

Abstract
The multi-domain transcriptional coactivators CBP/p300 integrate a multitude of signaling inputs, interacting with more than 400 proteins via one or more of their globular domains. While CBP/p300 function is typically considered in terms of these structured domains, about half of the protein consists of intrinsically disordered regions (IDRs) of varying length. However, these IDRs have only been thought of as linkers that allow flexible spatial arrangement of the structured domains, but recent studies have shown that similar IDRs mediate specific and critical interactions in other proteins. To examine the roles of IDRs in CBP, we performed yeast-two-hybrid screenings of placenta and lung cancer cDNA libraries, which demonstrated that the long IDR linking the KIX domain and bromodomain of CBP (termed ID3) can potentially bind to several proteins. The RNA-binding Zinc-finger protein 106 (ZFP106) detected in both libraries was identified as a novel substrate for CBP-mediated acetylation. Nuclear magnetic resonance (NMR) spectroscopy combined with cross-linking experiments and competition-binding assays showed that the fully disordered isolated ID3 transiently interacts with an IDR of ZFP106 in a fashion that disorder of both regions is maintained. These findings demonstrate that beside the linking function, ID3 can also interact with acetylation substrates of CBP.
Introduction
CREB-binding protein (CBP) and its homolog histone acetyltransferase p300 (p300) are multi-domain transcriptional regulators involved in diverse biological functions such as cell cycle, proliferation, differentiation, homeostasis, and apoptosis1, 2. Their function is mainly based on their interaction with a large variety of transcription factors and other regulatory proteins targeting their intrinsic histone acetyl transferase (HAT) activity on the chromatin and a broad range of partner proteins3, 4. Mutations affecting CBP/p300 genes have been implicated in Rubinstein-Taybi syndrome and several types of cancer, emphasizing their central role in orchestrating gene regulation2, 3.
CBP/p300 are about 2400 residues in length and contain several highly conserved domains and motifs, such as zinc-finger motifs PHD, RING, TAZ1, TAZ2 and ZZ, the CREB binding (KIX) domain, nuclear receptor coactivator binding domain (NCBD), bromodomain and HAT domain. Long intrinsically disordered regions (IDRs, Fig. 1a) connect the structured domains that mediate interactions with more than 400 partner proteins1. Although intrinsically disordered proteins/regions of proteins (IDPs/IDRs) have frequently been implicated in protein-protein interactions5, the function(s) of the IDRs in CBP/p300 has never been investigated in detail. Rather, it has been suggested that their role is to “connect” the folded, functional elements, enabling CBP/p300 to engage with distinct patterns of chromatin-bound transcription factors in assembling large signaling complexes, such as the enhanceosome6. However, it would be quite surprising if about half of the protein primary sequence would only have the function of “connecting” globular functional domains, and therefore it is likely that a variety of different unexplored functional roles are encoded in those long disordered regions. The N- and C-terminal IDRs in CBP/p300 may also enable long-range structural/functional communication within the protein, as exemplified by the repression of HAT activity by phosphorylation of N-terminal Ser 89 in p3007. HAT activity and substrate specificity are also regulated by the flanking domains of HAT (bromodomain, PHD, RING, ZZ and TAZ2 domain) through intramolecular interactions8.
Figure 1.
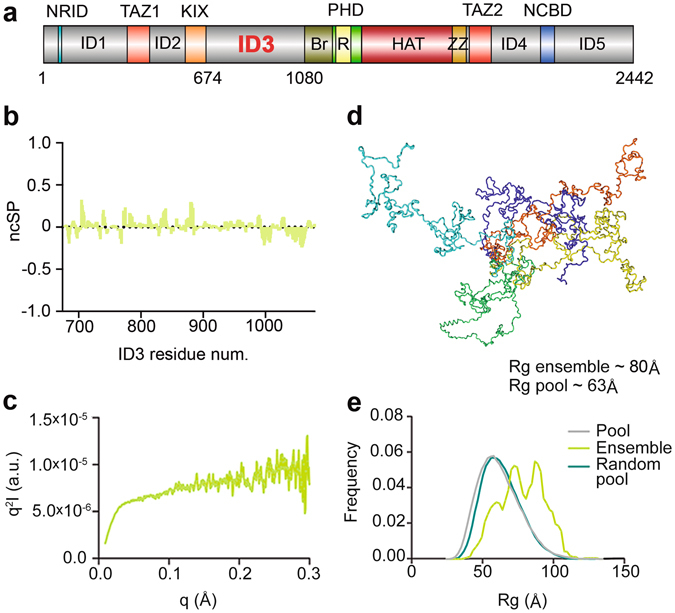
Domain 3 (ID3) of CBP is fully disordered. (a) CBP domain organization. CBP globular domains (colored; Br: Bromodomain, R: Ring) are connected by intrinsically disordered regions (IDRs, grey) of different lengths. The intrinsically disordered region 3 (ID3, aa674–1080) is located between the KIX domain and bromodomain of CBP. (b) Neighbor corrected structural propensities (ncSP) obtained from experimentally measured N, C′, Cα and Cβ chemical shifts. Positive and negative values correspond to α-helical and β-sheet propensities, respectively. (c) SAXS Kratky plot also shows an overall disordered state, with a minor tendency for local secondary structure formation. (d) Ensemble of selected conformations ID3 may adopt in solution. (e) Rg distributions of a random pool of ID3 conformations (teal), of the pool of ID3 conformations biased by experimental ncSP scores (grey) and the ensemble calculated by Flexible Meccano and selected by GAJOE (green).
To assess the possible binding and regulatory role(s) of IDRs in CBP/p300, we are conducting systematic studies on the structural state and binding function of the IDRs of CBP, including ID3 (located between the KIX domain and bromodomain), ID4 (located between the TAZ2 domain and NCBD domain)9 and ID5 (located at the C-terminal end of the molecule, past the NCBD domain; cf. Fig. 1a). Our ultimate aim with these studies is to develop a comprehensive structural-functional model of CBP/p3009, 10, that also includes the role(s) of IDRs and their interplay with globular domains. No specific interaction has been mapped to any of these regions yet6, 11. For ID3, we identified several potential new interaction partners in yeast two-hybrid (Y2H) screenings, among them the Zinc-finger protein 106 (ZFP106). Recent studies suggest that this nuclear protein, ZFP106, is involved in RNA processing and it has been associated with neuromuscular and neurodegenerative disorders12–15 such as amyotrophic lateral sclerosis (ALS). We identified ZFP106 as a novel substrate of CBP-mediated acetylation. Active, full-length CBP acetylated ZFP106 on several lysines residues located in two main clusters. The transient interaction between the two IDRs was confirmed by a combination of competition-binding assays, cross-linking experiments and NMR spectroscopy. Finally, we determined the regions of ID3 and ZFP106 involved in the fuzzy interaction by employing paramagnetic relaxation enhancement (PRE) NMR spectroscopy.
Results
ID3 is fully disordered
Although bioinformatics predictions suggest that ID3 is an IDR, its structural properties have never been experimentally characterized. To this end, we carried out the structural characterization of the domain by several techniques in reducing buffer at physiological salt concentration. Both in SDS-PAGE and size-exclusion chromatography (SEC), the apparent molecular weight (Mw) of ID3 is about 70 kDa, two times its actual Mw, which is often the first indication of structural disorder16. In far-ultraviolet (UV) circular dichroism, ID3 has a strong negative peak around 200 nm, typical of random coil proteins, with a slight tendency to local secondary structure formation16 (Supplementary Fig. S1b). Lack of long-lasting structural elements in ID3 allowed the employment of a heat-treatment step17 in the protein purification protocol, which increases the purity and stability of the sample. This was confirmed by comparing circular dichroism and 2D 1H-15N NMR spectra of ID3 obtained with and without this step (cf. Supplementary Fig. S1b and S1c), no significant difference between the two samples could be observed. Solution NMR has become the preferred technique for studying IDPs/IDRs18, 19 as it allows a detailed structural analysis of the polypeptides. To achieve the essentially complete sequence specific assignment (about 95%) of the 407 residues of ID3, including its 75 prolines (18%), the recently developed NMR strategy to study complex IDPs, which combines 1H and 13C detected high-resolution multi-dimensional NMR experiments18, 20–22 was necessary (Supplementary Fig. S1a and Tables S1 and S4). The chemical shift (CS) assignment of this disordered domain was facilitated by acquiring additional experiments on a shorter N-terminal fragment of ID3 (ID3-F1 from residue 674–848), which were used also to validate the assignment of the full-length linker. NMR was also used to characterize ID3 backbone dynamics by measuring 15N R 1, 15N R 2 relaxation rates and heteronuclear 1H-15N NOE values (hetNOE) (Supplementary Fig. S2). The domain showed high local mobility on the picoseconds to nanoseconds timescale within residue 725–750 and 775–800, displaying higher 1H-15N hetNOE and 15N R 2 values, which indicate slightly more structured conformations than in the rest of the polypeptidic chain. Furthermore, the scattered 15N R 2 values for some residues in the N-terminal region indicate the occurrence of some exchange processes18. Secondary-structure propensities (ncSP)23 calculated from experimental CS values24 suggested short regions of isolated transient α-helices (in the N-terminal half; approximate residue 674–874) or extended β-type conformations (in the C-terminal third; approximate residue 974–1080), highlighting the presence of potential binding sites (Fig. 1b).
The structural properties are different from those of ID49, for which two segments showing significant α-helical propensity were detected, indicating that each linker has specific structural and dynamic properties that are likely to have an impact on their function.
SAXS measurements [following removal of potential aggregates by high-pressure liquid chromatography (HPLC)] yielded an average radius of gyration (Rg) of 80 Å of the different conformers in solution (Fig. 1c and e), corresponding to a disordered state.
Ensemble description of ID3 structure
To investigate potential pre-formed structural elements in ID325, we combined data from SAXS and NMR experiments and calculated an ensemble of possible conformations that ID3 can adopt in solution. To this end, Flexible-Meccano was used with NMR-derived ncSP (Fig. 1b) as constraints to generate a random semi-pool of 9876 conformers26. For every conformer, theoretical SAXS scattering curves were calculated, compared to the experimental SAXS data and used for selecting the best fitting ensemble27 (Fig. 1d; for the fit between the experimental SAXS data and the selected pool, see Supplementary Fig. S3a). We observed a preference in the ensemble for more extended conformers, with a shift towards higher Rg values (Fig. 1e). Short dispersed α-helices observed in the ensemble (Fig. 1d) correspond to secondary structure elements derived from the ncSP input (Fig. 1b).
Potential binding motifs in the disordered ensemble of ID3
In order to highlight the potential role of ID3 in mediating protein-protein interactions, we also analyzed characteristic features of the sequence (Supplementary Fig. S4). In general, about half the residues of ID3 fall into potential binding sites predicted by ANCHOR28 and about half the sequence is highly conserved by DisCons29, suggesting the presence of multiple interaction sites within this disordered region of CBP. Several moderately populated helical regions in the N-terminal half appear as potential preformed binding sites, e.g. the highly conserved helical segment around residues 824–844 is predicted as a potential molecular recognition feature (MoRF). C-terminal regions of β propensity are highly conserved and have a conspicuous enrichment of post-translational modification (PTMs) sites, suggesting functional regulation of this region. The DynaMine30 profile also shows peaks in this region (Supplementary Fig. S4, underlined), which together predispose this region for mediating interaction(s) probably not based on induced folding31.
ID3 potentially interacts with several proteins
To find out if ID3 mediates protein-protein interactions, we applied a Y2H screening on two different cDNA libraries, one from human placenta (important for its broad spectrum of expressed genes) and another from human lung cancer (useful to observe potential disease-related interactions). We expected that besides general, housekeeping proteins, shared partners in the two different libraries may point to functionally important and potentially disease-associated interaction partners of CBP/p300 mediated by ID3, mitigating the effect of known high false positive hit rates of Y2H. A total of 37 proteins were classified as potential ID3 binders, including 4 candidates that have already been described to interact with CBP or p300 (HIPK132, PIAS133, PAIS334 and TDG35) (Fig. 2a). Surprisingly, sequence analysis of the interacting fragments and full-length proteins highlighted a preference of ID3 for binding disordered regions within interacting partners (Fig. 2b; for the statistical test indicating significance preference, see Supplementary Fig. S3b). Importantly, about half of the identified protein partners were involved in processes related to protein biogenesis, such as gene transcription, pre-mRNA maturation and protein biosynthesis. Among those, we decided to further investigate the transcriptional regulator ZFP106 (Q9H2Y7), because its role in immune response, muscle differentiation, DNA damage, splicing and motor and sensory neuron survival14, 36. Further, ZFP106 and CBP may be co-localized in nuclear speckles and be part of a larger complex regulating mRNA production or maturation12, 37, 38. ZFP106 was identified in both libraries and structural disorder is involved in recognition in both binding partners. In the placenta library, residues 1265–1521 of ZFP106 were identified, whereas a second, overlapping fragment (residue 1007–1448) was found in the lung cancer library (this clone was frame-shifted, which is probably compensated by translational frame-shift in yeast). Only one copy was found in each library, which is consistent with the low expression levels of ZFP106 in the respective tissue types38. For the detailed characterization of the ID3 – ZFP106 interaction, the shorter fragment 1265 to 1521, denominated as ZFP106-f, was selected.
Figure 2.
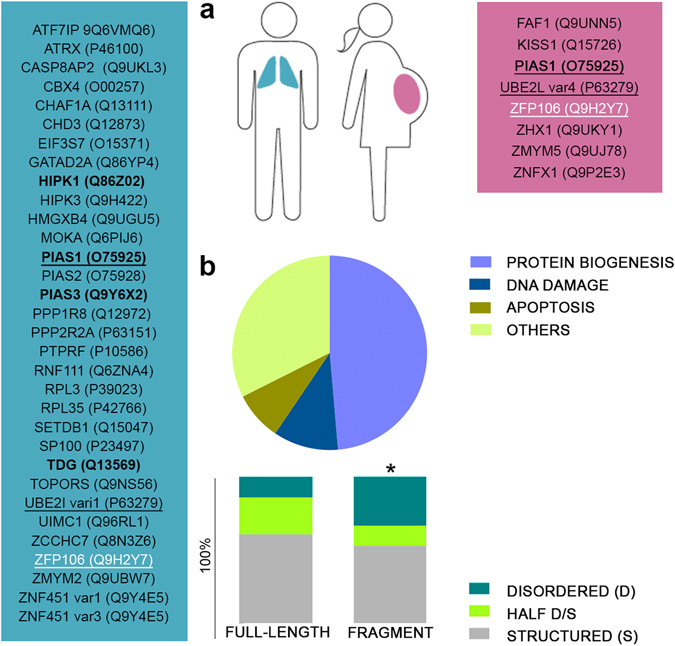
ID3 interacts with the transcription factor ZFP106. (a) Potential interaction partners of ID3 have been identified by a yeast 2-hybrid (Y2H) screen. Y2H interactions were classified according to the libraries in which they were detected: placenta (pink), lung cancer (blue), or both (black underlined). Four proteins have been described in previous studies to interact with one or more of the CBP’s globular domains32–35 (bold). ZFP106, selected for further studies, is marked in white. (b) Potential interactors were analyzed for their structural content (Structured: more than 60% of its sequence predicted to be structured; Disordered: more than 60% of its sequence predicted to be disordered; Half D/S: the rest; *Statistically significant preference of ID3 for binding disordered regions) and for their function. Disorder predictions were carried out with the IUPred webserver70, whereas gene ontology (GO) annotations and available literature were used to classify the proteins in four main groups: Protein biogenesis (including transcription, pre-mRNA maturation and protein biosynthesis), DNA damage, Apoptosis and Others.
The transcriptional regulator ZFP106 is acetylated by full-length CBP
CBP/p300 have recently been reclassified as lysine acetyltransferase 3 (KAT3) that can acetylate interacting protein partners, primarily transcription factors or coactivators1. To probe such a functional consequence of the interaction between CBP and ZFP106, we assayed the acetylation of ZFP106-f with active, full-length CBP. The reaction was performed in HAT assay buffer in the presence of reducing agents, at low protein concentrations. Western blot (WB) analysis with anti-acetylated-lysine antibody showed a band for the 50 kDa ZFP106-f, revealing ZFP106 as a novel substrate of CBP (cf. Fig. 3).
Figure 3.
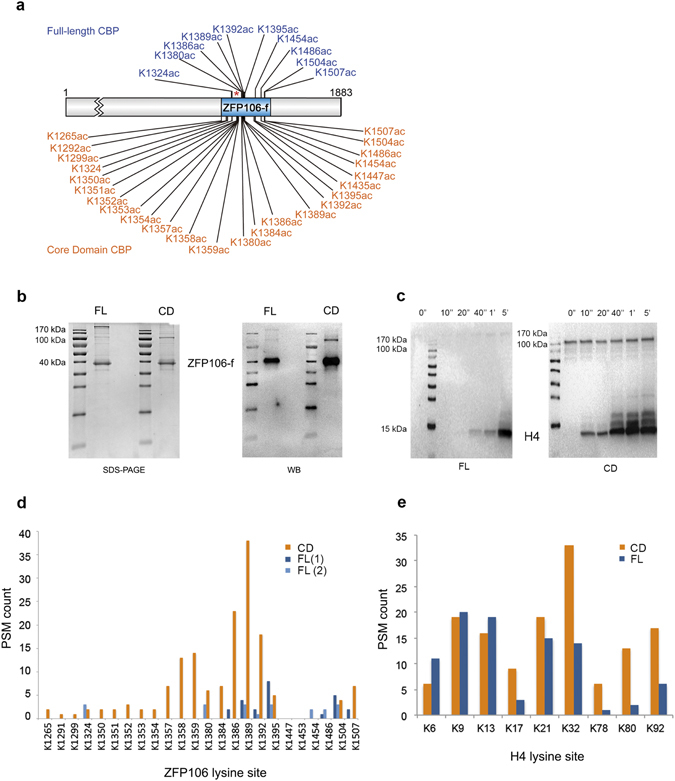
ZFP106 is specifically acetylated only by full-length CBP. (a) ZFP106-f was acetylated by either full-length (FL) or the catalytic core domain (CD) of CBP under comparable conditions, and the sites of acetylation were determined by MS. Specific acetylation pattern by full-length CBP (blue) is in contrast to the indiscriminate acetylation achieved by the core domain (orange). *Location of the lysine rich patch of ZFP106-f. (b) SDS-PAGE and WB developed with anti-acetylated-lysine antibody of ZFP106-f acetylated by the full-length or core domain of CBP. (c) Western blot developed with anti-acetylated-lysine antibody of histone 4 (H4) acetylated by the full-length or core domain of CBP. Time points of the reaction are presented and highlight a much higher activity for the CD compared to the FL enzyme. (d and e) The ratio of acetylated peptides corresponding to various lysines identified by MS using ZFP106-f (d) or H4 (e) as a substrate, acetylated with full-length CBP (dark blue: first MS analysis; light blue: second MS analysis) or its core domain (orange).
By MS, no basal acetylation (prior to CBP addition) of the ZFP106-f sample was detected, whereas CBP targeted lysines primarily within two main hot-spots in the C-terminal half of the protein, acetylating only ten lysines out of twenty-four (in about 5–10% of the population) (Fig. 3a). For the complete MS sequence coverage, two MS analyses (FL-1 and FL-2) were needed as each independent analysis covered only 62% and 90% of ZFP106-f amino acid sequence (Fig. 3d).
Acetylation of ZFP106-f by the CBP core domain alone is efficient but nonspecific
In order to determine if the targeting of lysine acetylation of ZFP106-f required specific protein-protein interaction in the context of full-length CBP, we compared the acetylation pattern of ZFP106-f and histone H4 (the classic CBP substrate) by the core domain (which contains the active HAT domain plus flanking small, globular domains: bromodomain, PHD, RING, ZZ and TAZ2, cf. Fig. 1a) and full-length CBP. Acetylation by the core domain reaches saturation much faster than acetylation by full-length CBP (e.g. H4 acetylation, Fig. 3c). With H4, there are already some differences between the ratio of acetylation of specific (functional N-terminal tail, K6 to K32) and non-functional (apparently non-functional, K78 to K92) lysines by the core domain and full-length CBP, i.e. full-length CBP has some capacity to discriminate between specific and non-specific sites. When ZFP106-f acetylation is studied this discrimination is much stronger; the core domain acetylates all the lysines available, while full-length CBP acetylating only a subset of them (about 10/24, Fig. 3a and d). Thus, the modification by the core domain can be considered indiscriminate, whereas by the full-length CBP “specific”. This can be interpreted as substrate targeting via specific protein-protein interaction between ZFP106-f and full-length CBP, involving its ID3 region.
Interaction of ID3 and ZFP106 assessed by competition-binding
The discovery of ZFP106-f as a novel substrate of CBP-mediated acetylation allowed us to ascertain the interaction of ID3 and ZFP106-f via competition-binding assays. To investigate the interaction in more detail, we carried out the competition assay by titrating recombinant ID3 into the acetylation reaction with active, full-length CBP. The free ID3 was found to compete with the ID3 of full-length CBP for binding the substrate, ZFP106-f, causing a significant concentration-dependent decrease in the acetylation of ZFP106-f (Fig. 4a). To provide evidence for the specificity of the interaction, we repeated the competition assays with an excess of ID3 of “scrambled” sequence, scrID3 (maintaining the original amino acid composition), which is an approach often used to provide evidence for the specificity of interactions of IDPs31. Scrambling abrogates all possible sequence-specific interaction, while maintaining the ones that only derive from physical features relying on amino acid composition. In addition, we generated an ID3 deletion mutant lacking the C-terminal region (ID3delCTR) (Supplementary Fig. S5), since this region was found to interact with ZFP (cf. section “Dissecting the fuzzy ID3-ZFP106-f interaction”). This construct also fails to have an impact on the acetylation level of ZFP106-f (Fig. 4a and Supplementary Fig. S5). Furthermore, in another control experiment with H4 as a substrate, ID3 did not induce a reduction of H4 acetylation, excluding the possibility that ID3 might directly inhibit CBP-HAT activity or bind to another CBP domain and alter its catalytic activity (Fig. 4a and Supplementary Fig. S5). Overall, these results show that ID3 specifically binds to ZFP106-f via defined sequence features.
Figure 4.
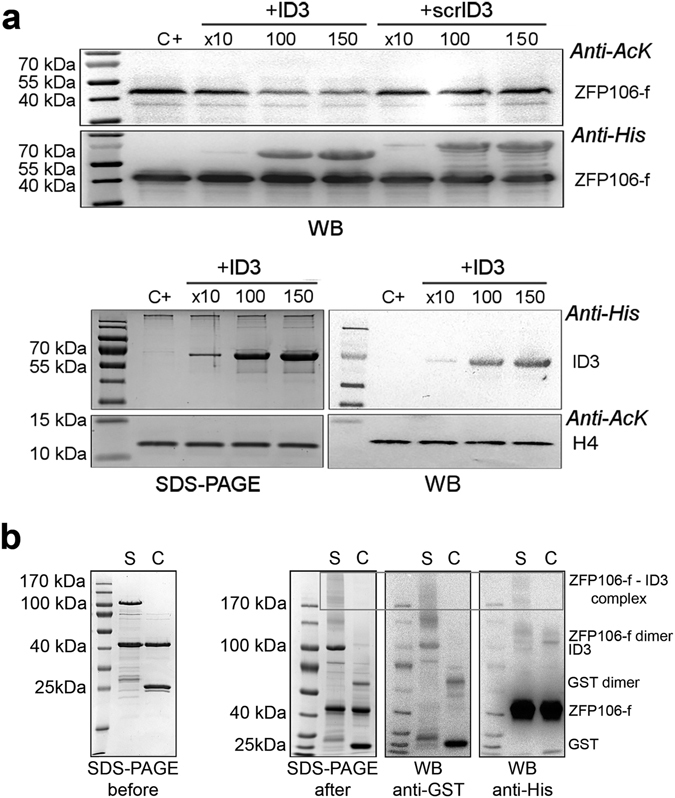
ID3 –ZFP106-f interaction assessed by competition binding assay and cross-linking. (a) Top: Competition assays with ZFP106-f as a substrate demonstrate that recombinant ID3 competes with ID3 in the full-length CBP for binding ZFP106-f, which results in a decrease of the ZFP106-f acetylation in the presence of an excess of ID3. However, addition of the same amount of scrambled ID3 (scrID3) has no effect. Bottom: as a control, the same experiment was run using histone 4 (H4) as a substrate; no change in H4 acetylation level could be detected after addition of an excess of ID3. Acetylation was followed by anti-Ac-Lys antibody, adding ID3 to the acetylation reaction in an excess of 10, 100 or 150 times to full-length CBP (18.5 pmol) and its effect was also compared to that of scrambled-ID3 (scrID3; control reaction) (C+: control reaction). (b) Cross-linked ZFP106-ID3 complex (S: sample reaction) is shown by the presence of high-Mw overlapping bands in the SDS-PAGE gel after cross-linking, anti-His WB and anti-GST WB (box). Under similar condition, ZFP106-GST (C: control reaction) were not cross-linked. Cross-linked complexes formed during the reaction are detailed on the right side of the WB and they have been determined by comparing signals between the anti-His and anti-GST WB (GST 25 kDa; ZFP106-f 40 kDa; GST dimer 62 kDa; ID3 100 kDa, ZFP106-f dimer 115 kDa, ZFP106-f – ID3 complex above 170 kDa).
Fuzzy interaction of ID3 with the transcriptional regulator ZFP106
In silico analysis and circular dichroism experiments (Fig. 5) showed that ZFP106-f is also largely disordered, which suggests a rather transient binding between the two IDRs that can occur either via a mutual (synergistic) folding39 or a “fuzzy” interaction31, in which both IDRs remain largely disordered. In fact, Y2H experiments are particularly suitable for detecting transient protein interactions40, whereas in co-IP and SEC experiments no strong or stable interaction was detected. Circular dichroism measurements confirmed the fuzziness of the complex, in which neither of the partners undergoes major structural rearrangements upon association (Fig. 5b). Next, we applied cross-linking with BS3 (bis(sulfosuccinimidyl)suberate)41 to capture the transient interaction of GST-tagged ID3 (or free GST as control) and His-tagged ZFP106-f (Fig. 4b). We observed on SDS-PAGE the formation of high-Mw complexes in which the presence (and thus interaction) of both protein partners was proven by two consecutive Western blots (with a membrane stripping step in between) with anti-His and anti-GST antibody to visualize high-Mw overlapping bands (box). It should be noted that in these experiments the Mw of cross-linked bands does not correspond to the sum of the Mw of the individual proteins, because cross-linked protein products migrate anomalously on the gel, as previously described42. In addition, ZFP106-f appears to be intermolecularly crosslinked to form homodimers of higher apparent Mw (about 115 kDa for a dimer of 80 kDa), and also intramolecularly, in multiple different ways, causing the band to appear as a smear of heterogeneous apparent Mw population (as a consequence of the high number of lysine residues in the protein). Specificity of the cross-linking was assessed in our control reaction (C), where under the same conditions no complex formation between GST and ZFP106-f could be observed (Fig. 4b and Supplementary Fig. S6).
Figure 5.
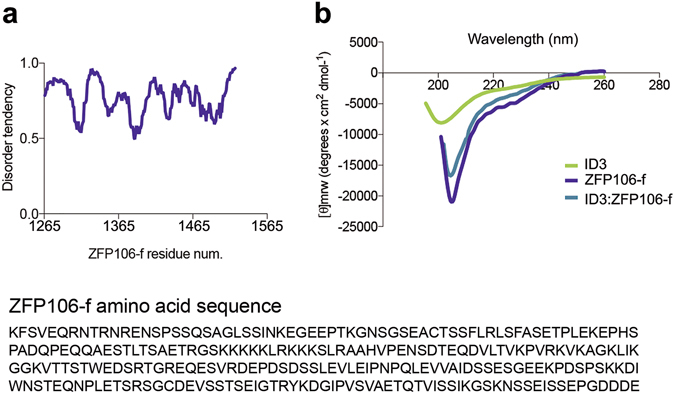
Fuzzy interaction between the two disordered regions. (a) Top: Disorder prediction of ZFP106-f using IUPred server indicates a high disorder tendency. Bottom: The ZFP106-f amino acid sequence used for the disorder prediction. (b) Circular dichroism spectra of ID3, ZFP106-f and 1:1 ratio of ID3:ZFP106-f. The spectrum of the complex resembles the individual spectra indicating no major changes in the structural content of the two partners upon association.
Dissecting the fuzzy ID3 - ZFP106-f interaction
To identify the residues of ID3 involved in the interaction with ZFP106-f, 2D 1H-15N NMR spectra of ID3 were recorded in the presence of increasing amounts of ZFP106-f. Only very minor and dispersed chemical shifts (CS) perturbations could be observed along the ID3 sequence (Supplementary Fig. S7a). Given the other lines of evidence for the interaction of the two proteins, this can be interpreted in terms of a fuzzy interaction with dispersed and heterogeneous interfaces between the two proteins. CS is not sensitive to this type of interaction, because measured CS values are linear, population-weighted averages of all protein species and conformations.
Therefore, to better characterize the interaction of the two proteins, we turned to paramagnetic resonance enhancement (PRE) measurements. By paramagnetically tagging one of the interaction partners, residues close to the interaction site of the second protein can be detected by decreases in signal intensity. Because PRE effects are averages weighted by the <r−6> -dependence of the paramagnetic effect, even a minor population with shorter distance between the nuclei and the paramagnetic probe (r) can have a larger - and thus observable - contribution to the signal measured43, 44. To outline the ID3 and ZFP106 regions involved in the interaction, PRE NMR experiments were carried out by labeling ZFP106-f at three different positions (M1, M2, M3) with an EDTA(Mn2+)-based paramagnetic tag (Fig. 6a) (for the diamagnetic references, wt ZFP106-f was used for the entire set of experiments).
Figure 6.
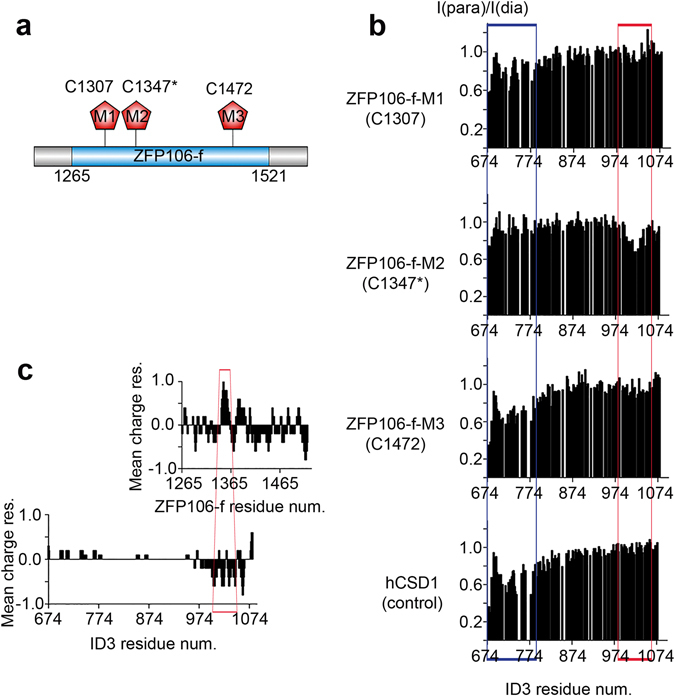
CTR of ID3 specifically interacts with the middle region of ZFP106-f. (a) Representation of the ZFP106 fragment identified in the Y2H assay (blue) and the three positions (M1, M2 and M3) where a paramagnetic label was attached to the protein. (b) I (para) /I(dia) plots of the 2D 1H-15N HSQC cross-peaks intensities of ID3 in the presence of the paramagnetically tagged single-Cys ZFP106-f mutants M1, M2 and M3 showing the site of interaction within ID3 (red box). A control experiment with an unrelated IDP, domain 1 of human Calpastatin (hCSD1), labeled with the same paramagnetic probe, is also shown to outline sticky region(s) of ID3 engaged in non-specific interaction (blue box). (c) Charge distribution plots of ID3 and ZFP106-f amino acid sequences calculated with the EMBOSS website (http://www.bioinformatics.nl/cgi-bin/emboss/charge). The regions involved in the interaction are highlighted (red): the middle region of ZFP106-f close to the M2 site, and the C-terminal 100 residues (CTR) of ID3.
The PREs observed with the three ZFP106-f constructs confirmed the interaction and highlighted two significantly affected ID3 regions, one in the N-terminal region (NTR, 674–780) and the other in the C-terminal region (CTR, 999–1050) (Fig. 6b). However, interaction with the NTR of ID3 is seen throughout all the PRE NMR experiments, irrespectively of the paramagnetic probe attachment site, and was also observed with an unrelated IDR (control experiment), human calpastatin domain 1 (hCSD1), tagged with the same paramagnetic probe (Fig. 6b and Supplementary Fig. S7e). The very same NTR region is the one mostly affected by chemical exchange as revealed by the high R 2 values (Supplementary Fig. S2b). Therefore, we concluded that the interaction with NTR is nonspecific. Further corroboration came with additional control experiments, in which the paramagnetic label free in solution (upon reduction of the disulfide bond between ZFP106-M2-cys and the label) also gave rise to same type of nonspecific signal in NTR of ID3, whereas it did not affect the signal at the CTR, confirming that the observed effects at the CTR of ID3 are due to the specific interaction of the two proteins (Supplementary Fig. S7d).
Moreover, the interacting region(s) of ZFP106-f can be delineated from the different effects the presence of the three ZFP106-f constructs has on ID3 signal intensities in the 2D 1H-15N NMR spectra (Fig. 6b). A strong signal decrease was observed with the ZFP106-f-M2, and much weaker effect with ZFP106-f-M1. The paramagnetic tag sites M1 and M2 are in relative close proximity to each other (Fig. 6a). In contrast, ZFP106-f-M3 caused no change in signal intensity in the CTR of ID3, suggesting an interaction site between residues 1300–1400 of ZFP106 (Fig. 6).
Interestingly, the region following the M2 label in ZFP106-f is enriched in lysines and carries a positive net charge, whereas the CTR of ID3 is highly negatively charged (Fig. 6c), indicating an electrostatically driven interaction. This was confirmed by NMR experiments performed in the presence of salt, which showed that at 150 mM NaCl the PRE effect was significantly reduced (Supplementary Fig. S7b and S7c). Specificity of the interaction could be concluded, though, from i) a still detectable binding at physiological salt concentrations, and ii) cancellation of the PRE effect on the CTR (but not by NTR) upon the application of DTT, which removes the paramagnetic label from the protein (Supplementary Fig. S7d).
In all, these PRE experiments provided evidence for a transient electrostatic interaction between the two proteins mediated by the negatively charged CTR of ID3 and a positively charged lysine-rich patch in the middle of ZFP106-f.
Discussion
CBP is a multi-domain transcription coactivator that mediates and integrates the action of hundreds of transcription factors and coactivators, which interact with its globular domains and target its activity on the chromatin and/or other proteins2, 6. The function of its long IDRs has been mostly described as that of passive linkers. Here, we focused our attention on possible additional roles of the 407 residues long ID3 located between the KIX domain and bromodomain of CBP.
SAXS, circular dichroism and NMR spectroscopy showed for ID3 a random coil state. By integrating 1H-detected and 13C-detected high-resolution multi-dimensional NMR experiments18, 20–22 we assigned about 95% of backbone chemical shifts, providing atomic resolution information on its structural and dynamic features. An ensemble of possible conformations calculated with experimentally derived ncSP scores and SAXS data supported the presence of several potential binding sites with mild local helical or β propensities, within a largely disordered module. The extended and flexible conformations of ID3 are expected to favor functions/interactions that take advantage of its dynamic properties. In accord, the specific binding role(s) of ID3 is supported by our Y2H studies in two cell-lines that uncovered dozens of potential ID3 binding partners. Among those, the transcriptional regulator ZFP106 was identified as a new acetylation substrate of CBP in vitro, which is in line with prior MS-based proteomic studies, which showed acetylation of ZFP106 on some of the lysine residues identified here (cf. PhosphoSitePlus45). The interaction of ZFP106-f with ID3 was confirmed by cross-linking and the observed competition between ID3 and full-length CBP in the acetylation reaction. Specificity of the interaction is underscored by the highly nonspecific acetylation of ZFP106-f by the core domain of CBP, which also strongly acetylates the lysine-rich patch located in the middle of ZFP106-f. PRE experiments, at low and high salt concentration, allowed us to determine the ID3 – ZFP106-f interacting regions, which include an extended negative patch of ID3 (roughly, residues S980-P1080) and a mostly positive patch of ZFP106-f (roughly, residues P1340-K1395). These experiments together with the HAT competition assays, in which the C-terminal charged region of ID3 has been deleted (Supplementary Fig. S5) confirmed the electrostatic nature of the interaction. The ZFP106-f lysine-rich patch involved in the interaction with ID3 is not acetylated by the full-length CBP, suggesting that ID3-ZFP106-f interaction may be involved in targeting specific lysine acetylation of ZFP106-f by “hiding/exposing” some ZFP106-f regions from/to the CBP-HAT domain. Specificity of this targeting relation is further corroborated by the similar behavior of H4, the archetypic substrate of CBP/p300. With H4, the core domain cannot distinguish between specific (N-terminal histone tails; K6 – K21) and nonspecific (K32 – K92) acetylation sites, whereas full-length CBP shows a strong preference for the former.
An intriguing aspect of our investigation is the transient character of this targeting interaction; such fuzzy and/or weak and transient complexes have been suggested to confer specificity without a structured state31, 46. For example, it has been shown that the weak, high-μM interaction of the cytoplasmic tails of T–cell receptors in T–cell signaling occurs via their disordered state, as suggested by circular dichroism and NMR47. The physiological regulatory relevance of an even weaker (“ultraweak”) interaction has been demonstrated in focal adhesion dynamics. Here, the interaction of the SH3 domain of Nck2 and the LIM4 domain of PINCH-1 has a Kd of 3 mM, still it is indispensable in integrin signaling, as suggested by genetic evidence48. The importance of transient, lowly-populated states that are still invisible to most structural biology techniques has also been demonstrated in encounter complexes49, RNA-protein recognition50 and the catalytic cycle of dihydrofolate reductase51. However, we cannot exclude the possibility that other domains of CBP or ZFP might be also involved in the interaction and influence affinity. This is also corroborated by the ID3 competition experiments, in which ID3 in a large molar excess (10x, 100x) is required to compete with binding and targeting of ID3 region within full-length CBP.
It is worth mentioning that the identified interaction regions are also rather long and may contain specific interaction motifs or charge distribution required for the interaction. Especially, ID3 sequence presents a characteristic and regular distribution of charges that has been conserved during evolution (cf. Fig. 6c and Supplementary Fig. S4). Similar binding features may also be present in long IDRs of other multi-complex proteins, such as bacterial RNAse E52, BRCA153, or UPF254. Moreover, the observed acetylation of ZFP106-f fits perfectly with CBP/p300 function in general: CBP/p300 integrate the action of hundreds of transcription factors and coactivators, the function of which is often regulated by acetylation2, 3. In this context, the highly significant enrichment of ID3 binding partners for transcription-regulatory and DNA-binding functions is notable4. Moreover, “in vivo” acetylation of ZFP106 was previously described by Li Y. and colleagues in 2006. High-throughput MS analysis of acetylated proteins present in colorectal carcinoma cell lines (SNU-C2B) identified four acetylation sites in ZFP106 (K1350-ac, K13510-ac, K1352-ac, K1358-ac) (Record ID: 11618 in PhosphoSitePlus45) within a region that is comprised in our construct (AA1265–1521). Interestingly, in our in vitro experiments those sites seem only to be acetylated by the highly active and nonspecific core domain but not by the full-length CBP.
In a cellular context, ZFP106 and CBP share roles in developmental pathways and diseases such as cancer. They also share a number of interaction partners, such as the tumor suppressors APC (Adenomatous polyposis coli protein) and Smad2 (Mothers against decapentaplegic homolog 2), which suggests that ZFP106 maybe involved in pathways such as TGFβ or Wnt signaling. In all, it seems reasonable that ZFP106 and CBP are part of a larger transcriptional complex that regulates expression of target genes involved in specific cellular processes, such as muscle cell differentiation or maintenance of the neuromuscular signaling12, 38, 55. Recent studies have shown that the nuclear ZFP106 protein that localized in nuclear speckles is required for normal assembly of the mammalian spliceosome, a process in which CBP/p300 also seems to participate12, 14, 56, 57.
In summary, our results suggest a possible additional role of an IDR in CBP, which may not only lead to a better description of the function of CBP/p300 and ZFP106, but also provides a precedence for addressing the function of long “linker” regions in large multi-domain signaling proteins.
Materials and Methods
Sequence analysis
A set of homologous ID3 amino acid sequences of various vertebrate species was assembled to study disorder propensities, sequence conservation, and the presence of potential binding sites. Cf. SI Materials and methods.
Scrambling the sequence of human ID3
For control purposes, a scrambled-ID3 sequence sharing 21% identity with wt ID3 was generated. (Supplementary Fig. S8). SI Materials and methods.
Cloning, mutagenesis and recombinant proteins
Full-length CBP (UniProt id Q92793), core domain of CBP (residues 1095–1849); ID3 of CBP, (residues 674–1080); ID3-F1 of CBP (residues 674–848); and Cys-to-Ser mutant ID3(C845S) obtained by site-directed mutagenesis of Cys845 of ID3, were cloned from the full-length construct. ZFP106-f (residues 1265–1521 of full-length ZFP106 (UniProt Q9H2Y7)). Scrambled-ID3 (scrID3), ID3delCTR and the three single-cysteine mutants of the ZFP106-f were cloned by GenScript (M1: C1472S (paramagnetic label on C1307), M2: insertion of Cys between Thr1346 – Arg1347 (paramagnetic label on insC1347*), M3: C1307S (paramagnetic label on C1472)). For further details, cf. SI Materials and methods.
Yeast two-hybrid screening
Y2H analysis was carried out by Hybrigenics Services S.A and described in SI Materials and methods. Identified partners were analyzed with IUPred (disorder level) and classified using GO annotation (biological process).
Recombinant protein expression, purification and labeling
Full-length (FL) CBP and its core domain (CD) were expressed in Sf9 cells, while the rest of the proteins were expressed in E. coli. Purification was achieved by a two- or three-step purification protocol including nickel (and Flag for the CBP constructs) affinity followed by size exclusion chromatography.
During ID3 purification a heat-treatment step of the lysate was done to increase the purity of the protein sample17, which was shown not to alter the secondary structure content of the protein (cf. Supplementary Fig. S1).
For the paramagnetic labeling, purified single-cysteine ZFP106-f mutants (M1, M2 and M3) and the hCSD1 (for control experiment) were mixed with the metal-containing probe (thiol-reactive label pEDTA). The reaction was carried out overnight at RT and labeled proteins were re-purified. For further details, cf. SI Materials and methods.
Determination of the paramagnetic labeling by intact mass spectroscopy analysis
Intact protein masses were measured by direct infusion in a microelectrospray ionization ion trap mass spectrometer (LTQ XL). The mass spectra were deconvoluted using the software ProMass Deconvolution. For further details, cf. SI Materials and methods.
Circular dichroism spectroscopy
Far-UV circular dichroism spectra of ID3, ZFP106-f and the complex (1:1 molar ratio) were recorded on a J-715 spectropolarimeter in reducing buffer of physiological salt concentration. For further details, cf. SI Materials and methods.
Small-angle X-ray scattering experiments
ID3 was injected into a size-exclusion column equilibrated with buffer of physiological salt concentration and SAXS data were collected continuously, selected frames corresponding to the main protein elution peak were averaged using FOXTROT58 (buffer scattering subtracted), following data processing/analysis using ATSAS suite59 and calculation of the radius of gyration by PRIMUS QT60. For further details, cf. SI Materials and methods.
Nuclear magnetic resonance experiments (NMR) and data processing
All the experiments and acquisition parameters regarding ID3 sequence-specific assignment, backbone dynamics and, PRE experiments are reported in SI Materials and methods and Tables S1–S5.
Sequence-specific assignment
All the multidimensional NMR experiments for sequence-specific assignment were performed on uniformly [13C,15N] labeled ID3 and ID3-F1 in reducing buffer of physiological salt concentration at pH 6.5, with 10% D2O added for the lock. The CS assignment of ID3 has been deposited in the BioMagResBank (BMRB, http://www.bmrb.wisc.edu) as entry 26841. For further details, cf. SI Materials and methods.
Backbone dynamics
Backbone dynamics (at 297 K) of uniformly 15N-labeled ID3 were studied by using 15N relaxation data obtained from 2D 1H-15N HSQC-edited experiments61, carried out using a buffer of physiological salt concentration at pH 6.5, with 10% D2O added for the lock. For further details, cf. SI Materials and methods.
Paramagnetic relaxation enhancement (PRE) experiments
For the PRE measurements, 2D 1H-15N HSQC spectra of the paramagnetic and diamagnetic samples were acquired62 on [13C,15N] ID3(C845S) and 1 molar equivalent (eq) of single-Cys ZFP106-f mutant EDTA(Mn2+)-tagged (paramagnetic samples). The diamagnetic references were done with wt ZFP106-f with DTT added (wt proteins are equivalent to their EDTA(Zn2+)-tagged version for PRE referencing62). The unrelated protein control experiments were done with 1 eq of wt hCSD1-EDTA(Mn2+) (paramagnetic sample) or wt hCSD1 (diamagnetic sample, DTT added). The reduced-label control experiments, the [13C,15N] ID3(C845S):ZFP106-f-M2 sample was measured in absence and presence of reducing agents (DTT). All NMR samples were measured at pH 6.5 and 10% D2O for the lock, NaCl was added to a final concentration of 150 mM. For further details, cf. SI Materials and methods.
NMR data processing
NMR data sets were processed using Bruker TopSpin 1.3 software or nmrPipe 63. NUS data sets were converted with nmrPipe and then processed using either the Multidimensional Fourier Transform (MFT) or the Sparse MFT (SMFT) algorithm. Both programs are available at http://nmr.cent3.uw.edu.pl. Data analysis was done with Sparky 64 or CcpNmr Analysis 65.
Secondary structure propensity calculation
Secondary structure propensity analysis was calculated from C′, Cα, Cβ and N chemical shifts (95% assigned) by using the neighbor-corrected structural propensity calculator (ncSP) tool66. For further details, cf. SI Materials and methods.
Ensemble modelling
Ensemble models of ID3 were based on the combination of SAXS data and ncSP derived from experimental chemical shift values. Flexible-Meccano26 and SCCOMP67 were used to generate a pool of 9876 semi-random conformations (with side-chains modeled). Then, CRYSOL27 and the GAJOE algorithm were employed27 to select subsets of the random pool that that agreed with the experimental SAXS data. For further details, cf. SI Materials and methods.
Acetylation Assay
In vitro ZFP106-f or H4 acetylation assays were performed in reducing buffer of physiological salt concentration at 20 or 30 °C using FL or CD of CBP. For the HAT competition assay, x10, 100 and 150 molar excess (relative to full-length CBP) of ID3, scrID3 and ID3delCTR were added to the acetylation reactions. The reactions were completed to saturation and the samples were analyzed by Western blot by anti-acetylated-lysine (Bioke) and anti-His (Sigma-Aldrich) antibodies. For the identification of acetylation sites, the proteins in solution were desalted and digested with either pepsin or endoglu-C, and analyzed by LC-MS/MS in a microelectrospray ionization ion trap mass spectrometer (LTQ XL, or Orbitrap XL) as described68, 69. For further details, cf. SI Materials and methods.
Cross-linking Assay
Proteins were mixed in approximate 1:1 ratio (concentrations adjusted by SDS-PAGE). Cross-linking reactions were carried out for 15 min at RT in PBS with 1 mM DTT (S: sample, ID3:ZFP106-f; C: control, GST:ZFP106-f). Crosslinker BS3 was added to a final concentration of 0.25 mM. For further details, cf. SI Materials and methods.
Electronic supplementary material
Acknowledgements
We thank Kris Pauwels for providing us with purified protein sample (hCSD1) and both Kris Pauwels and Cesyen Cedeño for the support and encouraging discussions all throughout this research project. Also, we would like to thank Didier Vertommen from MASSPROT facility (at the UCL) for his advice and contribution to this work. Finally, we thank also the staff at the Synchrotron SOLEIL SWING beamline for their assistance with the SAXS experiments. This work was supported by the Odysseus grant G.0029.12 from Research Foundation Flanders (FWO). The European Commission Projects Marie Curie ITN IDPbyNMR (Contract No. 264257), BioNMR (Contract No. 261863) and the use of resources of INSTRUCT, a Landmark ESFRI project, are also gratefully acknowledged as well as MIUR PRIN 2012SK7ASN.
Author Contributions
S.C-M.: Constructs design and cloning, sample preparation, acetylation assays, biophysical experiments and writing the article; A.P.: NMR experiments and data analysis, writing the article; S.K.: Constructs design and cloning, project design and supervision, writing article; M.V.: ID3 sequence analysis, computation; A.B.: FL and CD of CBP sample preparation and acetylation assays; P.L.: SAXS measurements and data processing, ensemble calculation; A.V.: PRE experiments design and supervision, writing article; K.G.: MS analysis; R.P. and I.F.: NMR project design and supervision, writing the article; P.T.: Overall project design and supervision, writing the article.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Sara Contreras-Martos & Alessandro Piai contributed equally to this work
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04611-x
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bedford DC, Brindle PK. Is histone acetylation the most important physiological function for CBP and p300? Aging (Albany NY) 2012;4:247–255. doi: 10.18632/aging.100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 3.Giles RH, Peters DJ, Breuning MH. Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 1998;14:178–183. doi: 10.1016/S0168-9525(98)01438-3. [DOI] [PubMed] [Google Scholar]
- 4.Soutoglou E, Katrakili N, Talianidis I. Acetylation regulates transcription factor activity at multiple levels. Mol Cell. 2000;5:745–751. doi: 10.1016/S1097-2765(00)80253-1. [DOI] [PubMed] [Google Scholar]
- 5.Tompa P, Schad E, Tantos A, Kalmar L. Intrinsically disordered proteins: emerging interaction specialists. Curr Opin Struct Biol. 2015;35:49–59. doi: 10.1016/j.sbi.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Dyson HJ, Wright PE. Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J Biol Chem. 2016;291:6714–6722. doi: 10.1074/jbc.R115.692020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan LW, Gambee JE. Phosphorylation of p300 at serine 89 by protein kinase C. J Biol Chem. 2000;275:40946–40951. doi: 10.1074/jbc.M007832200. [DOI] [PubMed] [Google Scholar]
- 8.Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev. 2015;115:2419–2452. doi: 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piai A, et al. Just a Flexible Linker? The Structural and Dynamic Properties of CBP-ID4 Revealed by NMR Spectroscopy. Biophys J. 2016;110:372–381. doi: 10.1016/j.bpj.2015.11.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tompa P. On the supertertiary structure of proteins. Nat Chem Biol. 2012;8:597–600. doi: 10.1038/nchembio.1009. [DOI] [PubMed] [Google Scholar]
- 11.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 12.Anderson DM, et al. Severe muscle wasting and denervation in mice lacking the RNA-binding protein ZFP106. Proc Natl Acad Sci USA. 2016;113:E4494–4503. doi: 10.1073/pnas.1608423113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castello A, et al. Insights into RNA Biology from an Atlas of Mammalian mRNA-Binding Proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 14.Joyce PI, et al. Deficiency of the zinc finger protein ZFP106 causes motor and sensory neurodegeneration. Hum Mol Genet. 2016;25:291–307. doi: 10.1093/hmg/ddv471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Celona, B. et al. Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife6, e19032 (2017). [DOI] [PMC free article] [PubMed]
- 16.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/S0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 17.Livernois AM, Hnatchuk DJ, Findlater EE, Graether SP. Obtaining highly purified intrinsically disordered protein by boiling lysis and single step ion exchange. Anal Biochem. 2009;392:70–76. doi: 10.1016/j.ab.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 18.Brutscher B, et al. NMR Methods for the Study of Instrinsically Disordered Proteins Structure, Dynamics, and Interactions: General Overview and Practical Guidelines. Adv Exp Med Biol. 2015;870:49–122. doi: 10.1007/978-3-319-20164-1_3. [DOI] [PubMed] [Google Scholar]
- 19.Gil S, et al. NMR spectroscopic studies of intrinsically disordered proteins at near-physiological conditions. Angew Chem Int Ed Engl. 2013;52:11808–11812. doi: 10.1002/anie.201304272. [DOI] [PubMed] [Google Scholar]
- 20.Bermel W, et al. High-dimensionality 13C direct-detected NMR experiments for the automatic assignment of intrinsically disordered proteins. J Biomol Nmr. 2013;57:353–361. doi: 10.1007/s10858-013-9793-z. [DOI] [PubMed] [Google Scholar]
- 21.Felli IC, Pierattelli R. Novel methods based on C-13 detection to study intrinsically disordered proteins. J Magn Reson. 2014;241:115–125. doi: 10.1016/j.jmr.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 22.Piai A, et al. “CON-CON” assignment strategy for highly flexible intrinsically disordered proteins. J Biomol Nmr. 2014;60:209–218. doi: 10.1007/s10858-014-9867-6. [DOI] [PubMed] [Google Scholar]
- 23.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamiola K, Acar B, Mulder FAA. Sequence-Specific Random Coil Chemical Shifts of Intrinsically Disordered Proteins. Journal of the American Chemical Society. 2010;132:18000–18003. doi: 10.1021/ja105656t. [DOI] [PubMed] [Google Scholar]
- 25.Fuxreiter M, Simon I, Friedrich P, Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol. 2004;338:1015–1026. doi: 10.1016/j.jmb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 26.Ozenne V, et al. Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics. 2012;28:1463–1470. doi: 10.1093/bioinformatics/bts172. [DOI] [PubMed] [Google Scholar]
- 27.Petoukhov MV, et al. New developments in the program package for small-angle scattering data analysis. J Appl Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dosztanyi Z, Meszaros B, Simon I. Bioinformatics. 2009. ANCHOR: web server for predicting protein binding regions in disordered proteins; pp. 2745–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varadi M, Guharoy M, Zsolyomi F, Tompa P. DisCons: a novel tool to quantify and classify evolutionary conservation of intrinsic protein disorder. BMC Bioinformatics. 2015;16:153. doi: 10.1186/s12859-015-0592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cilia E, Pancsa R, Tompa P, Lenaerts T, Vranken WF. From protein sequence to dynamics and disorder with DynaMine. Nat Commun. 2013;4:2741. doi: 10.1038/ncomms3741. [DOI] [PubMed] [Google Scholar]
- 31.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Aikawa Y, et al. Roles of HIPK1 and HIPK2 in AML1- and p300-dependent transcription, hematopoiesis and blood vessel formation. EMBO J. 2006;25:3955–3965. doi: 10.1038/sj.emboj.7601273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin X, Warner DR, Roberts EA, Pisano MM, Greene RM. Novel interaction between nuclear coactivator CBP and the protein inhibitor of activated Stat1 (PIAS1) J Interferon Cytokine Res. 2005;25:321–327. doi: 10.1089/jir.2005.25.321. [DOI] [PubMed] [Google Scholar]
- 34.Long J, Wang G, Matsuura I, He D, Liu F. Activation of Smad transcriptional activity by protein inhibitor of activated STAT3 (PIAS3) Proc Natl Acad Sci USA. 2004;101:99–104. doi: 10.1073/pnas.0307598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leger H, et al. A TDG/CBP/RARalpha ternary complex mediates the retinoic acid-dependent expression of DNA methylation-sensitive genes. Genomics Proteomics Bioinformatics. 2014;12:8–18. doi: 10.1016/j.gpb.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koscielny G, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014;42:D802–809. doi: 10.1093/nar/gkt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Mikecz A, Zhang SS, Montminy M, Tan EM, Hemmerich P. CREB-binding protein (CBP)/p300 and RNA polymerase II colocalize in transcriptionally active domains in the nucleus. Journal of Cell Biology. 2000;150:265–273. doi: 10.1083/jcb.150.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grasberger H, Ye H, Mashima H, Bell GI. Dual promoter structure of ZFP106: regulation by myogenin and nuclear respiratory factor-1. Gene. 2005;344:143–159. doi: 10.1016/j.gene.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 39.Demarest SJ, et al. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature. 2002;415:549–553. doi: 10.1038/415549a. [DOI] [PubMed] [Google Scholar]
- 40.Phizicky EM, Fields S. Protein-protein interactions: methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions: structural, functional, and network properties. Structure. 2010;18:1233–1243. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 42.Hajdu J, Solti M, Friedrich P. Cross-linking and coupling of rabbit muscle aldolase and glyceraldehyde-3-phosphate dehydrogenase by glutaraldehyde. Acta Biochim Biophys Acad Sci Hung. 1975;10:7–16. [PubMed] [Google Scholar]
- 43.Clore GM, Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anthis NJ, Clore GM. Visualizing transient dark states by NMR spectroscopy. Q Rev Biophys. 2015;48:35–116. doi: 10.1017/S0033583514000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hornbeck PV, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma R, Raduly Z, Miskei M, Fuxreiter M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015;589:2533–2542. doi: 10.1016/j.febslet.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 47.Sigalov A, Aivazian D, Stern L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry. 2004;43:2049–2061. doi: 10.1021/bi035900h. [DOI] [PubMed] [Google Scholar]
- 48.Vaynberg J, et al. Structure of an ultraweak protein-protein complex and its crucial role in regulation of cell morphology and motility. Mol Cell. 2005;17:513–523. doi: 10.1016/j.molcel.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 49.Volkov AN. Structure and Function of Transient Encounters of Redox Proteins. Acc Chem Res. 2015;48:3036–3043. doi: 10.1021/acs.accounts.5b00343. [DOI] [PubMed] [Google Scholar]
- 50.Borkar AN, et al. Structure of a low-population binding intermediate in protein-RNA recognition. Proc Natl Acad Sci USA. 2016;113:7171–7176. doi: 10.1073/pnas.1521349113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 52.Ait-Bara S, Carpousis AJ, Quentin Y. RNase E in the gamma-Proteobacteria: conservation of intrinsically disordered noncatalytic region and molecular evolution of microdomains. Mol Genet Genomics. 2015;290:847–862. doi: 10.1007/s00438-014-0959-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mark WY, et al. Characterization of segments from the central region of BRCA1: an intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J Mol Biol. 2005;345:275–287. doi: 10.1016/j.jmb.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 54.Clerici M, et al. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009;28:2293–2306. doi: 10.1038/emboj.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polesskaya A, et al. CBP/p300 and muscle differentiation: no HAT, no muscle. Embo Journal. 2001;20:6816–6825. doi: 10.1093/emboj/20.23.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuhn AN, Van Santen MA, Schwienhorst A, Urlaub H, Luhrmann R. Stalling of spliceosome assembly at distinct stages by small-molecule inhibitors of protein acetylation and deacetylation. Rna. 2009;15:153–175. doi: 10.1261/rna.1332609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Mikecz A, Zhang S, Montminy M, Tan EM, Hemmerich P. CREB-binding protein (CBP)/p300 and RNA polymerase II colocalize in transcriptionally active domains in the nucleus. J Cell Biol. 2000;150:265–273. doi: 10.1083/jcb.150.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.David G, Perez J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. Journal of Applied Crystallography. 2009;42:892–900. doi: 10.1107/S0021889809029288. [DOI] [Google Scholar]
- 59.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. ATSAS 2.1, a program package for small-angle scattering data analysis. Journal of Applied Crystallography. 2006;39:277–286. doi: 10.1107/S0021889806004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003;36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 61.Barbato G, Ikura M, Kay LE, Pastor RW, Bax A. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry. 1992;31:5269–5278. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- 62.Van de Water K, van Nuland NAJ, Volkov AN. Transient protein encounters characterized by paramagnetic NMR. Chem Sci. 2014;5:4227–4236. doi: 10.1039/C4SC01232A. [DOI] [Google Scholar]
- 63.Delaglio F, et al. Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. J Biomol Nmr. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 64.Goddard, T. D. Sparky - NMR Assignment and Integration Software.
- 65.Vranken WF, et al. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 66.Tamiola K, Mulder FA. Using NMR chemical shifts to calculate the propensity for structural order and disorder in proteins. Biochem Soc Trans. 2012;40:1014–1020. doi: 10.1042/BST20120171. [DOI] [PubMed] [Google Scholar]
- 67.Eyal E, Najmanovich R, McConkey BJ, Edelman M, Sobolev V. Importance of solvent accessibility and contact surfaces in modeling side-chain conformations in proteins. J Comput Chem. 2004;25:712–724. doi: 10.1002/jcc.10420. [DOI] [PubMed] [Google Scholar]
- 68.Ruys, S. P. D. et al. Identification of autophosphorylation sites in eukaryotic elongation factor-2 kinase (vol 442, pg 681, 2012). Biochem J443, 328–328 (2012). [DOI] [PMC free article] [PubMed]
- 69.Helsens K, et al. ms_lims, a simple yet powerful open source laboratory information management system for MS-driven proteomics. Proteomics. 2010;10:1261–1264. doi: 10.1002/pmic.200900409. [DOI] [PubMed] [Google Scholar]
- 70.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.