

Abstract
ideR, an essential gene of Mycobacterium tuberculosis, is an attractive drug target as its conditional knockout displayed attenuated growth phenotype in vitro and in vivo. To the best of our knowledge, no inhibitors of IdeR are identified. We carried out virtual screening of NCI database against the IdeR DNA binding domain followed by inhibition studies using EMSA. Nine compounds exhibited potent inhibition with NSC 281033 (I-20) and NSC 12453 (I-42) exhibiting IC50 values of 2 µg/ml and 1 µg/ml, respectively. We then attempted to optimize the leads firstly by structure based similarity search resulting in a class of inhibitors based on I-42 containing benzene sulfonic acid, 4-hydroxy-3-[(2-hydroxy-1-naphthalenyl) azo] scaffold with 4 molecules exhibiting IC50 ≤ 10 µg/ml. Secondly, optimization included development of energy based pharmacophore and screening of ZINC database followed by docking studies, yielding a molecule with IC50 of 60 µg/ml. More importantly, a five-point pharmacophore model provided insight into the features essential for IdeR inhibition. Five molecules with promising IC50 values also inhibited M. tuberculosis growth in broth culture with MIC90 ranging from 17.5 µg/ml to 100 µg/ml and negligible cytotoxicity in various cell lines. We believe our work opens up avenues for further optimization studies.
Introduction
Despite the availability of an effective frontline chemotherapeutic regimen comprising of four frontline drugs namely isoniazid, pyrazinamide, rifampicin and ethambutol, the morbidity and mortality rates associated with TB patients remain very high. Moreover, the lengthy treatment of 6–9 months often leads to non-compliance by the patients leading to an inexorable increase in the multi drug resistant and extremely drug resistant strains of Mycobacterium tuberculosis 1, 2. Thus, in view of the current scenario, the urgent need to identify novel potent inhibitory molecules against M. tuberculosis cannot be overemphasized.
Iron is an essential element required for the activity of numerous proteins involved in redox reactions and electron transport chain3. Down regulation of iron storage proteins such as transferrin and ferritins remains one of the critical immune responses generated by the host against the invading pathogen4–7. In response to this, M. tuberculosis upregulates its iron acquisition machinery, which synthesizes small molecules known as mycobactins and carboxymycobactins that function as iron chelators8, 9. These molecules bind iron from the host proteins and with the help of various transporters, this iron is then transported to the M. tuberculosis cytosol, where it is utilized for many crucial processes10–14. Although, iron is an essential element, it is toxic, if present, in higher amounts. Excess iron can react with peroxides to form free radicals via fenton reaction leading to cellular toxicity15. Thus, the pathogen requires a tight regulation of the intracellular levels of iron, which in M. tuberculosis is performed by the transcription factor IdeR. In the conditions of iron sufficiency in M. tuberculosis, iron binds and activates intracellular IdeR, which then represses the iron acquisition machinery of the pathogen and activates the synthesis of iron storage proteins. In the case of iron deficiency, IdeR can no longer bind to the target promoter region resulting in the production of mycobactins and repression of the synthesis of iron storage proteins16.
A conditional knockout of M. tuberculosis ideR gene displayed an attenuated phenotype when grown in vitro and in vivo suggesting the importance of IdeR for the growth and survival of M. tuberculosis 17. Besides, IdeR also plays a crucial role in protecting the cells against the oxidative stress generated by the host. These properties make IdeR an attractive drug target17. A series of studies carried out by Hol et al. on the crystal structures of M. tuberculosis IdeR in monomer and DNA bound forms identified Ser 37, Pro 39 and Gln 43 as few of the residues crucial for the binding of IdeR to the DNA molecule18–21. IdeR consists of two metal binding sites 1 and 2 and a dimerization domain which helps in the dimerization of IdeR monomers when iron is bound at the high affinity metal binding site 1 with a Kd less than 0.5 µM Fe2+. IdeR also carries a DNA binding domain which undergoes a conformational change when iron binds at the metal binding site 2 with a relatively lower affinity for iron having a Kd of 9.5 µM22. The distance between Cα atom of Gln 43 (an important residue present at the DNA binding helix) to metal binding site 1 is 26.7 Å and metal binding site 2 is 19.5 Å as calculated by Pohl et al.18. As is evident from the relative distances, the two iron binding sites are far away from the DNA binding helix of the protein IdeR. Studies of the crystal structure determination, iron binding kinetics and molecular dynamics simulation23 of IdeR have provided insights into the binding mechanism of IdeR with iron and its cognate DNA sequences and have established a starting point for carrying out structure based identification of potent inhibitory molecules against IdeR.
In this study, we took a structure based approach which included virtual screening followed by Electrophoretic Mobility Shift Assay (EMSA) to evaluate the inhibitory potential of various shortlisted molecules against the DNA binding ability of IdeR. Subsequently, the leads obtained from this approach were further utilized for (a) structure based similarity search approach which resulted in 4-hydroxy-3-[(2-hydroxy-1-naphthalenyl) azo] scaffold bearing molecules with potent in vitro inhibition and (b) energy based pharmacophore model generation followed by docking study which yielded a molecule with IC50 of 60 µg/ml and also provided an insight into the critical features required for IdeR based inhibition. Further, the molecules were evaluated against the growth of M. tuberculosis in broth culture followed by cytotoxicity studies in macrophage, kidney and hepatic cell lines resulting in several molecules that can be employed as starting points for carrying out further structure activity relationship studies to inhibit IdeR.
Results and Discussion
In silico screening against the DNA binding domain of IdeR
In order to carry out the structure based inhibitor identification, we filtered the NCI library containing 260,071 compounds based on the Lipinski rule of five and drug likeness using the online FAF-server24 which resulted in 95,748 compounds (http://fafdrugs3.mti.univ-paris-diderot.fr/). These were subsequently employed for docking studies by using Autodock 4.225. A number of IdeR monomer and DNA-bound crystal structures are available in PDB which provided key insights into the critical residues involved in the DNA binding which corroborate the results of footprinting experiments carried out by Gold et al.16. Figure 1a depicts the structure of IdeR homodimers bound to its cognate DNA molecule (PDB ID-1U8R). Based on the literature, Ser 37, Pro 39 and Gln 43 residues of the crystal structure of IdeR were identified to be important for binding to the DNA and were chosen as the grid center (docking site 1), which was used to dock the filtered NCI library (Fig. 1b). Based on the visual inspection of the interactions of the IdeR surface with DNA, a second docking site, adjacent to the docking site 1, was also selected with grid center at Ser 42 and Gln 43 (docking site 2) as depicted in Fig. 1c. It is to be noted that these two docking sites are present far apart from the metal binding sites (Fig. 1d–f). The relative spatial arrangement of the iron binding sites to the docking site 1 at the DNA binding helix is illustrated in Fig. 1d. Using these two sites, docking was performed and the compounds were shortlisted based on their free energies of binding. The average docking scores of the top scoring molecules from both the sites were in the range of −6 to −7 kcal/mol and are given in Table S1. Subsequently, a total of 123 compounds from site 1 and site 2 were procured from NCI and evaluated for their in vitro inhibitory potential to inhibit IdeR activity by employing EMSA.
Figure 1.
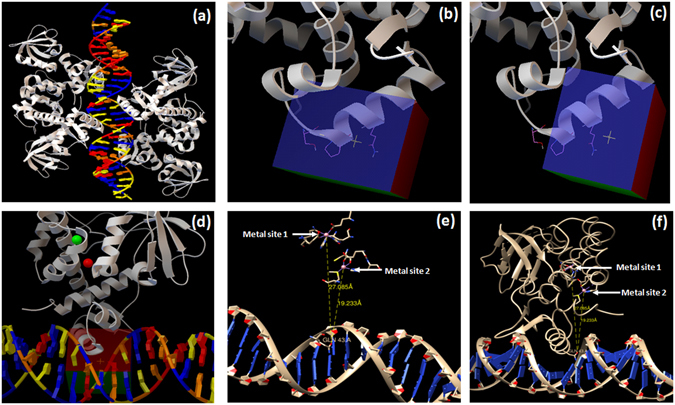
IdeR crystal structure and docking sites employed in this study. This figure depicts the spatial arrangement of IdeR, DNA and the docking sites. (a) IdeR homodimers bound to the cognate DNA sequence. (b) Docking site 1 at the DNA binding helix of IdeR. (c) Docking site 2 at the DNA binding helix of IdeR. (d) The spatial arrangement of docking site 1 with the metal binding site 1 (green) and site 2 (red) and cognate DNA sequence. (e) Distance between Gln 43 (present in the DNA binding helix) to the metal binding site 1 (27.085 Å) and site 2 (19.233 Å). It is evident from the distances depicted, the metal binding sites are far away from the DNA binding helix. (f) Distance between Gln 43 and metal binding sites along with the IdeR tertiary structure. (Images a to d were generated by using the software Autodock 4.225 and e, f were generated by using the software chimera43).
Inhibitory potential of the compounds against the DNA binding activity of IdeR
EMSA was employed to evaluate the ability of the shortlisted compounds to inhibit the DNA binding activity of IdeR. For this, ideR gene was expressed and IdeR was purified to near homogeneity by Ni-NTA chromatography. One hundred twenty three compounds were screened at a fixed concentration of 100 µg/ml wherein 18 compounds exhibited more than 40% inhibition of the DNA binding activity of IdeR as given in Table S1. Subsequently, IC50 values for these 18 compounds were determined by employing varying concentrations of the compounds ranging from 0.4 µg/ml to 100 µg/ml. Nine compounds exhibited IC50 values less than 25 µg/ml, which were further selected for pharmacophore development and screening work, structures of these compounds are given in Fig. 2. (Prefix I is given to the compounds identified from this initial NCI library screening). The two most potent compounds, compound I-20 (NSC 281033) and I-42 (NSC 12453) exhibited IC50 values of 2.4 µg/ml (Fig. 3a) and 1 µg/ml (Fig. 3b), respectively. Further, we assessed the inhibition of the IdeR DNA binding activity of top 6 inhibitors (I-8, I-20, I-21, I-34, I-39 and I-42) exhibiting IC50 ≤ 25 µg/ml in the presence of increasing concentrations of the DNA (0.8 pmoles to 12.8 pmoles) and the results clearly demonstrate that all the 6 compounds inhibit the activity by competing with DNA (Fig. 4 and Fig. S1). The competition studies for compounds I-20 and I-42, as shown in Fig. 4, demonstrate that increasing the DNA concentration alleviates the inhibition exerted by the compounds as is evident by increase in the percentage of bound DNA (lanes 2–7 for 2.5 µg/ml, lanes 9–14 for 5 µg/ml and lanes 16–21 for 10 µg/ml). Moreover, this competition is dose dependent as it was observed that at any fixed DNA concentration, the per cent bound DNA decreases as the amount of inhibitor is increased. The competition results for the rest of the four compounds are depicted in Fig. S1. We also carried out experiments to assess the effect of Fe2+ instead of Ni2+ on the binding of IdeR to the DNA as well as the binding inhibition of the molecules as shown in Fig. S2. We found that 100 µM iron leads to equivalent binding as is seen in 200 µM Ni2+ (Fig. S2, lanes 2 and 3). However, further increase in the iron concentration hampers the DNA binding activity of IdeR (Fig. S2, lane 4). This could be due to the high redox instability of Fe2+ in in vitro conditions as already reported in the literature26. Further, it was observed that there was no effect of iron on the inhibitory potential of the compounds I-42, I-20 and I-34 (Fig. S2, lanes 5–7). Some of the compounds used in this study belong to metal chelators groups, however, our competition assays involving six compounds showed that all of them exerted the observed inhibition by competing with DNA substrate. The docking studies indicated the interactions of compounds I-20 and I-42 with various residues present at the DNA binding helix. The predicted key interactions made by compound I-20 having a thiazolidine-benzyl scaffold include hydrogen bond between the hydroxyl group at the 3rd position of the benzyl ring with the carboxyl group of Gln 43 and the oxygen in the carboxylic acid attached to the thiazolidine ring interacts with the amino group of Ser 37 (Fig. 5a). The diazene group of compound I-42 which bears a benzyl-naphthalenyl scaffold is predicted to form a hydrogen bond with the hydroxyl group of Ser 42 and the oxygen atoms in the sulfonic acid is predicted to form a network of hydrogen bonding with the two amino groups of the side chain of Arg 60 (Fig. 5b). This provides key insights into a few other novel critical residues (apart from Ser 37, Pro 39 and Gln 43), that can be exploited while carrying out the structure activity relationship studies for inhibitor identification against IdeR. Compounds I-33 and I-34 containing a common dihydropyrazolo-diazepene scaffold exhibited an IC50 of 21.7 µg/ml and 6.1 µg/ml, respectively. The predicted key interactions made by compound I-34 include a hydrogen bond between nitrogen of the diazepine moiety with the amino group of Gln 43 and the oxygen atom of the carboxyl group attached to the diazepine moiety with the hydroxyl group of Ser 42 (Fig. 5c). Inhibitor I-108 exhibited an IC50 of 24 µg/ml and its predicted key interactions include formation of a network of hydrogen bond between oxygen atom of sulfonyl and pyrrolidine ring with amino group of Gln 43 and between a second oxygen atom of pyrrolidine ring with Arg 50 (Fig. 5d). Thus, the initial docking followed by EMSA provided few potent inhibitory molecules with IC50 ≤ 25 µg/ml and which make key interactions with the various residues of IdeR involved in its DNA binding activity.
Figure 2.
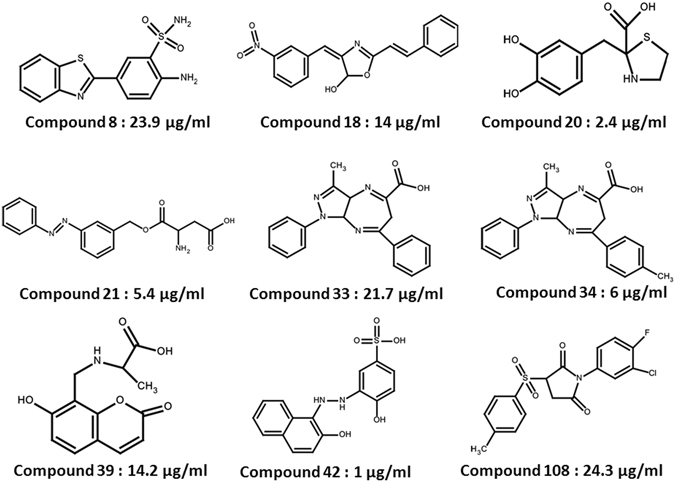
Structures of the most potent molecules and their respective IC50 values as determined by employing EMSA.
Figure 3.
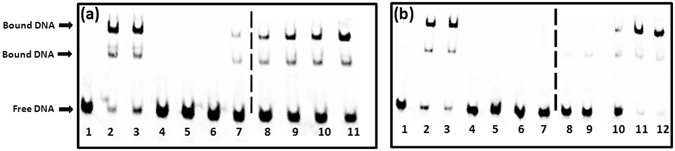
Dose response study of various molecules by employing EMSA. (a) Compound I-20, (b) Compound I-42. Dotted lines represent two different polyacrylamide gels. Lane 1-Free DNA, Lane 2-Protein+DNA, Lane 3-Protein+DNA+DMSO (−ve control), Lane 4–100 µg/ml, Lane 5–50 µg/ml, Lane 6–25 µg/ml, Lane 7–12.5 µg/ml, Lane 8–6.25 µg/ml, Lane 9–3.12 µg/ml, Lane 10–1.56 µg/ml, Lane 11–0.78 µg/ml, Lane 12–0.39 µg/ml (only in Fig. 3b). (For IC50 calculations, Protein+DNA and Protein+DNA+DMSO, control reactions were loaded in every gel and percentage of bound DNA for each inhibitor concentration was calculated by comparing the control reactions loaded in the same gel).
Figure 4.
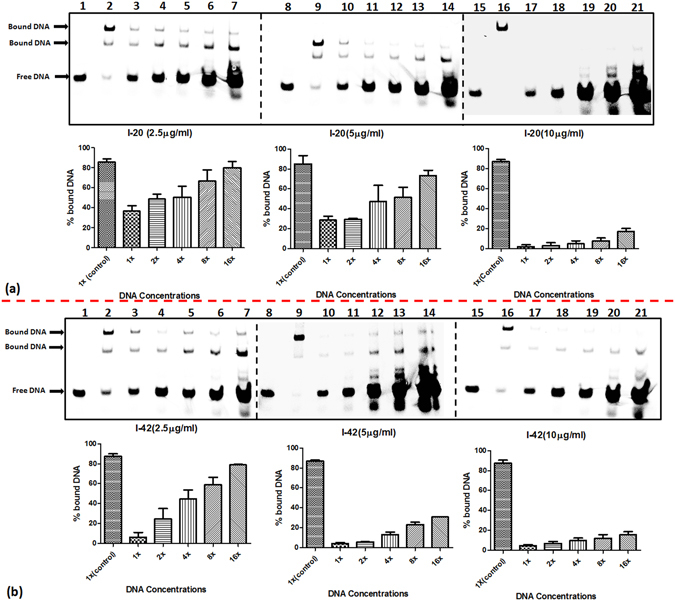
Competition experiments with compounds I-20 and I-42 and labeled DNA. Each gel depicts the alleviation of inhibition exhibited by compound I-20 (a) and compound I-42 (b) in the presence of increasing DNA. Lanes 1, 8, 15- free DNA (0.8 pmoles), lanes 2,9,16- IdeR+DNA (0.8 pmoles)+DMSO (vehicle control), lanes 3, 10, 17- IdeR+DNA (0.8 pmoles)+ inhibitor, lanes 4, 11, 18 - IdeR+ DNA (1.6 pmoles)+ inhibitor, lanes 5, 12, 19 - IdeR+DNA (3.2 pmoles)+ inhibitor, lanes 6, 13, 20 - IdeR+DNA (6.4 pmoles)+ inhibitor, lanes 7, 14, 21 - IdeR+DNA (12.8 pmoles)+ inhibitor. The bar diagram represents the percent bound DNA at varying DNA concentrations (x represents 0.8 pmoles of labeled DNA, dotted lines represent separate gels). The data is represented as Mean ± S.E.M by values obtained from two independent experiments.
Figure 5.
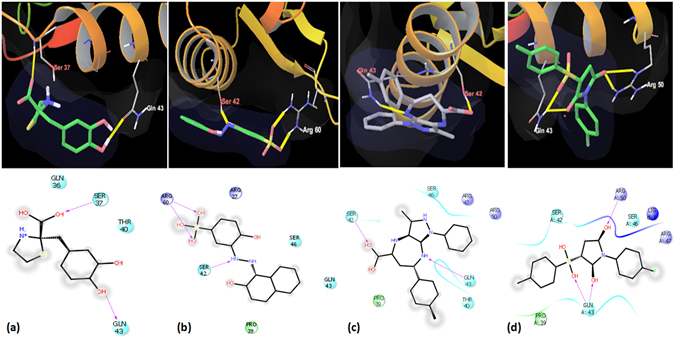
Binding mode analyses of (a) Inhibitor I-20- NSC 281033, (b) Inhibitor I-42-NSC 12453, (c) Inhibitor I-34- NSC 662444, (d) Inhibitor I-108- NSC 282699 (Yellow dash represents the Hydrogen bond between the ligand and protein residues).
Energy based Pharmacophore Generation, Docking and EMSA inhibition study
To provide key insights into the critical features required for inhibition of DNA binding ability of IdeR, an energy based pharmacophore model was developed by using e-pharmacophore script27, 28 available in the Schrodinger Software package. An energy based pharmacophore extracts and weighs interactions made by an inhibitor with the protein by using the descriptor information generated by the docking performed through Glide29–31 and provides a map of critical features contributing to the inhibitor-protein interaction. All the nine compounds (IC50 < 25 µg/ml) were docked at site 1 again by using Glide in the Schrodinger package. However, the docking scores of all the molecules were in the range of −1 to −2 kcal/mol as compared to −6 to −7 kcal/mol generated by Autodock 4.225, except compound I-20 which exhibited a score of −4.25 kcal/mol. Subsequently, the pharmacophore hypothesis was generated on the basis of the XP descriptor file generated by docking the compound I-20 by using Glide. This file was fed into the e-pharmacophore script and a pharmacophore model consisting of 5 features was generated which comprised of two hydrogen bond acceptors, two rings and one negative ionizable group. Multiple conformations of compound I-20 provided a map of its possible key interactions with the residues present on the DNA binding groove of IdeR as shown in Fig. 6a. The selection of each pharmacophore point was made on the basis of their proximity with the key residues involved in DNA binding i.e Hydrogen Bond Acceptor (A16) with Ser 37, Aromatic Ring (R48) with Gln 43, Hydrogen Bond Acceptor (A11) with Pro 39 (Fig. 6b). Two additional points were included namely Aromatic ring (R50) and Negative ionizable group (N38) which are shown to be interacting with Ala 28, Arg 60 and Arg 27 as shown in (Fig. 6c). The final pharmacophore model (Fig. 6d) was used to screen the drug like database of ZINC consisting of ~13 million compounds by using Phase module in Schrodinger package32, 33. This resulted in ~110,000 molecules with a filter of 4 out of 5 molecular features to be matched. 2-D and 3-D descriptors of these hits were generated by qikprop module in Schrodinger, which were subsequently used to screen these molecules on the basis of mycobacterial cell wall permeability by using MycPermCheck34 (http://www.mycpermcheck.aksotriffer.pharmazie.uni-wuerzburg.de/) online server which resulted in ~39,000 molecules. These molecules were docked at site 1 by employing Autodock 4.225 resulting in 35 molecules with free energy binding scores above −7 kcal/mol which were further shortlisted on the basis of their structural diversity by using tanimoto filter of 0.8 using the software Openbabel35. Subsequently, 22 molecules were selected, procured and tested for their in vitro inhibitory potential by employing EMSA out of which compound IA8 (ZINC 21711688, Ambinter ID, 20476786, prefix IA is given to this compound procured from Ambinter, which is identified in the pharmacophore based screening) (Fig. 6e) exhibited an IC50 of 61.3 µg/ml. A probable reason for the identification of poor inhibitors by this approach could be the chemical composition of the library screened. The nature of the library employed for pharmacophore and docking studies ultimately reflects upon the quality and potency of the inhibitors identified, however, a few diverse libraries are generally required to be screened for this purpose and indeed the parameters for selection of a library that will result into potent hits are difficult to decide a priori. Another probable reason for a low hit rate and weak IC50 value could be a low docking score (−4.25 kcal/mol) of compound I-20 on the basis of which the model was developed. Thus, the docking may not be able to completely capture the interactions that could corroborate with a potent IC50 value of 2.4 µg/ml. However, to the best of our knowledge, there is no published report of IdeR inhibitors and co-crystallized structures of inhibitors with IdeR which could have provided substantial information about the native conformation and key interactions of inhibitor bound with the protein. Such information has been the basis for developing energy based pharmacophore model in numerous studies36–39. Thus, this study presents an in vitro validated five point pharmacophore model which provides insights into the features required for IdeR inhibition and can be further optimized and used for future virtual screening endeavors.
Figure 6.
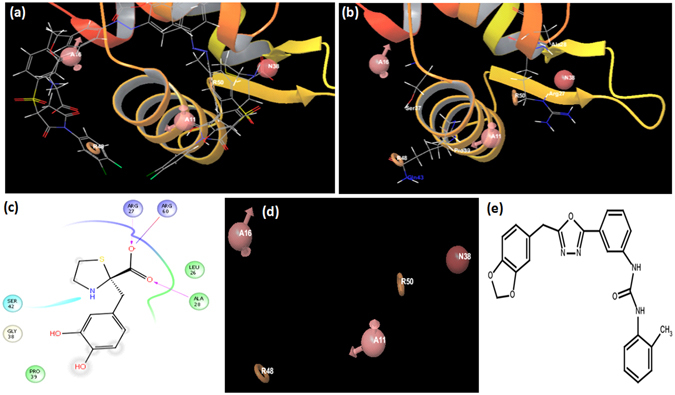
Energy Based Pharmacophore development. (a) Multiple conformations of I-20 (IC50-1 µg/ml) bound to the DNA binding surface of IdeR. (b) Various pharmacophore points in proximity with Ser 37, Pro 39, Gln 43, Arg 27 and Ala 28. (c) 2-D Interaction map of I-20 with various residues of the DNA binding surface of IdeR. (d) Final 5-Point pharmacophore taken for subsequent screening of the drug like database of ZINC. (A16 and A11 refers to Acceptor feature, R48 and R50 refers to Aromatic ring feature and N38 refers to negative ionizable group). (e) 2-D structure of IA-8 3-[3-[5-(1, 3-benzodioxol-5-ylmethyl)-1, 3, 4-oxadiazol-2-yl]phenyl]-1-(o-tolyl) urea with an IC50 of 60 µg/ml.
Structure similarity based search for analogs of the potent IdeR lead molecules
In order to identify more potent analogs of the compounds identified in the initial inhibitory screening, a structural similarity based screening of NCI library was carried out. For this,10 molecules namely (I-8, I-18, I-20, I-33, I-34, I-39, I-42, I-85, I-103, I-108) were taken as query molecules individually and the complete NCI database consisting of ~3,00,000 compounds was searched based on the 2-D fingerprint similarity as measured by the Tanimoto coefficient with a cut-off of 0.7 using the software Openbabel and similar analogs were shortlisted. In total, 66 molecules were procured from NCI and tested for their potential to inhibit DNA binding ability of IdeR by employing EMSA. Out of which, 17 molecules exhibited more than 40% inhibition at 100 µg/ml and were shortlisted for the determination of their IC50 values as given in Table S2. (Prefix IS is given to the similar analogs identified from this structure similarity based screening approach). Nine out of 16 molecules belonging to the class of compound 42 exhibited potent inhibition with IC50 of five molecules ≤ 15 µg/ml thereby establishing benzene sulfonic acid,4-hydroxy-3-[(2-hydroxy-1-naphthalenyl) azo] as a potent inhibitory scaffold and also provided insight into the key structural requirements for the inhibition of IdeR DNA binding ability. For eg, compound IS-2 and IS-12 bear a hydroxyl group at the position opposite to diazene moiety as compared to a phenyl ring in I-42 which emphasizes the fact that a bulkier aromatic group is required with a phenyl moiety as compared to a hydroxyl group which leads to a slight decrease in the potency as shown in Fig. 7a. Also, compound IS-8 with an IC50 of 0.66 µg/ml, essentially contains three repeating naphthalene azo sulfonic acid scaffold thereby highlighting the importance of this scaffold for IdeR inhibition. Compound IS-51 with an IC50 value of 16.5 µg/ml which is an analog of compound I-103 (IC50 of 33.3 µg/ml) exhibited a two-fold increase in potency as depicted in Fig. 7b. The presence of an extra diol-phenyl group in the core scaffold, which is present in the lead molecule I-103 is predicted to hinder the binding of the inhibitor with the protein as its absence in IS-51, results in better inhibition. Analogs of compound I-8 (IC50–23.9 µg/ml), also exhibited improved IC50 values with 14.49 µg/ml of IS-4, 20.94 µg/ml of IS-32 and a two fold increase in potency of IS-54, IC50 of 11.2 µg/ml as shown in Fig. 7c thereby suggesting a benzo-thiazol benzene sulfonic acid as another scaffold important for IdeR inhibition. This study thus made an attempt towards establishing three distinct scaffolds that are crucial for inhibiting the DNA binding activity of IdeR and can serve as a robust platform for the generation of more rationally designed analogs.
Figure 7.
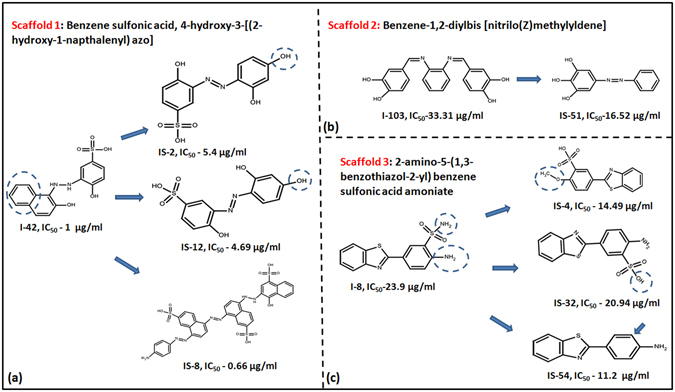
Structure similarity based identification of three potent scaffolds. This figure represents three scaffolds identified by testing analogs of compounds I-42 (a), I-103 (b) and I-8 (c) and their corresponding IC50 values.
Inhibition of M. tuberculosis in broth culture by using Resazurin Microtiter Assay (REMA)
All the molecules that inhibited the IdeR DNA binding activity in EMSA were tested for their ability to inhibit growth of M. tuberculosis in broth culture by using Resazurin Microtiter Assay (REMA)40, 41. Resazurin oxidation-reduction based method provides visible distinction as well as fluorescence based measurement of the viability of the cells. To evaluate the MIC90 of the compounds inhibiting the growth of M. tuberculosis in broth culture, serial dilutions of the compounds were employed with M. tuberculosis cells growing in MB7H9 medium. Compound I-108, which exhibited an IC50 of 24.3 µg/ml, also exhibited an MIC90 of 17.5 µg/ml against M. tuberculosis as shown in Table 1. Compounds I-33 and I-34 exhibiting an IC50 of 21.7 µg/ml and 6.1 µg/ml inhibited the growth of M. tuberculosis at 93.3 µg/ml and 86.6 µg/ml, respectively (Table 1). Also, compounds IS-4 and IS-54 (analogs of compound 8) exhibited an MIC90 of 100 µg/ml and 84.8 µg/ml respectively, although its parent analog, compound I-8 exhibited no inhibition of M. tuberculosis growth in MB7H9 medium (Table 1). It is to be noted that further investigations are required to correlate the whole cell inhibition exhibited by the five compounds with the inhibition of IdeR inside the cells, and the possibility of off-target effects cannot be negated.
Table 1.
Inhibitors exhibiting whole cell M. tuberculosis broth inhibition and cytotoxicity studies.
| Compound No. | NSC ID | MIC90 of compounds against M. tuberculosis (µg/ml) | Inhibition in THP1 (IC50 in µg/ml) | Inhibition in HEK (IC50 in µg/ml) | Inhibition in MDCK (IC50 in µg/ml) | Inhibition in HepG2 (IC50 in µg/ml) |
|---|---|---|---|---|---|---|
| I-33 | 662443 | 93.3 ± 8.16 | >200 | >200 | >200 | >200 |
| I-34 | 662444 | 86.6 ± 7.8 | >200 | >200 | >200 | >200 |
| I-108 | 282699 | 17.5 ± 3.06 | 167.5 ± 5.6 | >200 | >200 | >200 |
| IS-4 | 33755 | 100 | 165.8 ± 3.72 | 164.7 ± 1.24 | 172.8 ± 2.3 | 176.3 ± 1.6 |
| IS-54 | 34445 | 84.8 ± 8 | 181.6 ± 5.2 | 161.1 ± 4.22 | 174.3 ± 6.4 | 167.4 ± 3.78 |
| Rifampicin | 0.05–0.2 | >100 | >100 | >100 | >100 |
Cytotoxicity studies against mammalian cell lines
Since, M. tuberculosis primarily resides inside the host macrophages and in order for a compound to exhibit a good ADME/toxicity profile, it is important for it to be non-cytotoxic against hepatic and kidney cells of the host, thus, we analyzed the cytotoxicity of the compounds against THP-1 (human monocytic macrophage cell line), HepG2 (human liver cancer cell line), MDCK (Madine Darby Canine Kidney cell line) and HEK (Human Embryonic Kidney cell line) respectively. Compounds I-33 and I-34 exhibited no cytotoxicity up to 200 µg/ml (the maximum concentration tested) in all the four cell lines tested whereas compound I-108 exhibited no cytotoxicity in three of the four cell lines tested with slight toxicity in THP-1 cells. However, IS-4 and IS-54 exhibited slight toxicity in all the four cell lines tested, as given by their respective IC50 values shown in Table 1. Hence, I-33, I-34 and I-108 were further selected to evaluate their inhibitory potential against the growth of M. tuberculosis residing in macrophages.
Evaluation of the compounds to inhibit the intracellular M. tuberculosis
Based on the inhibitory potential of the compounds against the activity of IdeR, growth inhibition of M. tuberculosis in broth culture as well as evaluation of cytotoxicity in various mammalian cell lines, we shortlisted three molecules I-33, I-34 and I-108 with MIC90 values of 93 μg/ml, 86 μg/ml and 17.5 μg/ml, respectively, for the evaluation of the ability of these compounds to inhibit the growth of M. tuberculosis inside macrophages. PMA activated THP-1 cells were infected with M. tuberculosis at an MOI of 1:5 and were incubated in the presence of varying concentrations of the compounds (I-108–25 μg/ml to 150 μg/ml, I-33 and I-34–100 μg/ml to 500 μg/ml). The growth of the intracellular bacteria was analyzed on the agar plates. It was observed that in the presence of I-108, only a few pin pointed colonies could be seen at the concentrations of 150 μg/ml and 125 μg/ml, however, prominent growth of bacilli was seen at lower concentrations demonstrating that I-108 is able to limit the growth of intracellular bacteria, albeit, at the high concentrations of 150 μg/ml and 125 μg/ml. Importantly, I-108 is not toxic to the THP-1 cells at these concentrations, as evaluated by alamar blue assay (Fig. 8). Also, concentrations higher than 150 μg/ml were not included due to its toxicity to the THP-1 cells at higher concentrations. It is to be noted that the highest concentration employed for I-108 of 150 μg/ml is quite high for inhibition studies and any molecule inhibiting the growth of intracellular bacteria in this higher range is regarded only as a weak inhibitor. Therefore, inhibition exhibited by I-108 at higher concentrations of 150 μg/ml and 125 μg/ml should be considered only as a proof of concept and more rational modifications of this compound are required to improve its potency. Compounds I-33 and I-34 with MIC90 value of 93 μg/ml and 86 μg/ml, respectively, exhibited negligible inhibition of the growth of the intracellular bacteria as numerous small colonies were seen at 500 μg/ml (highest concentration tested) in the case of both I-33 and I-34, although, they were non-toxic to the THP-1 cells at this high concentration of 500 μg/ml, while at lower concentrations, the growth of the intracellular bacteria was unaffected (Fig. S3). Thus, I-33 and I-34 having weak MIC90 values of 93 μg/ml and 86 μg/ml, respectively, were unable to inhibit the growth of M. tuberculosis even at a very high concentration of 500 μg/ml. Rifampicin was used as a positive control. One of the possible reasons for the inability of I-33 and I-34 to inhibit the growth of intracellular bacteria as well as exert no cytotoxicity could be their poor permeability in the mammalian cells. Also, the determination of the inhibitory potential of these compounds by spotting on the agar plates is a qualitative but a relatively high throughput method and more detailed studies such as enumeration by CFU are required to determine the precise MIC99 values. However, these studies are quite labor extensive and thus, studies focused at first improving their inhibitory potential which would result in more potent analogs are warranted before undergoing such labor extensive studies. Thus, our study provides one molecule, I-108, which exhibited inhibitory potential against the activity of IdeR, growth inhibition of M. tuberculosis in vitro, low toxicity against various mammalian cell lines and also a moderate inhibition of growth of intracellular bacteria. Further optimization studies of this lead molecule can result into more potent compounds.
Figure 8.

Evaluation of inhibitory potential of compound I-108 against the growth of intracellular M. tuberculosis. (a) The influence of varying concentrations of I-108 on the growth of intracellular M. tuberculosis as analyzed on the agar plates. Panel a depicts the emergence of pin point colonies at the concentrations of 150 μg/ml and 125 μg/ml as compared to prominent growth of bacilli at lower concentrations of I-108. (b) Alamar blue assay to measure the viability of uninfected THP-1 cells in the presence of varying concentrations of I-108. The upper lane of panel b depicts the various concentrations of I-108 employed for the alamar blue assay (in μg/ml). Lanes 1–6 in panel a depicts following concentrations of I-108: Lane 1–150 μg/ml, Lane 2–125 μg/ml, Lane 3–100 μg/ml, Lane 4–50 μg/ml, Lane 5–25 μg/ml, Lane 6- -ve (only cells).
Conclusion
M. tuberculosis IdeR has remained an attractive drug target for almost a decade. Various genetic studies pertaining to its in vitro and in vivo essentiality, biochemical studies which include the characterization of IdeR such as crystal structure determination and iron and DNA binding kinetics, have laid a robust platform to carry out structure based drug discovery against IdeR. Our study involved the virtual screening followed by testing of 123 compounds in vitro by employing EMSA, which yielded few potent inhibitory compounds with IC50 ≤ 25 µg/ml. Subsequent energy based pharmacophore modeling followed by docking studies led to the identification of a five point pharmacophore model that provided key insights into the features responsible for IdeR inhibition and can be further optimized to carry out more virtual screening studies. A parallel structure similarity based approach led to the identification of a benzene sulfonic acid, 4-hydroxy-3-[(2-hydroxy-1-naphthalenyl) azo] scaffold and 2-amino-5-(1,3- benzothiazol-2-yl) benzene sulfonic acid class of compounds inhibiting the DNA binding activity of IdeR. The MIC and cytotoxicity studies provided few compounds such as I-108 that exhibited IC50 of 24.3 µg/ml, MIC90 of 17.5 µg/ml, negligible cytotoxicity in three of the four mammalian cell lines and was also able to limit the growth of intracellular bacteria, albeit at a high concentration of 125 µg/ml. Also, compound I-34 exhibited IC50 of 6 µg/ml, moderate MIC90 values of 86.6 µg/ml, negligible toxicity in various mammalian cell lines but was unable to limit the growth of intracellular bacteria even at a very high concentration of 500 µg/ml. Thus, I-108 can serve as a starting point for carrying out more rational modifications and structure activity relationship studies.
This study opens avenues for various studies related to the drug discovery efforts against Iron Dependent Regulator, an essential gene of M. tuberculosis. In our study, we identified molecules inhibitory to IdeR activity which is useful for further studies. Determination of IdeR co-crystallized structures bound to these inhibitors would provide information about the important residues crucial for determining the DNA binding activity of IdeR which in turn would help in the development of more potent inhibitors. Moreover, the molecules identified in this study can serve as a robust platform for carrying out more rational modification and structure activity relationship studies. Some of the compounds used in this study belong to metal chelators groups, however, our competition assays involving six compounds showed that all of them exerted the observed inhibition by competing with DNA substrate. Thus, our study follows a combination of various in silico approaches well supported and validated by subsequent in vitro experiments (pipeline depicted in Fig. 9) to identify various lead molecules against IdeR, an important drug target of M. tuberculosis.
Figure 9.
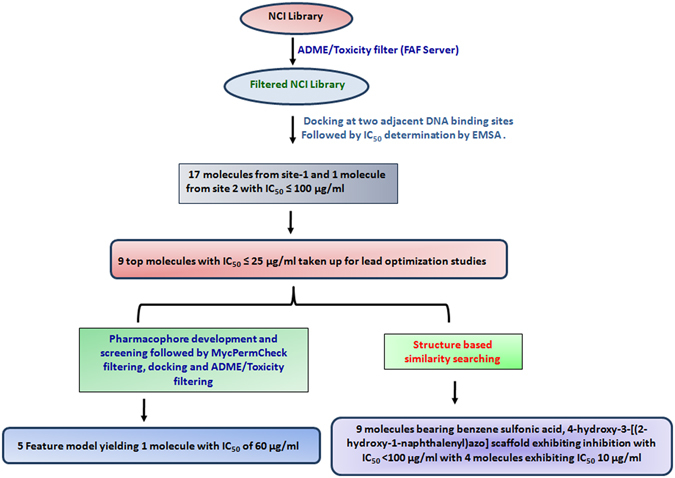
Flowchart of the strategy followed in this work.
Materials and Methods
Library generation and docking studies
Three dimensional structures of molecules belonging to the Open NCI Database consisting of 260,071 compounds were downloaded from the NCI website (http://cactus.nci.nih.gov/download/nci/). The downloaded.sdf files were converted into.smi format by Openbabel35 software and shortlisted based on the Lipinski Rule and ADME/Toxicity parameters by using FAF-Drugs server24 (http://fafdrugs3.mti.univ-paris-diderot.fr/). This shortlisted library was employed for the subsequent virtual screening studies. The three-dimensional crystal structure of IdeR (1U8R) was downloaded from the PDB database (http://www.rcsb.org/pdb/home/home.do) and Autodock 4.225 was used to perform the docking calculations. The structure was prepared by deleting the water molecules followed by the addition of the hydrogen atoms and the protein was saved in.pdbqt format. Genetic algorithm with default parameters was employed for the docking calculations. All the calculations were carried out on 48 node cluster of X2200 Sun Fire with dual core AMD processor with CENT-OS version 5. The compounds were procured from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Cloning of the gene, expression and purification of IdeR
The M. tuberculosis ideR gene was PCR amplified by using M. tuberculosis genomic DNA as the template. The sequence-5′gataatggtaccactagtcatatgaacgagttggttgatacc 3′ with NdeI restriction site at 5′ end was used as the forward primer and sequence-5′gataataagcttattatttttcgaactgcgggtggctccaagcgcttcagactttctcgaccttgac 3′ with HindIII restriction site was used in the reverse primer. The reverse primer also contained streptactin tag for purification. The amplified product was digested with NdeI and HindIII and cloned into pET28c digested with the same enzymes resulting in the construct pET28c/IdeR, which was subsequently used for carrying out the expression and purification studies. BL21 (λDE3) cells transformed with pET28c/IdeR were grown to an A600nm of 0.8–1.0 in LB media at 37 °C containing 25 µg/ml of kanamycin and synthesis of IdeR protein was induced by the addition of 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) at 37 °C. After induction for 3 hours, the E. coli cells were harvested by centrifugation at 4 °C, 6340 g for 10 minutes and resuspended in 30 ml lysis buffer (20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 1 mM phenylmethylsulfonylfluoride (PMSF), 2 mM β-mercaptoethanol, and 5 mM Imidazole). The resuspended cells were sonicated and the lysate was centrifuged at 12,000 g for 45 minutes at 4 °C and the supernatant was incubated with 1.5 ml Ni-NTA resin pre-equilibrated with buffer containing 20 mM Tris-HCl (pH 8.0), 50 mM NaCl and 5 mM imidazole for 1 hour. The flowthrough was collected and the resin was washed twice with 20 ml each of buffers (20 mM Tris-HCl (pH8.0), 50 mM NaCl) containing 5 mM and 10 mM imidazole and protein was eluted as 1 ml fractions by using the elution buffer (20 mM Tris-HCl, 50 mM NaCl and 250 mM imidazole). Protein estimation of the eluted fractions was carried out by Bradford assay, analyzed by SDS-PAGE on a 12.5% gel and samples were pooled and dialyzed against the buffer (20 mM Tris, 50 mM NaCl and 1 mM DTT) and concentrated by using an Amicon Ultra protein concentrator (Millipore, Billerica, MA, USA) with a 5 kDa filter and the purified protein was stored in aliquots at −80 °C.
Electrophoretic Mobility Shift Assay (EMSA)
A synthetic double stranded oligonucleotide belonging to intergenic region between MbtA and MbtB containing IdeR binding region with sequence-5′ccctgttagcacaggctgccctaattttagtg3′ (sense strand) and Cy5 fluorophore label at 5′ end of both the strands was procured from Integrated DNA Technologies, Coralville, Iowa, U.S.A and used as a substrate for the assay. Binding reactions included 20 mM Tris-HCl at pH 8.0, 1 mM DTT, 50 mM KCl, 5 mM MgCl2, 200 µM NiSO4, 0.05 mg ml−1 poly (dI-dC), 0.05 mg ml−1 BSA, 10% glycerol as reaction buffer and purified IdeR (0.35 µg) in a reaction volume of 20 µl and the reaction was carried out at room temperature for 30 minutes. Samples were electrophoresed on a 6% polyacrylamide gel containing 40 mM Tris-acetate (pH-8.0), 10% glycerol, 200 µM NiSO4 and the running buffer (Tris-acetate-40 mM and 200 µM NiSO4). Gels were run at 90 V at room temperature and the bands were visualized by using phosphorimager (FLA-9000, Fujifilm Corporation, Minato-ku, Tokyo, Japan) with Cy5 filter and each band intensity was quantified by using the Multi Gauge software (FujiFilm Corporation, Minato-ku, Tokyo, Japan).
In vitro Inhibition Assays
Compounds were dissolved in DMSO at a concentration of 5 mg/ml and screened at a fixed concentration of 100 µg/ml. Each reaction mix contained 0.35 µg IdeR and 0.4 µl of 5 mg/ml compound (final concentration of 100 µg/ml) in the reaction volume of 20 µl containing the reaction buffer with DMSO as the negative control. The reaction mix was incubated for 15 minutes at room temperature, followed by the addition of 0.8 picomoles of labeled DNA and further incubation was carried out for 30 minutes at room temperature. Subsequently, the reaction mix was loaded onto 6% polyacrylamide gel, electrophoresed and scanned by using FLA 9000. The molecules exhibiting more than 40% inhibition were further subjected to dose response studies with varying inhibitor concentrations ranging from 0.4 µg/ml to 100 µg/ml for the determination of their IC50 values. The concentration of the inhibitor resulting in 50% reduction in the bound DNA band was considered as the IC50 of that molecule and was calculated by using the following equation 1:
| 1 |
IC50 values were determined from the results of at least two independent tests and calculated from the inhibition curve using GraphPad Prism 5.
For competition experiments, % bound DNA was calculated by using the following equation 2:
| 2 |
Per cent bound DNA were calculated by using the results of atleast two independent tests and calculated from the One way ANOVA test by using the column bar graph of GraphPad Prism 5. The data is represented as Mean ± S.E.M. by values obtained from two independent experiments.
For lead Optimization studies
Strategy 1 (a): Energy Based Pharmacophore Development
Five point energy based pharmacophore model was developed by using the e-pharmacophore27, 28 script available with Schrodinger Package (Schrodinger, LLC, New York, NY) by using the standard pharmacophore features namely hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic group (H), negatively ionizable (N), positive ionizable (P) and aromatic ring (R). Energy minimized conformers with added hydrogens and stereoisomers were generated for the selected molecules by using LigPrep 2.5 module from Schrodinger package. These were docked at the active site of the protein by using Glide29–31 and an XP-descriptor file was generated which facilitated the generation of five-point pharmacophore. Subsequently, screening of ZINC drug like database was carried out by using Phase32, 33 (Phase, version 3.3, Schrodinger, LLC, New York, NY, 2011). Default settings were kept in place which included the generation of conformers and thorough sampling. Four out of 5 site points to be matched was set as criteria for filtering. All the calculations were carried out on an 8 node SunFire X2200 cluster with CENT-OS operating system.
1(b): Generation of the Molecular Descriptors through Qikprop
The hits retrieved from the energy based pharmacophore were subsequently utilized for the generation of 2-D and 3-D descriptors which included their topological as well as ADME/Toxicity based descriptors. Qikprop module available in the Schrodinger Suite was employed for this purpose.
1(c): Mycobacterium Cell wall permeability based filtering
To filter the molecules obtained from pharmacophore screening on the basis of mycobacterial cell wall permeability, an online server, MycPermCheck34 was employed (http://www.mycpermcheck.aksotriffer.pharmazie.uni-wuerzburg.de). Molecules above the cutoff of 0.82 were shortlisted which have a false positive rate of less than 10% as prescribed by the server.
1(d): Docking Studies by using Autodock4.2
The molecules filtered with MycPermCheck were docked at the active site by employing Autodock 4.225. The docking calculations for Autodock 4.225 were carried out on a Fujitsu RX300 S7 Server as master node and Fujitsu CX250 S1 servers as compute nodes (35 no. with CENT-OS version 6).
1(e): Structure similarity based filtering
Structural similarity based filtering was further employed to remove structurally similar molecules. Openbabel software was employed and the molecules above tanimoto coefficient of 0.8 cut-off were retained.
After subjecting the molecules to above-mentioned filters, 22 compounds were purchased from Ambinter c/o Greenpharma, New Orleans, France for subsequent inhibition studies.
Strategy 2: Structure similarity based search
The .sdf files of most potent molecules were used as query structures to find structurally similar analogs against the complete NCI library by using the software Openbabel. The tanimoto coefficient with cut-off 0.7 was set and the structurally similar molecules were procured for subsequent in vitro studies.
Resazurin microtiter assay (REMA) based whole cell assay for MIC90 determination
MIC90 determination was carried out against M. tuberculosis cells grown in MB7H9 media (Difco, Middlebrook from Becton Dickinson and Company, Sparks, MD, USA). Appropriate dilutions of the compounds were employed at a concentration range of 1.25 µg/ml–100 µg/ml and incubated with M. tuberculosis cells at a final A600nm of 0.02 in MB7H9 in an assay volume of 200 µl and MIC90 values were calculated by using REMA40, 41 as described previously42. Resazurin based assay gives quantitative measurement of the cell viability. Resazurin, a non fluorescent molecule which is blue in colour is reduced to fluorescent resorufin (pink in colour) in the presence of viable cells. The conversion of blue to pink is correlated with viability of the cells. The MIC90 is defined as the inhibitor concentration at which fluorescence intensity is decreased by 90% when compared with the appropriate controls. The values were calculated from three independent experiments and represented as Mean ± S.E.
Cytotoxicity Assays
The cytotoxicity of the compounds was determined by employing REMA using various mammalian cell lines such as THP-1 (human monocytic macrophage cell line), HEK (Human Embryonic Kidney cell line), MDCK (Madine darby canine kidney cell line) and HepG2 (Human hepatocellular carcinoma cell line). 1 × 104 cells/well were employed in a sterilized 96-well plate containing serial dilutions of compounds ranging from 1 µg/ml to 200 µg/ml concentration, in their respective media (RPMI for THP-1, DMEM for HEK and HepG2 and MEM for MDCK) with 10% fetal bovine serum and Antibiotic-Antimycotic mix (Invitrogen Corporation, Carlsbad, California, USA). IC50 is defined as the concentration of the compound that causes 50% reduction in the fluorescence intensity indicating 50% viable cells. IC50 values were determined from the results of at least two independent tests and Mean ± S.E.M was calculated from the inhibition curve using GraphPad Prism 5.
Evaluation of the compounds to inhibit the intracellular M. tuberculosis
To evaluate the inhibitory potential of the compounds against the growth of intracellular bacteria, M. tuberculosis H37Rv was grown till an Abs600nm of 1 to 1.2 and simultaneously, THP-1 cells were grown and activated by the addition of 30 nm phorbol myristic acetate (PMA) followed by incubation at 37 °C for 16 hours in the presence of 5% CO2. Media was discarded the next day and the adhered THP-1 cells were washed once with RPMI medium. The cells were scraped, harvested and counted by trypan blue exclusion staining. Simultaneously, M. tuberculosis cells were harvested at 4,500 rpm for 10 minutes and washed twice with MB7H9 medium. The M. tuberculosis cells were resuspended in RPMI medium and 0.5-mm glass beads were added followed by vortexing for 15 minutes to make a single cell suspension. The suspension was judiciously centrifuged to remove any clumps. Activated THP-1 cells were infected with M. tuberculosis in suspension at a multiplicity of infection (MOI) of 5:1 (bacteria/macrophage) at 37 °C with a constant shaking of 100 rpm. Following infection for two hours, the cells were harvested, washed and re suspended again in RPMI medium with addition of Amikacin (200 μg/ml) followed by incubation at 37 °C for 1 hour with constant shaking at 100 rpm to remove any extracellular bacilli. After the Amikacin treatment, cells were harvested, washed and 1 × 104 cells were seeded in each well containing appropriate dilutions of the compounds in a final volume of 250 μl (I-33 and I-34–100 to 500 μg/ml, I-108–25 μg/ml to 150 μg/ml, Rifampicin − 2.5 μg/ml to 10 μg/ml). Simultaneously, 1 × 104 cells of uninfected THP-1 cells were also incubated with the same corresponding dilutions of the compounds to measure the viability of uninfected THP-1 cells in the presence of compounds. The plates were then kept at 37 °C in the presence of 5% CO2 for five days and on the 5th day, media was removed from the wells followed by the addition of 100 μl sterile water for 2 hours at 37 °C to lyse the THP-1 cells. The cells were then vigorously pipetted and the entire volume of 100 μl was spotted on MB7H11 agar plates containing oleic acid-albumin-dextrose-catalase (OADC) followed by the incubation at 37 °C for three to four weeks to analyze the inhibition of bacterial growth. Also, 30 μl of 0.01% solution of resazurin was added to uninfected THP-1 plates and the growth measurement was analyzed the next day by alamar blue assay.
Electronic supplementary material
Acknowledgements
We thank the Drug Synthesis and Chemistry Branch, Development Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institute of Health, Bethesda, MD, for providing the compounds. Dr. N. Latha at Sri Venkateshwara college, University of Delhi South Campus and Dr. Nidhi Jatana at IGIB, Mathura road, New Delhi are acknowledged for providing access to the computational facility and usage of Schrodinger package for the pharmacophore modelling work and their intellectual inputs. Prof. Debi P. Sarkar at Dept. of Biochemistry, University of Delhi South Campus, New Delhi for providing HepG2 cell line and Dr. Murali Krishna Kaja and Dr. Anmol Chandele at ICGEB/EMORY vaccine centre for providing HEK cell line. We thank Priti Singh and Tannupriya Gosain for excellent technical help. DBT-supported Distributed Information Sub-Centre, Department of Biochemistry, University of Delhi South Campus, New Delhi is acknowledged for providing computational facilities. A.R is thankful to Department of Science and Technology, India for the J.C Bose fellowship. This work was supported by financial grant from Department of Biotechnology, Government of India (Grant Number – BT/01/COE/05/06-II).
Author Contributions
A.R., G.K. and A.K.T. conceived and designed the experiments. A.R. performed the experiments and analyzed the data. A.R., G.K. and A.K.T. wrote the manuscript. A.K.T. and G.K. provided overall supervision throughout the study.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04748-9
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Garima Khare, Email: garima1822@yahoo.co.in.
Anil K. Tyagi, Email: aniltyagi@south.du.ac.in
References
- 1.Nachega JB, Chaisson RE. Tuberculosis drug resistance: a global threat. Clin. Infect. Dis. 2003;36:24–30. doi: 10.1086/344657. [DOI] [PubMed] [Google Scholar]
- 2.Raviglione M, Smith I. XDR tuberculosis-implications for global public health. N. Engl. J. Med. 2007;356:656–659. doi: 10.1056/NEJMp068273. [DOI] [PubMed] [Google Scholar]
- 3.Crichton RR, Charloteaux-wauters M. Iron transport and storage. Eur. J. Biochem. 1987;164:485–506. doi: 10.1111/j.1432-1033.1987.tb11155.x. [DOI] [PubMed] [Google Scholar]
- 4.Weinberg ED. Iron withholding: a defense against infection and neoplasia. Physiol. Rev. 1984;64(1):65–102. doi: 10.1152/physrev.1984.64.1.65. [DOI] [PubMed] [Google Scholar]
- 5.Wright DG, Gallin JI. Secretory responses of human neutrophils: Exocytosis of specific (secondary) granules by human neutrophils during adherence in vitro and during exudation in vivo. J. Immunol. 1979;123(1):285–94. [PubMed] [Google Scholar]
- 6.Muller D, Edwards ML, Smith DW. Changes in iron and transferrin levels and body temperature in experimental airborne legionellosis. J. Infect. Dis. 1983;147(2):302–7. doi: 10.1093/infdis/147.2.302. [DOI] [PubMed] [Google Scholar]
- 7.Leffell MS, Spitznagel JK. Fate of human lactoferrin and myeloperoxidase in phagocytizing human neutrophils: effects of immunoglobulin G subclasses and immune complexes coated on latex beads. Infect. Immun. 1975;12(4):813–20. doi: 10.1128/iai.12.4.813-820.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snow GA. Mycobactins: iron-chelating growth factors from mycobacteria. Bacteriol. Rev. 1970;34(2):99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gobin J, et al. Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc. Natl. Acad. Sci. 1995;92(11):5189–93. doi: 10.1073/pnas.92.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez GM, Smith I. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J. Bacteriol. 2006;188(2):424–30. doi: 10.1128/JB.188.2.424-430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farhana A, et al. Mechanistic insights into a novel exporter-importer system of Mycobacterium tuberculosis unravel its role in trafficking of iron. PLoS One. 2008;3(5):e2087. doi: 10.1371/journal.pone.0002087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wells RM, et al. Discovery of siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog. 2013;9(1):e1003120. doi: 10.1371/journal.ppat.1003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madigan CA, et al. Lipidomic analysis links mycobactin synthase K to iron uptake and virulence in Mycobacterium tuberculosis. PLoS Pathog. 2015;11(3):e1004792. doi: 10.1371/journal.ppat.1004792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones CM, et al. Self-poisoning of Mycobacterium tuberculosis by interrupting siderophore recycling. Proc. Natl. Acad. Sci. 2014;111(5):1945–50. doi: 10.1073/pnas.1311402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keyer K, Imlay JA. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. 1996;93(24):13635–40. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gold B, Rodriguez GM, Marras SA, Pentecost M, Smith I. The Mycobacterium tuberculosis IdeR is a dual functional regulator that controls transcription of genes involved in iron acquisition, iron storage and survival in macrophages. Mol. Microbiol. 2001;42(3):851–65. doi: 10.1046/j.1365-2958.2001.02684.x. [DOI] [PubMed] [Google Scholar]
- 17.Pandey R, Rodriguez GM. IdeR is required for iron homeostasis and virulence in Mycobacterium tuberculosis. Mol. Microbiol. 2014;91(1):98–109. doi: 10.1111/mmi.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pohl E, Holmes RK, Hol WG. Crystal structure of the iron-dependent regulator (IdeR) from Mycobacterium tuberculosis shows both metal binding sites fully occupied. J.Mol.Biol. 1999;285(3):1145–56. doi: 10.1006/jmbi.1998.2339. [DOI] [PubMed] [Google Scholar]
- 19.Feese MD, Ingason BP, Goranson-Siekierke J, Holmes RK, Hol WG. Crystal structure of the iron-dependent regulator from Mycobacterium tuberculosis at 2.0-A resolution reveals the Src homology domain 3-like fold and metal binding function of the third domain. J.Biol.Chem. 2001;276(8):5959–66. doi: 10.1074/jbc.M007531200. [DOI] [PubMed] [Google Scholar]
- 20.Wisedchaisri G, Holmes RK, Hol WG. Crystal structure of an IdeR-DNA complex reveals a conformational change in activated IdeR for base-specific interaction. J.Mol.Biol. 2004;342(4):1155–69. doi: 10.1016/j.jmb.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 21.Wisedchaisri G, et al. Crystal structures, metal activation, and DNA-binding properties of two-domain IdeR from Mycobacterium tuberculosis. Biochemistry. 2007;46(2):436–47. doi: 10.1021/bi0609826. [DOI] [PubMed] [Google Scholar]
- 22.Chou CJ, et al. Functional studies of the Mycobacterium tuberculosis iron-dependent regulator. J. Biol. Chem. 2004;279(51):53554–61. doi: 10.1074/jbc.M407385200. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh S, Chandra N, Vishveshwara S. Mechanism of Iron-Dependent Repressor (IdeR) Activation and DNA binding: A molecular dynamics and protein structure network study. PLoS Comput Biol. 2015;11(12):e1004500. doi: 10.1371/journal.pcbi.1004500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miteva MA, et al. FAF-Drugs: free ADME/tox filtering of compound collections. Nucleic Acids Res. 2006;34:W738–44. doi: 10.1093/nar/gkl065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris GM, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- 26.Schmitt MP, Holmes RK. Cloning, sequence, and footprint analysis of two promoter/operators from Corynebacterium diphtheria that are regulated by the diphtheria toxin repressor (DtxR) and iron. J Bacteriol. 1994;176(4):1141–9. doi: 10.1128/jb.176.4.1141-1149.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salam NK, Nuti R, Sherman W. Novel Method for Generating Structure-Based Pharmacophores Using Energetic Analysis. J. Chem. Inf. Model. 2009;49:2356–2368. doi: 10.1021/ci900212v. [DOI] [PubMed] [Google Scholar]
- 28.Loving K, Salam NK, Sherman W. Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Aided Mol. Des. 2009;23:542–554. doi: 10.1007/s10822-009-9268-1. [DOI] [PubMed] [Google Scholar]
- 29.Friesner RA, et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring.1. Method and assessment of Docking Accuracy. J. Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 30.Halgren TA, et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring.2. Enrichment Factors in Database Screening. J. Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 31.Friesner RA, et al. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J.Med.Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 32.Dixon SL, et al. PHASE: A New Engine for Pharmacophore Perception, 3D QSAR Model Development, and 3D Database Screening. 1. Methodology and Preliminary Results. J. Comput. Aided Mol. Des. 2006;20:647–671. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- 33.Dixon SL, Smondyrev AM, Rao SN. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006;67:370–372. doi: 10.1111/j.1747-0285.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 34.Merget B, Zilian D, Muller T, Sotriffer CA. MycPermCheck: the Mycobacterium tuberculosis permeability prediction tool for small molecules. Bioinformatics. 2013;29(1):62–8. doi: 10.1093/bioinformatics/bts641. [DOI] [PubMed] [Google Scholar]
- 35.O’Boyle NM, et al. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Therese PJ, et al. Multiple e-pharmacophore modeling, 3D-QSAR, and high-throughput virtual screening of hepatitis C virus NS5B polymerase inhibitors. J Chem Inf Model. 2014;54(2):539–52. doi: 10.1021/ci400644r. [DOI] [PubMed] [Google Scholar]
- 37.Palakurti R, Sriram D, Yogeeswari P, Vadrevu R. Multiple e-Pharmacophore Modeling Combined with High-Throughput Virtual Screening and Docking to Identify Potential Inhibitors of β-Secretase (BACE1) Mol Inform. 2013;32(4):385–98. doi: 10.1002/minf.201200169. [DOI] [PubMed] [Google Scholar]
- 38.Natarajan P, et al. E-pharmacophore-based virtual screening to identify GSK-3β inhibitors. J Recept Signal Transduct Res. 2016;36(5):445–58. doi: 10.3109/10799893.2015.1122043. [DOI] [PubMed] [Google Scholar]
- 39.Katari SK, et al. Inhibitor design against JNK1 through e-pharmacophore modeling docking and molecular dynamics simulations. J Recept Signal Transduct Res. 2016;24:1–14. doi: 10.3109/10799893.2016.1141955. [DOI] [PubMed] [Google Scholar]
- 40.Palomino JC, et al. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003;46:3616–3619. doi: 10.1128/AAC.46.8.2720-2722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin A, Camacho M, Portaels F, Palomino JC. Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple and inexpensive method. Antimicrob. Agents Chemother. 2003;47:3616–3619. doi: 10.1128/AAC.47.11.3616-3619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khare G, Kumar P, Tyagi AK. Whole cell screening-based identification of inhibitors against the intraphagosomal survival of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013;57(12):6372–6377. doi: 10.1128/AAC.01444-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pettersen EF, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.