparathyroidism, sensorineural deafness, and renal dysplasia. N Engl J Med 327: 1069-1074, 1992. [DOI] [PubMed] [Google Scholar]
- 2. Hasegawa T, Hasegawa Y, Aso T, et al. . HDR syndrome (hypoparathyroidism, sensorineural deafness, renal dysplasia) associated with del(10)(p13). Am J Med Genet 73: 416-418, 1997. [DOI] [PubMed] [Google Scholar]
- 3. Van Esch H, Groenen P, Nesbit MA, et al. . GATA3 haplo-insufficiency causes human HDR syndrome. Nature 406: 419-422, 2000. [DOI] [PubMed] [Google Scholar]
- 4. Labastie MC, Catala M, Gregoire JM, Peault B. The GATA-3 gene is expressed during human kidney embryogenesis. Kidney Int 47: 1597-1603, 1995. [DOI] [PubMed] [Google Scholar]
- 5. Grote D, Souabni A, Busslinger M, Bouchard M. Pax 2/8-regulated Gata 3 expression is necessary for morphogenesis and guidance of the nephric duct in the developing kidney. Development 133: 53-61, 2006. [DOI] [PubMed] [Google Scholar]
- 6. Sun Q, Cheng D, Zhang M, He Q, Chen Z, Liu Z. Predominance of intraglomerular T-bet or GATA3 may determine mechanism of transplant rejection. J Am Soc Nephrol 22: 246-252, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grote D, Boualia SK, Souabni A, et al. . Gata3 acts downstream of beta-catenin signaling to prevent ectopic metanephric kidney induction. PLoS Genet 4: e1000316, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Belge H, Dahan K, Cambier JF, et al. . Clinical and mutational spectrum of hypoparathyroidism, deafness and renal dysplasia syndrome. Nephrol Dial Transplant 2016(Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 9. Ferraris S, Del Monaco AG, Garelli E, et al. . HDR syndrome: a novel “de novo” mutation in GATA3 gene. Am J med Genet A 149A: 770-775, 2009. [DOI] [PubMed] [Google Scholar]
- 10. Chenouard A, Isidor B, Allain-Launay E, Moreau A, Le Bideau M, Roussey G. Renal phenotypic variability in HDR syndrome: glomerular nephropathy as a novel finding. Eur J Pediatr 172: 107-110, 2013. [DOI] [PubMed] [Google Scholar]
- 11. Muroya K, Hasegawa T, Ito Y, et al. . GATA3 abnormalities and the phenotypic spectrum of HDR syndrome. J Med Genet 38: 374-380, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nesbit MA, Bowl MR, Harding B, et al. . Characterization of GATA3 mutations in the hypoparathyroidism, deafness, and renal dysplasia (HDR) syndrome. J Biol Chem 279: 22624-22634, 2004. [DOI] [PubMed] [Google Scholar]
- 13. Ali A, Christie PT, Grigorieva IV, et al. . Functional characterization of GATA3 mutations causing the hypoparathyroidism-deafness-renal (HDR) dysplasia syndrome: insight into mechanisms of DNA binding by the GATA3 transcription factor. Hum Mol Genet 16: 265-275, 2007. [DOI] [PubMed] [Google Scholar]
- 14. Yang Z, Gu L, Romeo PH, et al. . Human GATA-3 trans-activation, DNA-binding, and nuclear localization activities are organized into distinct structural domains. Mol Cell Biol 14: 2201-2212, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan RW, Lai FM, Li EK, et al. . Imbalance of Th1/Th2 transcription factors in patients with lupus nephritis. Rheumatology (Oxford) 45: 951-957, 2006. [DOI] [PubMed] [Google Scholar]
- 16. Tindemans I, Serafini N, Di Santo JP, Hendriks RW. GATA-3 function in innate and adaptive immunity. Immunity 41: 191-206, 2014. [DOI] [PubMed] [Google Scholar]
- 17. Holdsworth SR, Kitching AR, Tipping PG. Th1 and Th2 T helper cell subsets affect patterns of injury and outcomes in glomerulonephritis. Kidney Int 55: 1198-1216, 1999. [DOI] [PubMed] [Google Scholar]
- 18. Bhimma R, Coovadia HM. Hepatitis B virus-associated nephropathy. Am J Nephrol 24: 198-211, 2004. [DOI] [PubMed] [Google Scholar]