

Abstract
A cure for epilepsy is currently not available, and seizure genesis, seizure recurrence, and resistance to antiseizure drugs remain serious clinical problems. Studies show that the blood–brain barrier is altered in animal models of epilepsy and in epileptic patients. In this regard, seizures increase expression of blood–brain barrier efflux transporters such as P-glycoprotein (P-gp), which is thought to reduce brain uptake of antiseizure drugs, and thus, contribute to antiseizure drug resistance. The goal of the current study was to assess the viability of combining in vivo and ex vivo preparations of isolated brain capillaries from animal models of seizures and epilepsy as well as from patients with epilepsy to study P-gp at the blood–brain barrier. Exposing isolated rat brain capillaries to glutamate ex vivo upregulated P-gp expression to levels that were similar to those in capillaries isolated from rats that had status epilepticus or chronic epilepsy. Moreover, the fold-increase in P-gp protein expression seen in animal models is consistent with the fold-increase in P-gp observed in human brain capillaries isolated from patients with epilepsy compared to age-matched control individuals. Overall, the in vivo/ex vivo approach presented here allows detailed analysis of the mechanisms underlying seizure-induced changes of P-gp expression and transport activity at the blood–brain barrier. This approach can be extended to other blood–brain barrier proteins that might contribute to drug-resistant epilepsy or other CNS disorders as well.
Keywords: P-glycoprotein, blood–brain barrier, status epilepticus, epilepsy, efflux transporter, pilocarpine
Graphical abstract
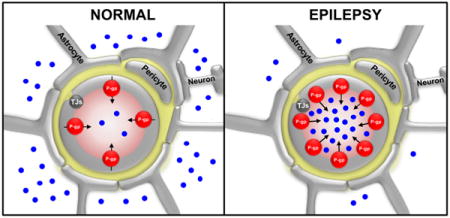
INTRODUCTION
Epilepsy is the most common neurological disorder that affects more than 65 million individuals worldwide.1,2 One major clinical problem in epilepsy therapy is drug resistance (refractory or intractable epilepsy), where patients do not, or only poorly, respond to treatment with antiseizure drugs (ASDs).3 The causes and mechanisms that contribute to ASD resistance are largely unknown and are an area of current research. The drug efflux transporter hypothesis is one of several hypotheses that has been postulated regarding the cause of ASD resistance.4–6
Drug efflux transporters at the blood–brain barrier determine brain penetration of many CNS-active drugs, including some ASDs.7,8 Consequently, increased blood–brain barrier efflux transporter expression and activity in conditions such as brain cancer, HIV encephalitis, and epilepsy limits drug brain uptake and thus decreases drug efficacy in these disorders.6 The major drug efflux transporter at the blood–brain barrier is P-glycoprotein (P-gp, ABCB1), a 180 kDa membrane protein that functions as an ATP-driven efflux pump.9,10 P-gp transports a wide spectrum of structurally diverse and unrelated hydrophobic and amphipathic compounds, and has been shown to handle ASDs such as phenytoin, phenobarbital, felbamate, lamotrigine, topiramate, and levetiracetam.11–18
Increased P-gp protein expression levels have been detected in epileptogenic tissue from patients diagnosed with refractory epilepsy as well as in several rodent models of epilepsy; this phenomenon has been linked to poor brain uptake of ASDs.6,19–31 The recent PET study by Feldmann et al.20 provided the strongest evidence thus far that P-gp is relevant in ASD-resistant patients. This study included 14 patients with refractory temporal lobe epilepsy (TLE), eight patients with controlled TLE, and 13 healthy controls. Brain uptake of the P-gp substrate [11C]verapamil was reduced in patients with refractory TLE relative to healthy controls. This study is the first direct in vivo evidence of P-gp overactivity in patients with refractory epilepsy. These findings were recently confirmed by Bauer et al.19 Based on these observations, it has been postulated that increased expression levels of blood–brain barrier efflux transporters contribute to ASD resistance in patients with epilepsy.19,21 Thus, upregulation of P-gp expression and activity levels at the blood–brain barrier by epileptic seizures may explain the most reliable predictor of ASD-resistant epilepsy: seizure frequency before onset of antiseizure pharmacotherapy.32–34
Previously, we used freshly isolated brain capillaries as an ex vivo model of the blood–brain barrier to study P-gp protein expression levels in whole cell preparations or brain capillary crude membrane preparations.32,35,36 We also used isolated brain capillaries to study P-gp transport activity by measuring accumulation of the fluorescent P-gp substrate NBD-cyclosporin A (NBD-CSA) in the capillary lumen with confocal microscopy.32,37,38 In addition, we used isolated brain capillaries to identify the signaling pathways involved in upregulating P-gp at the blood–brain barrier by various stimuli, among them seizures and status epilepticus (SE).32,34,39 We found that seizure-induced release of glutamate signals through the N-methyl D-aspartate receptor (NMDAR), cyclooxygenase-2 (COX-2), prostanoid E receptor 1 (EP1), and the transcription factor NF-кB resulted in increased expression of P-gp in brain capillaries.32,34,39 However, the major limitation in these experiments was that seizure activity has to be artificially mimicked by exposing brain capillaries from naїve animals to glutamate.
In previous studies using these protocols, we and others demonstrated that SE increases P-gp protein expression levels in endothelial cells of brain slices.32,34,40 Such protein expression data, however, does not provide information on P-gp transport activity, which according to previous studies may be one of multiple factors that controls ASD brain uptake.41,42 While P-gp stored in intracellular vesicles of endothelial cells can be immunohistochemically detected in brain sections, only P-gp localized in the luminal plasma membrane of brain capillary endothelial cells actively contributes to transport. Thus, only functional P-gp contributes to ASD efflux, which affects ASD brain uptake, likely reducing ASD efficacy.6,41
In the present study, we dosed rats with the muscarinic receptor agonist pilocarpine to induce SE following different protocols (high dose pilocarpine model, fractionated pilocarpine dosing model). We isolated brain capillaries from these rats after acute SE and from chronic epileptic rats and found increased P-gp protein expression and transport activity levels that were independent of the rat model used. Exposing isolated brain capillaries to glutamate also increased P-gp protein expression and transport activity. The increase of P-gp protein expression across the different animal models is comparable to the increase observed in human brain capillaries isolated from individuals with epilepsy who experienced generalized seizures compared to brain capillaries isolated from seizure-free control individuals.
Our study demonstrates that a combined in vivo/ex vivo approach of isolating brain capillaries from animal models of seizures and epilepsy and using isolated brain capillaries from patients with epilepsy is a viable approach to study P-gp at the blood–brain barrier. Importantly, this approach can be extended to other blood–brain barrier proteins that might contribute to ASD-resistant epilepsy or other CNS disorders as well.
MATERIALS AND METHODS
Chemicals
Pilocarpine, lithium chloride, methyl-scopolamine, and all other chemicals were purchased from Sigma (St. Louis, MO, USA). Diazepam was obtained from Hospira (Lake Forest, IL, USA). Mouse monoclonal C219 antibody against P-gp was obtained from Signet Laboratories (Dedham, MA, USA); Human P-gp protein fragment and mouse monoclonal β-actin antibody was from Abcam (Cambridge, MA, USA). The β-actin antibody used for WES multiplex protein detection was purchased from Cell Signaling Technology (Danvers, MA, USA). The Wes Master Kit was from ProteinSimple (San Jose, CA, USA). [N-ε (4-Nitrobenzofurazan-7-yl)-D-Lys8]-cyclosporin A (NBD-CSA) was custom-synthesized by R. Wenger (Basel, Switzerland;43). PSC833 was a gift from Novartis (Basel, Switzerland).
Human Brain Tissue Samples
Human brain tissue samples (inferior parietal lobule) were obtained from the UK-ADC tissue bank. Case inclusion criteria for this study were enrollment in the UK-ADC longitudinal autopsy cohort,44 short post-mortem interval (≤4 h), and a final consensus diagnosis of normal cognition determined by the UK-ADC neuropathologists, neuropsychologists, and neurologists. Cases were classified into two groups: (1) normal without seizures (n = 2) and (2) normal with seizures (n = 2). A classification of “normal” denotes a consensus diagnosis of normal cognition and CERAD rating of “criteria not met”.45 All brain tissue samples were from female individuals, whose average age at death was 84 ± 0 years (group 1, no seizures, post-mortem interval: 2.8 h ± 0.4 h) and 91.5 ± 0.7 years (group 2, seizures, post-mortem interval: 4.0 h ± 1.4h).
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee and carried out in strict accordance with AAALAC regulations, the US Department of Agriculture Animal Welfare Act, and the Guide for the Care and Use of Laboratory Animals of the NIH. After arrival in the animal facility, female Wistar Unilever rats (180–200 g, 8–10 weeks, Charles River Laboratories, Portage, MI, USA) were housed under controlled environmental conditions (22–24 °C, 40% relative humidity, 12 h dark/light cycle) with free access to tap water and standard Laboratory Rodent Diet 5001 (LabDiet, Richmond, IN, USA). Animals were allowed to adapt to the new environment for at least 1 week before experiments. All epileptic animals were monitored three times daily: once in the morning and once in the evening by laboratory staff, and once during the day by animal facility staff.
Epileptic animals that displayed symptoms that went beyond of exhibiting regular seizures were monitored more closely and obtained individualized care. Specifically, animals that lost weight were given Critical Care animal feed (Oxbow Animal Health, Murdock, NE, USA). Animals that did not recover and kept losing weight were euthanized by CO2 inhalation followed by decapitation. At the end of the experiments, all rats were euthanized by CO2 inhalation followed by decapitation.
Induction of Status Epilepticus
Status epilepticus in female Wistar rats was chemically induced using the muscarinic receptor agonist pilocarpine following two protocols.
Protocol 1: High Dose Pilocarpine Status Epilepticus Protocol.46
All rats (control group n = 6; pilocarpine group n = 24) received methyl-scopolamine (1 mg/kg i.p.). Thirty minutes after methyl-scopolamine, control rats received saline and rats in the pilocarpine group received 340 mg/kg pilocarpine i.p. Within 20–60 min of pilocarpine dosing, 50% of animals (12 of 24 rats) experienced ongoing generalized convulsive seizures (status epilepticus, SE). SE was continuously monitored and terminated after 90 min with diazepam (10 mg/kg, i.p.); diazepam administration was repeated after 10 min if seizure activity continued. Of the 12 rats where pilocarpine dosing induced SE, three animals survived (75% mortality). Of the 12 rats where pilocarpine dosing did not induce SE, eight animals survived and were used as pilocarpine control animals. All three groups of rats (control rats (n = 6), rats that received pilocarpine but did not develop SE (n = 8), and rats with pilocarpine-induced SE (n = 3)) were euthanized 48 h after SE induction to isolate brain capillaries.
Protocol 2: Fractionated Pilocarpine Dose Status Epilepticus Protocol.47
Fourteen hours prior to pilocarpine dosing, all rats (control group, n = 6; pilocarpine group, n = 24) were given lithium chloride (127 mg/kg) by i.p. injection; 30 min prior to pilocarpine dosing, all rats were given methyl-scopolamine (1 mg/kg) by i.p. injection. To induce SE, rats in the pilocarpine group (n = 24) received pilocarpine (10 mg/kg) by repeated i.p. injections every 30 min until the onset of ongoing generalized convulsive seizures (SE) that were comparable to those observed in the high-dose pilocarpine model. The maximum number of pilocarpine injections was limited to 12 per animal (maximum dose per animal: 120 mg/kg). Of the 24 rats that received fractionated pilocarpine dosing, 20 rats developed SE. As under protocol 1, SE in rats was continuously monitored and terminated after 90 min by i.p. injection of diazepam (10 mg/kg). Of the 20 rats experiencing SE, 16 animals survived (20% mortality). Of the four rats where pilocarpine did not induce SE, all four animals survived and were used as pilocarpine control animals. All three groups of rats (control rats (n = 6), rats that received pilocarpine but did not develop SE (n = 4), and rats with pilocarpine-induced SE (n = 16)) were euthanized 48 h after SE induction followed by isolation of brain capillaries.
Chronic Epilepsy Model and Video-Monitoring
To generate rats with chronic epilepsy (recurrent, spontaneous seizures), animals underwent SE induction as described above under “Protocol 2: Fractionated Pilocarpine Dose Status Epilepticus Protocol”. Instead of euthanasia, rats that underwent SE induction were kept until spontaneous and recurrent generalized seizures (epilepsy) occurred about 3 months after SE. Spontaneous recurrent generalized seizures in rats were assessed by continuous (24/7) video-monitoring over a four-week period. Video data were collected using Bosch WZ16N4080 compact integrated day-night cameras in infrared mode that were connected to an Exacq 1608-12-1000 16-camera DVR. Recorded video data were analyzed in a double-blind fashion using ExacqVision Pro VMS Software v4 (Fishers, IN, USA) to determine seizure occurrence/frequency, duration, and severity. Seizure severity was graded with a class-ranking system developed by Racine (1972) based on the following types of behavior: either unilateral or bilateral forearm clonus (class 3), rearing (class 4), and rearing and loss of postural control (class 5). Only rats with spontaneous, recurrent seizures were used for the following studies.
Brain Capillary Isolation
Brain capillaries from rats and human brain tissue were isolated according to previously described protocols.36,38,48 Rats were euthanized by CO2 inhalation and decapitated; brains were immediately harvested and collected in ice-cold PBS buffer (2.7 mM KCl, 1.46 mM KH2PO4, 136.9 mM NaCl, 8.1 mM Na2HPO4, supplemented with 5 mM D-glucose and 1 mM Na-pyruvate, pH 7.4). Brains were dissected by removing meninges, choroid plexus, and white matter, and homogenized in PBS. The brain homogenate was mixed with Ficoll (final concentration 15%) and centrifuged at 5800g for 20 min at 4 °C. The capillary pellet was resuspended in 1% BSA-PBS buffer and passed over a glass bead column (glass bead diameter 0.4–0.6 mm; Sartorius AG, Göttingen, Germany). Brain capillaries were agitated in 1% BSA-PBS, and the brain capillary containing supernatant was collected, centrifuged, and washed with PBS. Freshly isolated brain capillaries were used for transport experiments and plasma membrane isolation followed by Western blotting and analysis with Wes.
Frozen human brain tissue was slowly thawed within 10 min and then cleaned from meninges prior to homogenization. All following steps were conducted according to the protocol for rat brain capillary isolations mentioned above.
P-gp Transport Activity
P-gp transport activity in isolated brain capillaries was assessed as described previously.36,38,48 Capillaries were incubated in confocal imaging chambers for 1 h at RT with 2 μM of the fluorescent P-gp substrate, NBD-cyclosporin A (NBD-CSA). For each treatment, confocal images of 10–15 capillaries were acquired using a Nikon C1 laser scanning confocal microscope unit (Nikon TE2000 inverted microscope, 40× oil immersion objective, numerical aperture = 1.3, 488 nm line of a Argon laser; Nikon Instruments Inc., Melville, NY, USA). Confocal images were analyzed by quantitating NBD-CSA fluorescence intensity in the capillary lumen using NIH ImageJ software, version 1.40g (NIH, Bethesda, MD, USA) as previously described.37 Specific luminal NBD-CSA fluorescence was calculated as the difference between total luminal fluorescence and fluorescence in the presence of the P-gp-specific inhibitor PSC833 as previously reported.37,38,49,50
Tissue Sampling, Capillary Membrane Isolation, and Western Blotting
To determine P-gp expression in the parahippocampal cortex and the hippocampus, we used two control animals, two animals that received pilocarpine but did not develop SE, and two animals that experienced SE. Animals were euthanized as described above. Parahippocampal cortex and hippocampus tissue were harvested and immediately snap frozen in liquid nitrogen. Isolated brain capillaries were homogenized in mammalian tissue lysis buffer (Sigma, St. Louis, MO, USA) containing protease inhibitor cocktail (Roche, Mannheim, Germany). Homogenized brain capillary samples were centrifuged at 10,000g for 15 min at 4 °C, followed by a centrifugation of the denucleated supernatants at 100,000g for 90 min at 4 °C. Pellets (crude brain capillary plasma membranes) were resuspended and protein concentrations were determined using the Bradford assay. Western blots were performed using the NuPage electrophoresis and blotting system from Invitrogen (Carlsbad, CA, USA35). Blotting membranes were incubated overnight with antibody to P-gp (1:100, 1 μg/mL) and β-actin (1:1000, 1 μg/mL), washed, and incubated with the corresponding horseradish peroxidase-conjugated ImmunoPure secondary antibody (1:15,000, Pierce, Rockford, IL, USA). Proteins were detected using SuperSignal West Pico Chemoluminescent substrate (Pierce, Rockford, IL, USA), and protein bands were visualized with a BioRad Gel Doc XRS imaging system (BioRad, Hercules, CA, USA).
Simple Western Assay
In addition to Western blotting, P-gp was analyzed and quantitated with a Simple Western assay using the Wes instrument (ProteinSimple, San Jose, CA, USA). The Simple Western assay is a capillary electrophoresis technique that automates protein loading, separation, immunoprobing, washing and detection, and allows absolute protein quantitation.1,51,52 For the assay, reagents of the Wes Master Kit (ProteinSimple, San Jose, CA) were used, and all steps of the assay were performed according to the manufacturer’s protocol. Samples were diluted with WES Master Kit sample buffer, mixed 1:4 with Wes fluorescent master mix to a final concentration of 0.005 μg/μL, and heated at 70 °C for 10 min. Samples, primary antibodies, Wes Master Kit blocking buffer, secondary antibody, washing buffer, and chemiluminescent substrate were dispensed in a microplate provided by the manufacturer. A Wes Master Kit capillary cartridge and the prepared microplate were placed into the Wes instrument, which processed all assay steps automatically using default settings. Briefly, capillaries were loaded with both stacking and separation matrices followed by sample loading. During capillary electrophoresis, proteins were separated by size and then immobilized to the capillary wall. P-gp and β-actin were identified with primary antibodies against P-gp (C219, Thermo-Scientific, 1:150) and β-actin (β-actin antibody, Cell Signaling Technology, 1:20), followed by immunodetection using Wes Master Kit horseradish peroxidase-conjugated antimouse secondary antibody and chemiluminescent substrate. Protein signal and molecular weight were automatically reported by the Compass software (version 2.6.5; ProteinSimple, San Jose, CA, USA). Using Compass software, electropherograms were generated for each capillary (treatment group) and each protein (P-gp, β-actin) and the area under the curve, which represents the signal intensity of the chemiluminescent reaction and is proportional to the amount of target protein in a respective capillary, was analyzed for P-gp and β-actin.
Statistical Analysis
Data are presented as means ± SEM. One-way analysis of variance (ANOVA) or two-tailed unpaired Student’s t test was used to evaluate differences between controls and treated groups; differences were considered to be statistically significant at p < 0.05.
RESULTS
Glutamate Increases P-gp Expression and Activity in Isolated Rat Brain Capillaries
We exposed brain capillaries isolated from naïve rats to 50–100 μM glutamate for 30 min and measured P-gp protein expression and transport activity after 6 h. This protocol was designed to mimic conditions in vivo, where glutamate is released during seizures. In epileptic patients, glutamate levels in the brain after seizures are mostly in the lower micromolar range.53–55 However, glutamate concentrations of up to 75 μM have been measured in the epileptogenic hippocampus in patients.53,54 In preliminary experiments using isolated brain capillaries, we did not observe changes in P-gp protein expression and transport activity levels at glutamate concentrations below 25 μM. Therefore, we exposed isolated rat brain capillaries to 50 and 100 μM glutamate and used Western blotting to assess P-gp protein expression in isolated brain capillary membranes. We found a concentration-dependent increase in P-gp expression levels with 50 and 100 μM glutamate compared to untreated control capillaries (Figure 1A; MW P-gp, 180 kDa; MW β-actin, 42 kDa). To determine P-gp transport activity we used a previously established assay32,37,38,49,50 and exposed isolated, intact brain capillaries to the fluorescent cyclosporin A derivative, NBD-CSA, and monitored its accumulation in brain capillary lumens as a measure of P-gp transport activity. We observed a concentration-dependent increase in NBD-CSA accumulation in capillary lumens from capillaries exposed to glutamate compared to control capillaries (Figure 1B), indicating increased P-gp transport activity. Image analysis revealed that 50 μM glutamate increased P-gp expression levels by 78% (1.8-fold) and 100 μM glutamate increased P-gp expression levels by 178% (2.8-fold; Figure 1C). This observation is consistent with increased P-gp protein expression levels and our previously published data.32,34,39 To test if glutamate mediates the increase in P-gp expression and activity through a mechanism involving transcription and translation, we performed experiments with the transcription inhibitor actinomycin D and the translation inhibitor cycloheximide. Figure 1D,E show that both actinomycin D and cycloheximide prevented the glutamate-mediated increase of P-gp protein expression and transport function. These data suggest that the glutamate-mediated increase of P-gp involves both transcription and translation.
Figure 1.
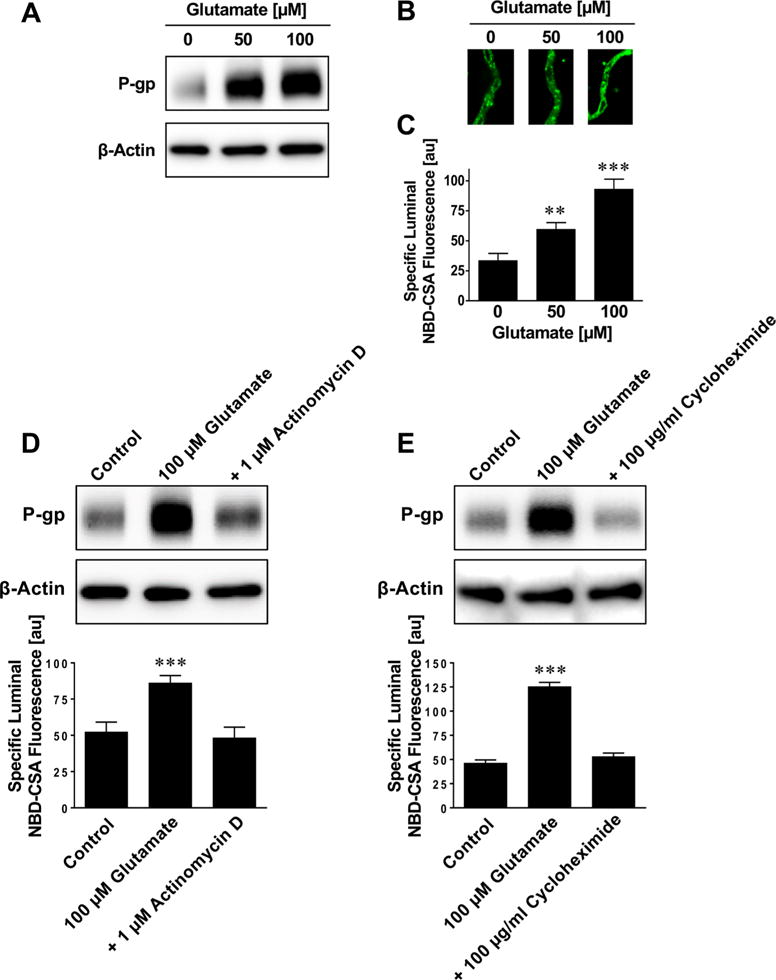
Glutamate upregulates P-glycoprotein in brain capillaries ex vivo. (A) Western blot showing that glutamate increases P-gp protein expression levels in crude membranes isolated from brain capillaries. β-Actin was used as protein loading control. Isolated brain capillaries were exposed to 50–100 μM glutamate for 30 min then incubated in glutamate-free medium for 5 1/2 h followed by capillary membrane isolation. (B) Representative confocal microscopy images of isolated brain capillaries that were first exposed to 50–100 μM glutamate for 30 min followed by 5 1/2 h in glutamate-free medium, and then exposed to the fluorescent P-gp substrate NBD-cyclosporin A (NBD-CSA; marker for P-gp transport activity). NBD-CSA fluorescence in the capillary lumen increases with increasing glutamate concentration, indicating an increase in P-gp transport activity. (C) Specific NBD-CSA fluorescence in the capillary lumen increases with increasing glutamate concentration. Data were obtained through analysis of the confocal images in B. Specific NBD-CSA fluorescence is the difference between total luminal fluorescence and fluorescence in the presence of the specific P-gp inhibitor PSC833, thus representing specific P-gp transport activity. (D) Actinomycin D, a transcription inhibitor, and (E) cycloheximide, a translation inhibitor, block the effect of glutamate. Data are mean ± SEM (n =10–15 capillaries per treatment group from one brain capillary isolation; pooled tissue from 10 rats). Units are arbitrary fluorescence units (scale, 0–255). Statistical comparison (one-way ANOVA): **, significantly higher than controls, p < 0.01; ***, significantly higher than controls, p < 0.001.
Status Epilepticus Increases P-gp Expression and Activity in Rats
We assessed two protocols to chemically induce SE in rats with the muscarinic receptor agonist pilocarpine. Using a “high dose pilocarpine” protocol, female rats were given one dose of 340 mg/kg pilocarpine i.p. to induce status epilepticus (Figure 2A).46 Brain capillaries and brain capillary membranes were isolated 48 h after SE to determine P-gp protein expression and transport activity levels. Figure 2B,C show increased P-gp expression and transport activity levels in brain capillaries from SE rats compared to capillaries from control rats and rats that received pilocarpine but did not develop SE.
Figure 2.
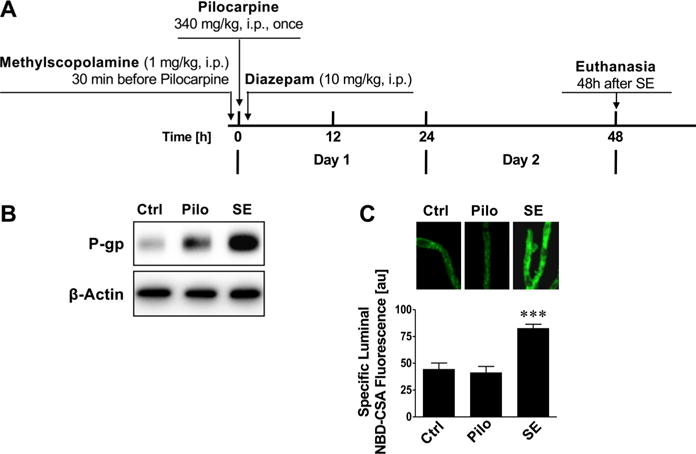
Status epilepticus (high dose SE model) upregulates P-glycoprotein in brain capillaries in vivo. (A) High dose pilocarpine protocol to induce status epilepticus. (B) Western blot showing that P-gp protein expression levels are increased in brain capillary membranes of animals that received one high dose of pilocarpine to induce status epilepticus (SE) compared to control animals that did not receive pilocarpine. P-gp protein expression levels are also slightly increased in brain capillary membranes from rats that received pilocarpine but did not develop SE. This increase is likely due to the few seizures these animals experienced (not a full SE), suggesting a “dose response-like” relation between seizures (glutamate release) and P-gp protein expression levels. β-Actin was used as protein loading control. (C) P-gp transport activity determined as NBD-CSA fluorescence accumulation in brain capillary lumens is increased in capillaries from rats that developed SE. Data are mean ± SEM (n =10–15 capillaries per treatment group; pooled tissue from all rats per group; control group n = 6 rats, pilocarpine group n = 8 rats, status epilepticus group n = 3 rats). Units are arbitrary fluorescence units (scale, 0–255). Statistical comparison (one-way ANOVA): ***, significantly higher than controls, p < 0.001.
Using the fractionated lithium-pilocarpine protocol, rats were repeatedly administered a low pilocarpine dose (10 mg/kg, i.p.) until the onset of SE (Figure 3A).47 In this protocol, each animal received an individualized dose of pilocarpine until SE onset, which minimized the risk of overdosing with pilocarpine that could potentially result in adverse side effects and higher mortality (high dose model, 75% mortality rate; fractionated model, 20% mortality rate). As in the high dose pilocarpine protocol, in the fractionated pilocarpine protocol brain capillaries were isolated 48 h after SE to determine P-gp expression and transport activity levels. SE induced by fractionated pilocarpine dosing also increased P-gp protein expression and transport activity levels in brain capillaries compared to brain capillaries from control rats and rats that did not develop SE (Figure 3B,C. Using Western blotting, we also demonstrated increased P-gp protein expression levels in whole tissue samples taken from the rat parahippocampal cortex (Figure 3D). To determine the duration of the SE effect on P-gp expression and activity levels in brain capillaries, one group of rats was kept for 2 weeks after SE induction before brain tissue was harvested and brain capillaries were isolated. P-gp protein expression and activity in brain capillaries from these rats was back to control levels 2 weeks after SE induction (Figure 3B,C). These findings are in agreement with our previous studies, where we demonstrated increased P-gp protein expression levels in sections of these brain regions using immunohistochemistry.32,34,39 Note that the slight increase in P-gp signal in the Western blots in Figures 2B and 3B for pilocarpine control rats, which experienced several seizures but not a full SE, suggests a dose response-like relation between seizures (glutamate release) and P-gp protein expression levels. Pilocarpine itself had no effect on P-gp protein expression or transport activity levels in isolated brain capillaries ex vivo (Figure 3E,F). Together, these data show that pilocarpine-induced seizures, but not pilocarpine itself, increased P-gp protein expression and transport activity levels in both brain capillary membranes and whole brain tissue. Further the data show that P-gp upregulation is independent of the pilocarpine SE induction protocol that was used.
Figure 3.
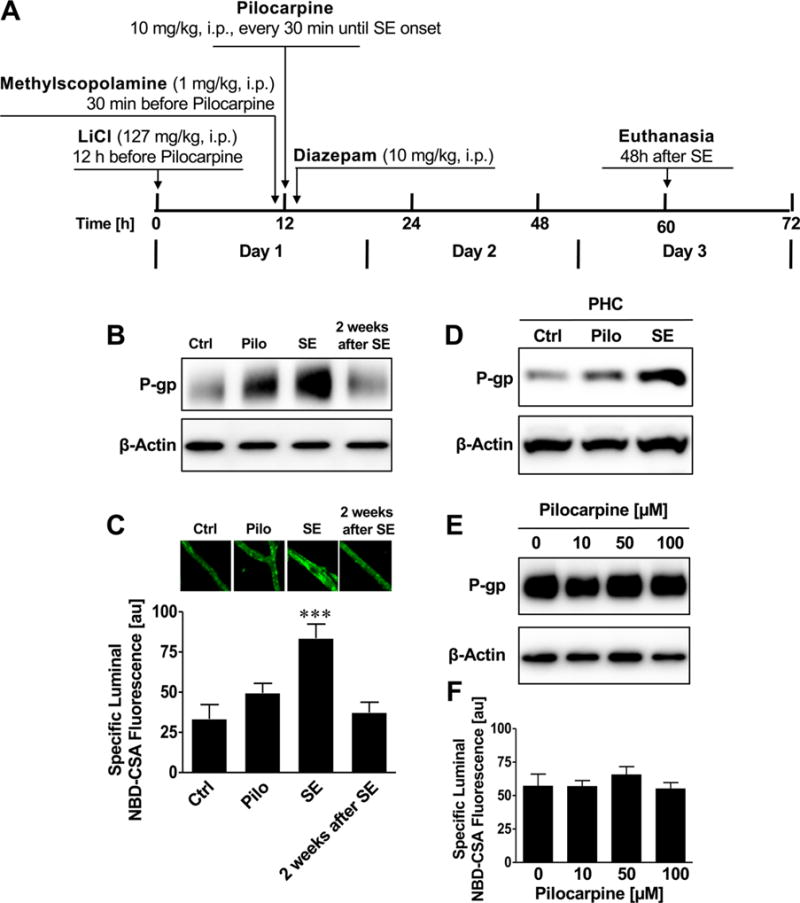
Status epilepticus (fractionated dose SE model) upregulates P-glycoprotein in brain capillaries in vivo. (A) Fractionated pilocarpine dose protocol to induce status epilepticus. (B) Western blot showing that P-gp protein expression levels are increased in brain capillaries from rats that experienced SE induced by fractionated pilocarpine dosing. The Western blot also shows that 2 weeks after SE induction this effect was reduced and P-gp expression levels were again at control levels. β-Actin was used as protein loading control. (C) P-gp transport activity determined as NBD-CSA fluorescence accumulation in brain capillary lumens is increased in capillaries from rats that developed SE, but this effect is normalized 2 weeks after SE. (D) Western blot showing that P-gp protein expression levels are increased in the parahippocampal cortex (PHC) from rats that experienced SE induced by fractionated pilocarpine dosing. β-Actin was used as protein loading control. (E,F) Western blot and P-gp transport activity assay showing that pilocarpine does not affect P-gp protein expression and transport activity levels. Isolated brain capillaries were exposed to 10–100 μM pilocarpine for 6 h. Data are mean ± SEM (n =10–15 capillaries per treatment group; pooled tissue from all rats per group; control group n = 6 rats, pilocarpine group n = 4 rats, status epilepticus group n = 16 rats). Units are arbitrary fluorescence units (scale, 0–255). Statistical comparison (one-way ANOVA): ***, significantly higher than controls, p < 0.001.
Chronic Epilepsy Increases P-gp Expression and Activity in Rats
We used a rat chronic epilepsy model that has been described as an ASD-resistant epilepsy model and resembles human ASD-resistant complex focal epilepsy.56,57 To generate chronic epilepsy (CE) in rats, we induced SE as described above and shown in Figure 4A. Only animals experiencing SE with generalized seizures for 90 min were selected for the following procedures. Twelve weeks after SE, rats were continuously video-monitored (24/7) for 4 weeks to identify spontaneous recurrent seizures and confirm chronic epilepsy. Supporting Information shows a representative video of a rat experiencing a seizure. The recording is analyzed for seizure frequency, duration, and severity; seizure severity was categorized using the Racine Scale.58 Table 1 shows that CE rats had on average 51 ± 14.7 seizures/week that lasted in average 24 ± 4.3 s and had an average seizure severity of 4.7 ± 0.14 (Racine scale, range: 1–5; 0, no seizures; 1, least severe seizures; 5, most severe seizures).58
Figure 4.
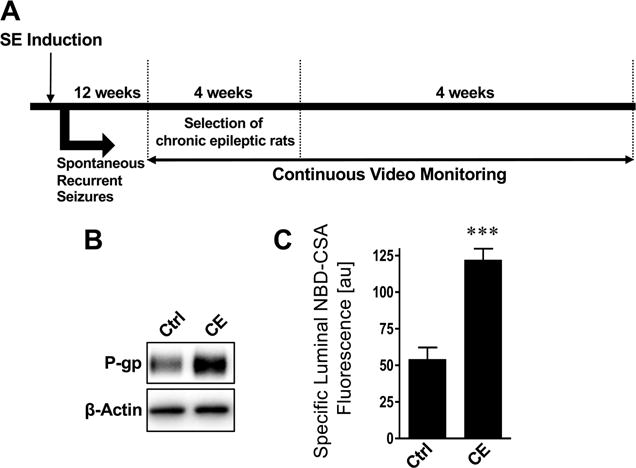
P-glycoprotein is upregulated in isolated brain capillaries from chronic epileptic rats. (A) Schematic showing the protocol to generate chronic epileptic rats using the fractionated pilocarpine dose approach. (B) Western blot and (C) transport activity assay showing that P-gp protein expression and transport activity levels are increased in brain capillaries isolated from CE rats. Data are mean ± SEM (n = 10–15 capillaries per treatment group; pooled tissue from all rats per group; control group n = 10 rats, chronic epileptic group n = 5 rats). Units are arbitrary fluorescence units (scale, 0–255). Statistical comparison (one-way ANOVA): ***, significantly higher than controls, p < 0.001.
Table 1.
Seizure Frequency, Duration, and Severity of Chronic Epileptic Ratsa
| seizures/week | seizure duration (s) | seizure severity (Racine scale 0–5) | |
|---|---|---|---|
| control rats | 0 | 0 | 0 |
| chronic epileptic rats | 51 ± 14.7 | 24 ± 4.3 | 4.7 ± 0.14 |
The table summarizes seizure frequency, duration, and severity (using the Racine scale58) of chronic epileptic rats over the course of 4 weeks based on video-analysis. Data are mean ± SEM (control group, n =10 rats; chronic epileptic group, n = 18 rats).
Three months after confirming chronic epilepsy, CE rats were euthanized and brain capillaries were isolated. Protein expression levels of the efflux transporter P-gp were increased in capillaries from CE rats compared to capillaries from control rats (Figure 4B). Consistent with this finding, P-gp transport activity levels in brain capillaries from CE rats were 2.3-fold higher compared to control capillaries (126% higher than controls; Figure 4C). These data suggest that the mechanisms underlying the changes in P-gp protein expression and transport activity are not different between the acute SE models (Figures 2 and 3) and the CE model (Figure 4).
P-gp Expression and Activity in Brain Capillaries in Animal Models and Human Epilepsy
In the present study, we used four different models: (1) ex vivo glutamate model, (2) high-dose pilocarpine rat SE model, (3) fractionated pilocarpine rat SE model, and (4) rat chronic epilepsy model. In addition, we isolated brain capillaries from post-mortem brain samples of patients with epilepsy. To assess if upregulation of P-gp in isolated brain capillaries was comparable between the various models and human epilepsy, we analyzed P-gp protein expression with the Simple Western size-separation assay, a novel and unique capillary electrophoresis technique that automates protein loading, separation, immunoprobing, washing, and detection, and allows absolute protein quantitation.1,51,52
Figure 5A shows an image of a size-based WES assay for P-gp and β-actin that were detected in brain capillary membranes isolated from normal adult post-mortem brain tissue. Bands for P-gp were detected at 183.7 ± 0.6 kDa and bands for β-actin were detected at 49.3 ± 0.6 kDa. Figure 5B shows the corresponding electropherograms of lanes 1–3 for P-gp and β-actin; an overlay is shown in Figure 5C. Signal strength is represented as peak area and can be quantitated.
Figure 5.
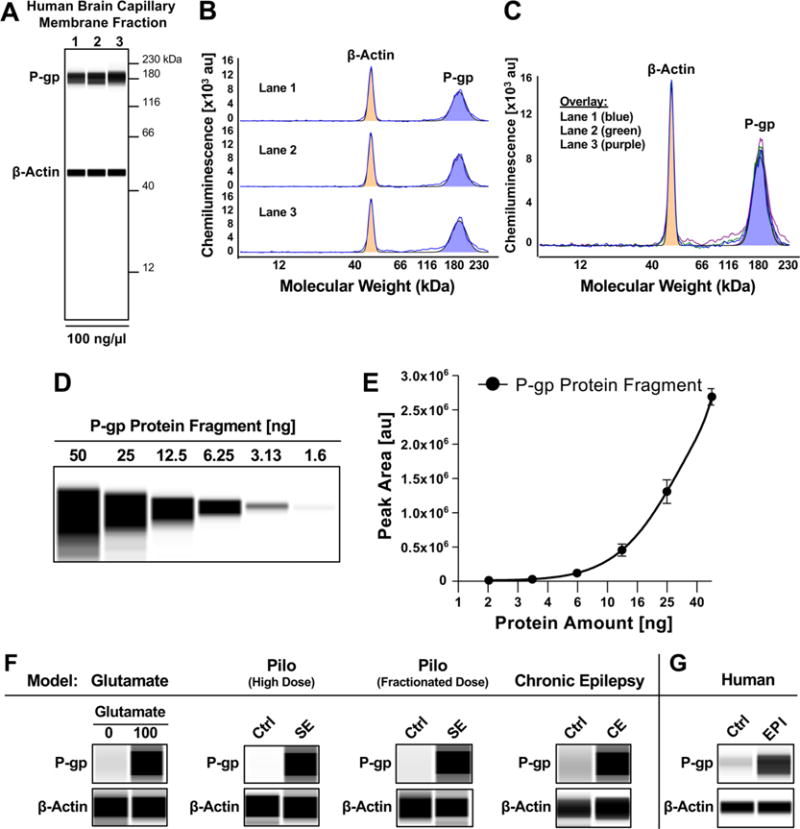
P-gp expression and activity in brain capillaries in animal models and human epilepsy. (A) Wes images showing P-gp and β-actin protein expression in isolated human brain capillaries. (B) Electropherogram for each lane and C) overlay of lanes 1–3. (D) Concentration series and (E) standard curve for human P-gp protein fragment used for quantitative analyses. (F) Wes images showing upregulation of P-gp protein expression levels in all four models: (1) ex vivo glutamate model, (2) high-dose SE model, (3) fractionated SE model, and (4) CE model. (G) Increased P-gp protein expression in brain capillaries isolated post-mortem from brains of individuals that experienced generalized seizures compared to seizure-free control individuals. β-actin was used as protein loading control.
The variation coefficient (%CV) between peak areas for lanes 1–3 is less than 1% for P-gp and less than 8% for β-actin. This is lower than the published variation coefficient (10%) for size-based WES59 and indicates that peaks for P-gp and β-actin are reproducible and can be compared between lanes. These data also demonstrate that the WES assay allows us to detect and quantify P-gp and β-actin simultaneously in human brain capillary samples at the nanogram level.
Using a human P-gp protein fragment (amino acids: 1036–1280, Abcam, Cambridge, MA, USA) we generated a standard curve for each run by plotting protein content [ng] against chemiluminescence. Figures 5D shows the WES image, and Figure 5E shows the corresponding standard curve for P-gp.
Figure 5F shows representative WES images for P-gp and β-actin for all four different models used in this study. Consistent with the data obtained by Western blotting, P-gp is upregulated in capillaries treated with glutamate (2.3-fold) as well as in capillaries isolated from the epilepsy models (high dose pilocarpine SE model (1.7-fold), fractionated pilocarpine dose SE model (1.9-fold), CE model (1.7-fold)) and the human brain tissue from patients with epilepsy (1.7-fold). Thus, the fold-change of chemiluminescence for P-gp obtained with the WES system is consistent with the data obtained with our standard Western blot method (Figure 1A).
We also calculated the total P-gp protein amount based on the standard curve and normalized to β-actin (Table 2). P-gp protein expression levels were upregulated from 100 ± 2.5% in untreated control capillaries to 154 ± 2.1% in glutamate-treated capillaries, from 100 ± 1% in control rats to 138 ± 9% in the high-dose pilocarpine rat SE model, from 100 ± 1% in control rats to 151 ± 13% in the fractionated pilocarpine dose rat SE model, and from 100 ± 14% to 140 ± 3.5% in the chronic epilepsy rat model. The fold-changes are summarized in Table 2.
Table 2.
Quantitative Analysis of P-gp Upregulation Using Simple Western Assaya
| glutamate
|
pilo (high dose)
|
pilo (fractionated dose)
|
CE
|
human
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ctrl | Glu | Ctrl | SE | Ctrl | SE | Ctrl | CE | Ctrl | EPI | |
| P-gp (ng) | 6.68 ± 0.17 | 10.6 ± 0.14 | 3.95 ± 0.04 | 5.54 ± 0.36 | 3.27 ± 0.03 | 5.00 ± 0.46 | 4.48 ± 0.63 | 6.27 ± 0.16 | 7.5 ± 0.2 | 10.5 ± 0.5 |
| ratio (treated/control) | 1.58 ± 0.04 | 1.40 ± 0.09 | 1.52 ± 0.44 | 1.40 ± 0.20 | 1.4 ± 0.22 | |||||
The table shows protein content (ng) per sample and the ratio of P-gp upregulation in all four models that were combined with capillary isolation: (1) ex vivo glutamate model (p < 0.00001), (2) high-dose SE model (p < 0.019), (3) fractionated SE model (p < 0.0002), and (4) chronic epilepsy model (p < 0.009). Data are mean ± SD (n = 3), two-tailed unpaired Student’s t-test was used to evaluate differences between controls and treated groups.
Importantly, the overall effect observed for P-gp in capillaries exposed to glutamate compared to the effect detected in capillaries isolated from SE and CE animals appears to be consistent. Moreover, we found that P-gp protein levels were upregulated from 100 ± 6% in capillaries isolated from seizure-free control individuals to 140 ± 12% in capillaries isolated from patients with epilepsy that experienced generalized seizures (Figure 5G). Please note that the fold-change for the total protein amount ranges from 1.4–1.6 in all four models (Table 2), which is slightly lower than the calculated fold-change of chemiluminescence for P-gp (1.7–2.3). This discrepancy is due to the exponential nature of the standard curve (Figure 5E).
Together, this is the first study to demonstrate that the increase in blood–brain barrier P-gp levels in multiple animal models of seizures/epilepsy is comparable to those at the blood— brain barrier of patients with epilepsy that experienced seizures.
DISCUSSION
In the present study we introduce a combined in vivo/ex vivo approach to assess P-gp protein expression and transport activity levels in models of seizures and epilepsy in the rat, as well as in brain capillaries from patients with epilepsy. Our data show that exposing brain capillaries to glutamate ex vivo and that acute seizures (SE) and chronic epilepsy (spontaneous recurrent seizures) in vivo upregulate both P-gp protein expression and transport activity. We demonstrate that glutamate increases P-gp protein expression and transport activity in a concentration-dependent manner, which is consistent with our previous findings and the current literature (Figure 1).32,34,39,60,61 A similar increase in P-gp expression and activity was found in brain capillaries isolated from rats 2 days after SE induction with pilocarpine (Figures 2 and 3). The magnitude of P-gp upregulation, both protein expression and transport activity levels, was independent of the protocol used to induce SE, in that high-dose pilocarpine administration vs fractionated pilocarpine administration resulted in a comparable effect. Further, isolated brain capillaries from CE animals showed increased P-gp levels comparable to levels we found in capillaries exposed to glutamate ex vivo and in capillaries isolated from the pilocarpine seizure models (Figure 4). These data indicate that seizure activity, and not the mode of seizure induction, determines the effect on P-gp. This conclusion is also supported by the finding that pilocarpine by itself at a concentration of up to 100 μM does not affect P-gp expression and activity levels, ruling out that pilocarpine has a direct effect on P-gp upregulation (Figure 3E,F). When comparing the increase of P-gp expression and transporter activity in capillaries isolated from SE and CE animals with the increase observed in isolated brain capillaries exposed to glutamate, the overall effect assessed with the WES system appears to be comparable and consistent (glutamate, 1.5-fold; SE (high dose), 1.4-fold; SE (fractionated dose), 1.5-fold; CE, 1.4-fold; Figure 5 and Table 2). This supports previous findings that the driving factor of P-gp upregulation is glutamate flooding of the extracellular space during seizure activity.32,60 Consistent with this are our data for the control, pilocarpine, and SE groups shown in Figure 3B,C that were generated from brain capillaries isolated 48h after SE (glutamate stimulus), where P-gp expression and activity are upregulated. However, in brain capillaries from animals that were euthanized 2 weeks after SE and did not experience seizures during this 2-week period (no glutamate stimulus), P-gp expression and activity levels were back to control levels. In contrast, in the chronic epilepsy model where animals had spontaneous recurrent seizures (constant glutamate stimulus), P-gp expression and activity are upregulated compared to control animals (Figure 4B,C). Together, these data support that glutamate released during seizures is responsible for P-gp upregulation.
Notably, the fold-change (1.4–1.5) observed in the animal models is also comparable to the fold-change found in brain capillaries isolated from patients with epilepsy vs seizure-free control individuals (1.4-fold change, Figure 5G). This indicates that the increase in P-gp protein expression levels at the blood— brain barrier in animal models is comparable to the increase in P-gp expression in brain capillaries from patients with epilepsy.
There are several aspects to note regarding the human brain samples used in this study. First, the short post-mortem interval (≤4 h) between death and brain collection indicates the high quality of the samples. Second, brain samples from patients with epilepsy and control individuals were matched for both age and brain region. Finally, published studies in this field used brain tissue resected from the seizure focus, i.e., tissue that was diseased.24,30,62,63 These studies also lack a comparison with age-and brain region-matched post-mortem samples from control individuals. In contrast, the data from human samples presented here are based on a valid comparison between diseased and nondiseased control samples that also matched for age and brain region. Despite the high level of consistency among models used here, the study was limited by the number of available human brain samples (n = 2). Another limitation of the present study is the high age of the individuals brain tissue samples were collected from (control individuals, 84 ± 0 years; epilepsy patients, 91.5 ± 0.7 years). Further studies may be required to investigate the impact age has on seizure-induced P-gp upregulation.
One challenge of working with isolated brain capillaries is the small amount of tissue available because brain capillaries occupy only approximately 1% of the brain volume,64 which results in only a small amount of protein that is available for analysis. This challenge can be overcome by using the WES system that allows detection of P-gp protein expression at the nanogram level. This new technique is of particular interest when working with human tissue where the availability of brain tissue is usually limited to small amounts. Using the WES system, we quantitated total P-gp protein amount in brain capillary membranes, something that cannot be easily accomplished by traditional Western blotting. The fold-change of chemiluminescence for P-gp in the presented epilepsy models assessed by the WES system ranges from 1.7- to 2.3-fold, this number is comparable to the 2-fold increase of P-gp obtained by densitometric analysis of traditional Western blots that we previously published.32 However, the fold-change for the total protein amount of the same data determined with the WES system is 1.4–1.6 (Table 2), which is slightly lower. This discrepancy is due to the exponential nature of the standard curve.
Recently some of the mechanisms underlying seizure-induced P-gp upregulation have been identified, yet the dynamics and the exact time-course of transporter upregulation remains unknown. In this regard, expression of mdr1 mRNA after kainic acid-induced limbic seizures in hippocampi of mice has been measured by qPCR.65 Mdr1 mRNA expression was increased by 85% 3–24 h after SE induction, and at 72 h, mdr1 mRNA expression was back to control levels. In another study, mdr1a and mdr1b mRNA expression was measured after pilocarpine-induced SE in rats.66 Unexpectedly, 6 and 24 h after SE onset, mdr1a/b mRNA was decreased in the hippocampus, amygdala, and piriform cortex, followed by normalization to control levels at approximately 24–48 h. Mdr1 mRNA levels were increased and peaked either 4 days after SE or continued to increase depending on the brain region.66 In the present study, we show that P-gp protein expression levels were increased 48 h after SE induction and were back to control levels 2 weeks after SE induction. This observation is consistent with previous findings suggesting that seizure-induced transporter upregulation per se is transient in nature.66 However, in patients with refractory epilepsy who suffer from uncontrolled seizures, one would expect that frequently recurring seizures result in a state of continuously increased P-gp levels. Additional studies are needed to understand the time-dependent regulation of transporter mRNA, protein, and activity after seizures/SE in detail, particularly in brain tissue from patients with epilepsy.
Moreover, using immunohistochemistry, other groups found that other efflux transporters like BCRP, Mrp1, and Mrp2 are also overexpressed in brain capillaries shortly after SE, during the latent period, and in chronic epileptic rats,31 and experiments with P-gp and BCRP knockout mice suggest that BCRP contributes to ASDs efflux.22 Together, these data suggest that other transporters could potentially work in concert with P-gp in limiting brain uptake of ASDs.
Clearly, blood–brain barrier P-gp expression and transport activity in epilepsy are regulated by complex molecular mechanisms involving seizure-induced glutamate release and downstream inflammatory signaling resulting in transcription and translation of the transporter.8,32,60 One approach to discern the mechanism by which glutamate triggers P-gp upregulation is utilizing isolated brain capillaries ex vivo. The major disadvantage of this approach is the need to artificially mimic changes in the epileptic brain, i.e., increased glutamate levels associated with seizures. This was accomplished by exposing isolated brain capillaries to 100 μM glutamate for 30 min, conditions that likely do not represent seizure conditions in vivo. However, here we demonstrate that this limitation may be overcome by combining established in vivo animal seizure models followed by isolating brain capillaries from these models or from the epileptic human brain and analyzing capillaries ex vivo, thus creating a unique and powerful in vivo/ex vivo approach. There are additional advantages to this approach: First, glutamate can be used as a tool to mimic glutamate flooding in the brain parenchyma during seizures.32,34,39 Second, P-gp protein expression and transport activity levels can both be measured under controlled conditions at the same time.32,34,39 Lastly experiments with pharmacological inhibitors with transgenic and/or knockout mice and with isolated brain capillaries from human brain can be performed to unravel the signaling mechanism(s) involved in transporter regulation.32
Together optimized and validated preclinical tools that correctly predict and analyze pathophysiological changes at the blood–brain barrier are in demand.67 The results of the current study show that isolated brain capillaries can be used as such a tool and that isolating brain capillaries from animal models of disease and the human epileptic brain is a viable approach to studying blood–brain barrier function in CNS disorders. This approach can also easily be extended to studying other blood— brain barrier proteins, including other drug efflux transporters,22,31 metabolizing enzymes,68–70 and tight junction proteins.71–73 In this regard, recent reports show that expression levels of metabolizing enzymes are also changed in epilepsy. For example, CYP2E1 is upregulated in microvessels after SE in brain slices from mice,68 UGT1A4 is upregulated in human brain endothelial cells isolated from temporal lobe resections from patients with epilepsy,69 and CYP3A4 is upregulated and colocalized with P-gp in endothelial cells isolated from resected brain tissue samples from patients with refractory epilepsy.70 Further, the approach presented here could also be used to study the underlying mechanism(s) of blood–brain barrier dysfunction, in particular barrier leakage, which is another important characteristic of epilepsy.71–73
CONCLUSIONS
We have developed a new approach to studying molecular mechanisms at the blood–brain barrier in epilepsy in detail by combining established acute seizure and chronic epilepsy animal models used in epilepsy research with the isolation of brain capillaries from these animal models as well as from post-mortem brain tissue of patients with epilepsy. This in vivo/ex vivo combination approach may better allow translating results to the in vivo situation and potentially to human pathophysiology.
Supplementary Material
Acknowledgments
We thank and acknowledge Peter Nelson and Sonya Anderson at the UK-ADC Brain Tissue Bank for providing all human brain tissue samples (NIH grant number: P30 AG028383 from the National Institute on Aging). We thank Paula Thomason for editorial assistance and all our lab members for proofreading the manuscript. The project described was supported by grant number 1R01NS079507 from the National Institute of Neurological Disorders and Stroke (to B.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health.
ABBREVIATIONS
- ASDs
antiseizure drugs
- P-gp
P-glycoprotein
- i.p
intraperitoneal
- NBD-CSA
[N-ε-(4-nitrobenzofurazan-7-yl)-D-cyclosporin A
- SE
status epilepticus
- CE
chronic epilepsy
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.6b00770.
Rat with chronic epilepsy experiencing a generalized seizure (AVI)
ORCID
Bjoern Bauer: 0000-0003-3867-2251
Notes
The authors declare no competing financial interest.
References
- 1.Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life-time epilepsy: a meta-analytic approach. Epilepsia. 2010;51(5):883–90. doi: 10.1111/j.1528-1167.2009.02481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thurman DJ, Beghi E, Begley CE, Berg AT, Buchhalter JR, Ding D, Hesdorffer DC, Hauser WA, Kazis L, Kobau R, Kroner B, Labiner D, Liow K, Logroscino G, Medina MT, Newton CR, Parko K, Paschal A, Preux PM, Sander JW, Selassie A, Theodore W, Tomson T, Wiebe S, Epidemiology, I. C.o. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia. 2011;52(Suppl 7):2–26. doi: 10.1111/j.1528-1167.2011.03121.x. [DOI] [PubMed] [Google Scholar]
- 3.Brodie MJ. Antiepileptic drug therapy the story so far. Seizure. 2010;19(10):650–5. doi: 10.1016/j.seizure.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 4.Kwan P, Brodie M. Potential role of drug transporters in the pathogenesis of medically intractable epilepsy. Epilepsia. 2005;46(2):224–35. doi: 10.1111/j.0013-9580.2005.31904.x. [DOI] [PubMed] [Google Scholar]
- 5.Loscher W. Mechanisms of drug resistance. Epileptic Disord. 2005;7(Suppl 1):S3–9. [PubMed] [Google Scholar]
- 6.Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6(8):591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- 7.Brandt C, Bethmann K, Gastens AM, Loscher W. The multidrug transporter hypothesis of drug resistance in epilepsy: Proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2006;24(1):202–11. doi: 10.1016/j.nbd.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 8.van Vliet EA, Zibell G, Pekcec A, Schlichtiger J, Edelbroeķ PM, Holtman L, Aronica E, Gorter JA, Potschka H. COX-2 inhibition controls P-glycoprotein expression and promotes brain delivery of phenytoin in chronic epileptic rats. Neuropharmacology. 2010;58(2):404–12. doi: 10.1016/j.neuropharm.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta, Biomembr. 1976;455(1):152–62. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 10.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22(47):7468–85. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 11.Cucullo L, Hossain M, Rapp E, Manders T, Marchi N, Janigro D. Development of a humanized in vitro blood–brain barrier model to screen for brain penetration of antiepileptic drugs. Epilepsia. 2007;48(3):505–16. doi: 10.1111/j.1528-1167.2006.00960.x. [DOI] [PubMed] [Google Scholar]
- 12.Kwan P, Sills GJ, Brodie MJ. The mechanisms of action of commonly used antiepileptic drugs. Pharmacol Ther. 2001;90(1):21–34. doi: 10.1016/s0163-7258(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 13.Lazarowski A, Czornyj L, Lubienieki F, Girardi E, Vazquez S, D’Giano C. ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia. 2007;48(Suppl5):140–9. doi: 10.1111/j.1528-1167.2007.01302.x. [DOI] [PubMed] [Google Scholar]
- 14.Luna-Tortos C, Fedrowitz M, Loscher W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology. 2008;55(8):1364–75. doi: 10.1016/j.neuropharm.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 15.Potschka H, Fedrowitz M, Loscher W. P-Glycoprotein-mediated efflux of phenobarbital, lamotrigine, and felbamate at the blood–brain barrier: evidence from microdialysis experiments in rats. Neurosci Lett. 2002;327(3):173–6. doi: 10.1016/s0304-3940(02)00423-8. [DOI] [PubMed] [Google Scholar]
- 16.Potschka H, Fedrowitz M, Loscher W. Multidrug resistance protein MRP2 contributes to blood–brain barrier function and restricts antiepileptic drug activity. J Pharmacol Exp Ther. 2003;306(1):124–31. doi: 10.1124/jpet.103.049858. [DOI] [PubMed] [Google Scholar]
- 17.Potschka H, Loscher W. In vivo evidence for P-glycoprotein-mediated transport of phenytoin at the blood–brain barrier of rats. Epilepsia. 2001;42(10):1231–40. doi: 10.1046/j.1528-1157.2001.01901.x. [DOI] [PubMed] [Google Scholar]
- 18.Zhang C, Kwan P, Zuo Z, Baum L. The transport of antiepileptic drugs by P-glycoprotein. Adv Drug Delivery Rev. 2012;64(10):930–42. doi: 10.1016/j.addr.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Bauer M, Karch R, Zeitlinger M, Liu J, Koepp MJ, Asselin MC, Sisodiya SM, Hainfellner JA, Wadsak W, Mitterhauser M, Muller M, Pataraia E, Langer O. In vivo P-glycoprotein function before and after epilepsy surgery. Neurology. 2014;83(15):1326–31. doi: 10.1212/WNL.0000000000000858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feldmann M, Asselin MC, Liu J, Wang S, McMahon A, Anton-Rodriguez J, Walker M, Symms M, Brown G, Hinz R, Matthews J, Bauer M, Langer O, Thom M, Jones T, Vollmar C, Duncan JS, Sisodiya SM, Koepp MJ. P-glycoprotein expression and function in patients with temporal lobe epilepsy: a case-control study. Lancet Neurol. 2013;12(8):777–85. doi: 10.1016/S1474-4422(13)70109-1. [DOI] [PubMed] [Google Scholar]
- 21.Ma A, Wang C, Chen Y, Yuan W. P-glycoprotein alters blood–brain barrier penetration of antiepileptic drugs in rats with medically intractable epilepsy. Drug Des, Dev Ther. 2013;7:1447–54. doi: 10.2147/DDDT.S52533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakanishi H, Yonezawa A, Matsubara K, Yano I. Impact of P-glycoprotein and breast cancer resistance protein on the brain distribution of antiepileptic drugs in knockout mouse models. Eur J Pharmacol. 2013;710(1–3):20–8. doi: 10.1016/j.ejphar.2013.03.049. [DOI] [PubMed] [Google Scholar]
- 23.Aronica E, Gorter JA, Jansen GH, van Veelen CW, van Rijen PC, Leenstra S, Ramkema M, Scheffer GL, Scheper RJ, Troost D. Expression and cellular distribution of multidrug transporter proteins in two major causes of medically intractable epilepsy: focal cortical dysplasia and glioneuronal tumors. Neuroscience. 2003;118(2):417–29. doi: 10.1016/s0306-4522(02)00992-2. [DOI] [PubMed] [Google Scholar]
- 24.Dombrowski. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia. 2001;42:1501–1506. doi: 10.1046/j.1528-1157.2001.12301.x. [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann K, Gastens AM, Volk HA, Loscher W. Expression of the multidrug transporter MRP2 in the blood–brain barrier after pilocarpine-induced seizures in rats. Epilepsy Res. 2006;69(1):1–14. doi: 10.1016/j.eplepsyres.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Yang Z, Yang J, Yang H. Increased P-glycoprotein expression and decreased phenobarbital distribution in the brain of pentylenetetrazole-kindled rats. Neuropharmacology. 2007;53(5):657–63. doi: 10.1016/j.neuropharm.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 27.Marchi N, Guiso G, Rizzi M, Pirker S, Novak K, Czech T, Baumgartner C, Janigro D, Caccia S, Vezzani A. A pilot study on brain-to-plasma partition of 10,11-dyhydro-10-hydroxy-5H-dibenzo-(b,f)azepine-5-carboxamide and MDR1 brain expression in epilepsy patients not responding to oxcarbazepine. Epilepsia. 2005;46(10):1613–9. doi: 10.1111/j.1528-1167.2005.00265.x. [DOI] [PubMed] [Google Scholar]
- 28.Rambeck B, Jurgens UH, May TW, Pannek HW, Behne F, Ebner A, Gorji A, Straub H, Speckmann EJ, Pohlmann-Eden B, Loscher W. Comparison of brain extracellular fluid, brain tissue, cerebrospinal fluid, and serum concentrations of antiepileptic drugs measured intraoperatively in patients with intractable epilepsy. Epilepsia. 2006;47(4):681–94. doi: 10.1111/j.1528-1167.2006.00504.x. [DOI] [PubMed] [Google Scholar]
- 29.Sisodya. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain. 2002;125:22–31. doi: 10.1093/brain/awf002. [DOI] [PubMed] [Google Scholar]
- 30.Tishler DM, Weinberg KI, Hinton DR, Barbaro N, Annett GM, Raffel C. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia. 1995;36(1):1–6. doi: 10.1111/j.1528-1157.1995.tb01657.x. [DOI] [PubMed] [Google Scholar]
- 31.van Vliet EA, van Schaik R, Edelbroek PM, Redeker S, Aronica E, Wadman WJ, Marchi N, Vezzani A, Gorter JA. Inhibition of the multidrug transporter P-glycoprotein improves seizure control in phenytoin-treated chronic epileptic rats. Epilepsia. 2006;47(4):672–80. doi: 10.1111/j.1528-1167.2006.00496.x. [DOI] [PubMed] [Google Scholar]
- 32.Bauer B, Hartz AM, Pekcec A, Toellner K, Miller DS, Potschka H. Seizure-induced up-regulation of P-glycoprotein at the blood–brain barrier through glutamate and cyclooxygenase-2 signaling. Mol Pharmacol. 2008;73(5):1444–53. doi: 10.1124/mol.107.041210. [DOI] [PubMed] [Google Scholar]
- 33.Loscher W, Schmidt D. Epilepsy: perampanel-new promise for refractory epilepsy? Nat Rev Neurol. 2012;8(12):661–2. doi: 10.1038/nrneurol.2012.222. [DOI] [PubMed] [Google Scholar]
- 34.Pekcec A, Unkruer B, Schlichtiger J, Soerensen J, Hartz AM, Bauer B, van Vliet EA, Gorter JA, Potschka H. Targeting prostaglandin E2 EP1 receptors prevents seizure-associated P-glycoprotein up-regulation. J Pharmacol Exp Ther. 2009;330(3):939–47. doi: 10.1124/jpet.109.152520. [DOI] [PubMed] [Google Scholar]
- 35.Bauer B, Hartz AM, Fricker G, Miller DS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood–brain barrier. Mol Pharmacol. 2004;66(3):413–9. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 36.Bauer B, Hartz AM, Miller DS. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood–brain barrier. Mol Pharmacol. 2007;71(3):667–75. doi: 10.1124/mol.106.029512. [DOI] [PubMed] [Google Scholar]
- 37.Hartz AM, Bauer B, Block ML, Hong JS, Miller DS. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood–brain barrier. FASEB J. 2008;22(8):2723–33. doi: 10.1096/fj.08-106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartz AM, Bauer B, Fricker G, Miller DS. Rapid modulation of P-glycoprotein-mediated transport at the blood–brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol Pharmacol. 2006;69(2):462–70. doi: 10.1124/mol.105.017954. [DOI] [PubMed] [Google Scholar]
- 39.Zibell G, Unkruer B, Pekcec A, Hartz AM, Bauer B, Miller DS, Potschka H. Prevention of seizure-induced up-regulation of endothelial P-glycoprotein by COX-2 inhibition. Neuropharmacology. 2009;56(5):849–55. doi: 10.1016/j.neuropharm.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 40.Bankstahl JP, Loscher W. Resistance to antiepileptic drugs and expression of P-glycoprotein in two rat models of status epilepticus. Epilepsy Res. 2008;82(1):70–85. doi: 10.1016/j.eplepsyres.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 41.Bartmann H, Fuest C, la Fougere C, Xiong G, Just T, Schlichtiger J, Winter P, Boning G, Wangler B, Pekcec A, Soerensen J, Bartenstein P, Cumming P, Potschka H. Imaging of P-glycoprotein-mediated pharmacoresistance in the hippocampus: proof-of-concept in a chronic rat model of temporal lobe epilepsy. Epilepsia. 2010;51(9):1780–90. doi: 10.1111/j.1528-1167.2010.02671.x. [DOI] [PubMed] [Google Scholar]
- 42.Feldmann M, Koepp M. P-glycoprotein imaging in temporal lobe epilepsy: in vivo PET experiments with the Pgp substrate [11C]-verapamil. Epilepsia. 2012;53(Suppl 6):60–3. doi: 10.1111/j.1528-1167.2012.03704.x. [DOI] [PubMed] [Google Scholar]
- 43.Schramm U, Fricker G, Wenger R, Miller DS. P-glycoprotein-mediated secretion of a fluorescent cyclosporin analogue by teleost renal proximal tubules. Am J Physiol. 1995;268(1 Pt 2):F46–52. doi: 10.1152/ajprenal.1995.268.1.F46. [DOI] [PubMed] [Google Scholar]
- 44.Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66(12):1136–46. doi: 10.1097/nen.0b013e31815c5efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41(4):479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 46.Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9(3):315–35. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- 47.Glien M, Brandt C, Potschka H, Voigt H, Ebert U, Loscher W. Repeated low-dose treatment of rats with pilocarpine: low mortality but high proportion of rats developing epilepsy. Epilepsy Res. 2001;46(2):111–9. doi: 10.1016/s0920-1211(01)00272-8. [DOI] [PubMed] [Google Scholar]
- 48.Hartz AM, Bauer B, Fricker G, Miller DS. Rapid regulation of P-glycoprotein at the blood–brain barrier by endothelin-1. Molecular pharmacology. 2004;66(3):387–94. doi: 10.1124/mol.104.001503. [DOI] [PubMed] [Google Scholar]
- 49.Hartz AM, Miller DS, Bauer B. Restoring blood–brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol Pharmacol. 2010;77(5):715–23. doi: 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartz AM, Zhong Y, Wolf A, LeVine H, 3rd, Miller DS, Bauer B. Abeta40 Reduces P-Glycoprotein at the blood–brain Barrier through the Ubiquitin-Proteasome Pathway. J Neurosci. 2016;36(6):1930–41. doi: 10.1523/JNEUROSCI.0350-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beccano-Kelly DA, Kuhlmann N, Tatarnikov I, Volta M, Munsie LN, Chou P, Cao LP, Han H, Tapia L, Farrer MJ, Milnerwood AJ. Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Front Cell Neurosci. 2014;8:301. doi: 10.3389/fncel.2014.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Neill RA, Bhamidipati A, Bi X, Deb-Basu D, Cahill L, Ferrante J, Gentalen E, Glazer M, Gossett J, Hacker K, Kirby C, Knittle J, Loder R, Mastroieni C, Maclaren M, Mills T, Nguyen U, Parker N, Rice A, Roach D, Suich D, Voehringer D, Voss K, Yang J, Yang T, Vander Horn PB. Isoelectric focusing technology quantifies protein signaling in 25 cells. Proc Natl Acad Sci U S A. 2006;103(44):16153–8. doi: 10.1073/pnas.0607973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ronne-Engstrom E, Hillered L, Flink R, Spannare B, Ungerstedt U, Carlson H. Intracerebral microdialysis of extracellular amino acids in the human epileptic focus. J Cereb Blood Flow Metab. 1992;12(5):873–6. doi: 10.1038/jcbfm.1992.119. [DOI] [PubMed] [Google Scholar]
- 54.Ueda Y, Tsuru N. Simultaneous monitoring of the seizure-related changes in extracellular glutamate and gamma-aminobutyric acid concentration in bilateral hippocampi following development of amygdaloid kindling. Epilepsy Res. 1995;20(3):213–9. doi: 10.1016/0920-1211(94)00081-7. [DOI] [PubMed] [Google Scholar]
- 55.Wilson CL, Maidment NT, Shomer MH, Behnke EJ, Ackerson L, Fried I, Engel J., Jr Comparison of seizure related amino acid release in human epileptic hippocampus versus a chronic, kainate rat model of hippocampal epilepsy. Epilepsy Res. 1996;26(1):245–54. doi: 10.1016/s0920-1211(96)00057-5. [DOI] [PubMed] [Google Scholar]
- 56.Muller CJ, Bankstahl M, Groticke I, Loscher W. Pilocarpine vs. lithium-pilocarpine for induction of status epilepticus in mice: development of spontaneous seizures, behavioral alterations and neuronal damage. Eur J Pharmacol. 2009;619(1–3):15–24. doi: 10.1016/j.ejphar.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 57.Raza M, Pal S, Rafiq A, DeLorenzo RJ. Long-term alteration of calcium homeostatic mechanisms in the pilocarpine model of temporal lobe epilepsy. Brain Res. 2001;903(1–2):1–12. doi: 10.1016/s0006-8993(01)02127-8. [DOI] [PubMed] [Google Scholar]
- 58.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–94. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 59.Chen JQ, Wakefield LM, Goldstein DJ. Capillary nanoimmunoassays: advancing quantitative proteomics analysis, biomarker assessment, and molecular diagnostics. J Transl Med. 2015;13:182. doi: 10.1186/s12967-015-0537-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bankstahl JP, Hoffmann K, Bethmann K, Loscher W. Glutamate is critically involved in seizure-induced overexpression of P-glycoprotein in the brain. Neuropharmacology. 2008;54(6):1006–16. doi: 10.1016/j.neuropharm.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 61.Zhu HJ, Liu GQ. Glutamate up-regulates P-glycoprotein expression in rat brain microvessel endothelial cells by an NMDA receptor-mediated mechanism. Life Sci. 2004;75(11):1313–22. doi: 10.1016/j.lfs.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 62.Avemary J, Salvamoser JD, Peraud A, Remi J, Noachtar S, Fricker G, Potschka H. Dynamic regulation of P-glycoprotein in human brain capillaries. Mol Pharmaceutics. 2013;10(9):3333–41. doi: 10.1021/mp4001102. [DOI] [PubMed] [Google Scholar]
- 63.Salvamoser JD, Avemary J, Luna-Munguia H, Pascher B, Getzinger T, Pieper T, Kudernatsch M, Kluger G, Potschka H. Glutamate-Mediated Down-Regulation of the Multidrug-Resistance Protein BCRP/ABCG2 in Porcine and Human Brain Capillaries. Mol Pharmaceutics. 2015;12(6):2049–60. doi: 10.1021/mp500841w. [DOI] [PubMed] [Google Scholar]
- 64.Pardridge WM. blood–brain barrier genomics and the use of endogenous transporters to cause drug penetration into the brain. Curr Opin Drug Discov Devel. 2003;6(5):683–91. [PubMed] [Google Scholar]
- 65.Rizzi M, Caccia S, Guiso G, Richichi C, Gorter JA, Aronica E, Aliprandi M, Bagnati R, Fanelli R, D’Incalci M, Samanin R, Vezzani A. Limbic seizures induce P-glycoprotein in rodent brain: functional implications for pharmacoresistance. J Neurosci. 2002;22(14):5833–9. doi: 10.1523/JNEUROSCI.22-14-05833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuteykin-Teplyakov K, Brandt C, Hoffmann K, Loscher W. Complex time-dependent alterations in the brain expression of different drug efflux transporter genes after status epilepticus. Epilepsia. 2009;50(4):887–97. doi: 10.1111/j.1528-1167.2008.01916.x. [DOI] [PubMed] [Google Scholar]
- 67.Krajcsi P, Jani M, Toth B, Erdo F, Kis E, Beery E, Sziraki I. Efflux transporters in the blood–brain interfaces-in vitro and in vivo methods and correlations. Expert Opin Drug Metab Toxicol. 2012;8(4):419–31. doi: 10.1517/17425255.2012.668184. [DOI] [PubMed] [Google Scholar]
- 68.Boussadia B, Ghosh C, Plaud C, Pascussi JM, de Bock F, Rousset MC, Janigro D, Marchi N. Effect of status epilepticus and antiepileptic drugs on CYP2E1 brain expression. Neuroscience. 2014;281:124–34. doi: 10.1016/j.neuroscience.2014.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghosh C, Hossain M, Puvenna V, Martinez-Gonzalez J, Alexopolous A, Janigro D, Marchi N. Expression and functional relevance of UGT1A4 in a cohort of human drug-resistant epileptic brains. Epilepsia. 2013;54(9):1562–70. doi: 10.1111/epi.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghosh C, Marchi N, Desai NK, Puvenna V, Hossain M, Gonzalez-Martinez J, Alexopoulos AV, Janigro D. Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia. 2011;52(3):562–71. doi: 10.1111/j.1528-1167.2010.02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Vliet EA, Otte WM, Gorter JA, Dijkhuizen RM, Wadman WJ. Longitudinal assessment of blood–brain barrier leakage during epileptogenesis in rats. A quantitative MRI study. Neurobiol Dis. 2014;63:74–84. doi: 10.1016/j.nbd.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 72.Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, Hallene K, Diglaw T, Franic L, Najm I, Janigro D. Seizure-promoting effect of blood–brain barrier disruption. Epilepsia. 2007;48(4):732–42. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marchi N, Granata T, Ghosh C, Janigro D. blood–brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia. 2012;53(11):1877–86. doi: 10.1111/j.1528-1167.2012.03637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.