

Abstract
When challenged by hemodynamic stress, aging hearts respond differently to young hearts. Preclinical models of heart disease should take into account the effects of age. However, in the transverse aortic constriction (TAC) model of pressure-overload cardiomyopathy, the larger aorta of aging mice has not previously been taken into account. First, we studied the aortic size in mice, and found that the aortic cross-sectional area (CSA) is 28% larger in aging mice than in young adult mice (P=0.001). We then performed TAC to make the same proportional reduction in CSA in young and aging mice. This produced the same pressure gradient across the constriction and the same rise in B-type natriuretic peptide expression. Young mice showed acute deterioration in systolic function assessed by pressure-volume loops, progressive LV remodeling on echocardiography, and a 50% mortality at 12 weeks post-TAC. In contrast, aging mice showed no acute deterioration in systolic function, much less ventricular remodeling and were protected from death. Aging mice also showed significantly increased levels of matrix metalloproteinase-3 (MMP-3; 3.2 fold increase, P<0.001) and MMP-12 (1.5-fold increase, P<0.001), which were not seen in young mice. Expression of tissue inhibitor of MMP-1 (TIMP-1) increased 8.6-fold in aging hearts vs 4.3-fold in young hearts (P<0.01). In conclusion, following size-appropriate TAC, aging mice exhibit less LV remodeling and lower mortality than young adult mice. This is associated with induction of protective ECM changes.
Keywords: Aging, heart failure, pressure overload, cardiac fibrosis
Introduction
Cardiovascular disease remains the leading cause of death globally [1]. Heart disease increases in prevalence with increasing age [2], underscoring the importance of studying preclinical models of aging and heart disease. The changes seen in the left ventricle (LV) in response to increased hemodynamic load, including LV hypertrophy, fibrosis and dysfunction are also seen in aging hearts [3-6] in the absence of hemodynamic stress. Prior studies examining the effects of pressure overload in aging hearts [7,8] have shown that aging hearts are not able to cope with the increased hemodynamic stress as well as their young counterparts, and that aging mice have worse outcomes. However, it is known that greater degrees of proportional aortic constriction are associated with worse ventricular function [9]. The aortic size is larger in aging mice, and therefore ligating around the same size needle in smaller young aortas and larger aging aortas will produce a proportionally greater degree of constriction in the aging aorta. This may bias against the aging mice. Therefore, we propose that transverse aortic constriction (TAC) studies involving aging mice should take aortic size into account and induce similar proportional constriction of the aorta in the young and aging groups.
We therefore studied aging hearts following size-appropriate surgical TAC in order to determine the effects of aging in response to pressure overload. We show that aging hearts induce a cardioprotective MMP profile, are protected from LV remodeling and this results in a significant reduction in mortality in the aging mice.
Materials and methods
Animals were handled according to the NIH guidelines and the guidelines of the Institutional Animal Care and Use Committee of the University of California, San Francisco, who specifically approved this study. Male C57BL/6J mice were used for all studies. Young mice were 3 months, aging were 18 months of age. The baseline differences between these ages have previously been characterized by our group [6].
Transverse aortic constriction surgery
Mice were anesthetized with inhaled 2% isoflurane, intubated and ventilated. Pedal withdrawal reflex was assessed every 15 minutes to ensure the anesthesia depth was adequate. Local infiltration of 0.25% bupivicaine at the surgical site was administered once prior to the surgical procedure. A subcutaneous or intraperitoneal injection of 0.05-0.1 mg/kg buprenorphine was administered once at the time of the procedure. The chest was opened and the transverse aorta ligated between the carotid arteries with a 6/0 polypropylene suture over a 27G needle for young mice and a 26G needle for aging mice. The needles were then removed, the chest was closed and the animal allowed to recover. Additional buprenorphine injections were given to the mice post-operatively, as required.
Echocardiography
To define the difference in size of the aorta between young and aging mice, a cohort of mice underwent echocardiography to define the aortic size. Under inhaled 1.5% isoflurane anesthesia, the ascending aortic diameter was measured in young (n=5) and aging (n=10) mice. Aortic cross sectional area (CSA) was determined using the formula
CSA = π × (d/2)2
where d = diameter of the ascending aorta.
Echocardiography was performed prior to and after TAC surgery under anesthesia with 1.5% inhaled isoflurane. Left ventricular (LV) volumes (end-systolic (ESV) and end-diastolic (EDV) volumes) were measured as previously described [6,10,11]. Analyses of the echocardiography images were performed by an investigator who was blinded to the identity of the treatment groups. LV ejection fraction was calculated as
LVEF = [(LVEDV-LVESV)/LVEDV] × 100
Tissue preparation and analysis
To assess the early and late effects of TAC, animals were sacrificed at either 3 days or 13 weeks post-TAC under general anesthesia with 1.5% isoflurane. Once under general anesthesia, hearts (n=6 per age group) were arrested in diastole with a single intravenous injection of concentrated KCl and the hearts removed and perfused with 10% neutral buffered formalin via the ascending aorta and then paraffin embedded. Sections from the mid-ventricular level were examined for fibrosis using Masson’s trichrome staining [6,12,13]. Quantification of fibrous tissue was performed on photomicrographs and quantified by using ImagePro (Media Cybernetics Inc, Bethesda, MD) software.
Protein analysis
At both the early and late time-points, another group of hearts were excised from young (n=3) and aging (n=3) mice, and perfused with cold PBS. The left ventricles were flash frozen in liquid nitrogen and stored in -80°C. For each sample, the LV was homogenized in RIPA buffer with PMSF, complete protease inhibitor cocktail and phosphatase inhibitor cocktail added. Both the RIPA buffer and PMSF were from Santa Cruz Biotechnology and protease and phosphatase inhibitor cocktail tablets from Roche. The lysates were quantified by Pierce BCA protein assay. 15 μg of protein for each sample was separated by 14% Tris-Glycine gels (Invitrogen) and transferred to PDVF membranes (Biorad) overnight at 4°C at 90 mA. The membrane was blocked with 10% (w/v) non-fat dry milk in Tris-Buffered Saline Tween (50 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20) at room temperature for 1 h. Membranes were then incubated overnight at 4°C with primary antibodies then washed 3 times with TBST and incubated with anti-rabbit HRP conjugated antibody for 1 h at room temperature. The immunoblots were visualized using ECL plus detection system on Amersham Hyperfilm ECL chemiluminesence film (GE Healthcare).
RNA analysis
Total RNA was isolated from the left ventricles of young (n=3) and aging (n=3) unoperated mice, young (n=3) and aging (n=3) mice day 3 post-TAC, and young (n=3) and aging (n=3) mice week 13 post-TAC by TRIzol reagent (Invitrogen). Trace genomic DNA in total RNA was removed by DNase I and RNeasy Mini Kit (Qiagen). cDNA was generated from 1 ug of total RNA by using SuperScript III First-Strand Synthesis kit (Invitrogen). Microarrays were performed using Affymetrix 1.0 Mouse gene chip. To minimize the number of mice used, we used the RNA extracted from young (n=3) and aging (n=3) mice from our prior study as baseline. Importantly, although we used RNA from the same animals, we used different samples of this RNA and the microarray analysis was repeated for this experiment, to avoid a potential batch effect. The results were very consistent with the prior data [6] which strengthens our data. Data were normalized using robust multi-array average (RMA) method. Control and low performing probesets (those with intensity values below a threshold across all samples, the threshold was taken to be the global lowest 25th percentile of intensity values) were excluded from analysis. Also, based on Affymetrix’s annotation information, only those probesets which were part of the main design of the array and perfectly matching only one sequence were considered for analysis of differential expression. 20648 out of 35557 probesets remained after filtering.
Hemodynamic studies
To demonstrate the gradient across the TAC suture, we measured the LV pressure and the pressure in the descending aorta. This was performed in young and aging mice, at baseline and after TAC. Mice were anesthetized with inhaled 2% isoflurane, intubated and ventilated. The chest was opened and LV pressure measured by apical puncture with a fluid filled transducer. Once hemostasis was achieved, the abdominal cavity was opened in the same mouse and the descending aorta visualized. This was then cannnulated and the aortic pressure measured.
To assess the effect of TAC on LV function early, we performed echocardiography and invasive pressure-volume loop analysis on young (n=6) and aging (n=6) mice at baseline and young (n=6) and aging (n=6) on day 3 after TAC. Under isoflurane anesthesia, mice underwent carotid artery cutdown and cannulation. A 1.2 F transducer (Transonic Scisense, London, Ontario, Canada) was advanced to the LV. Pressure-volume loops were generated at baseline, during inferior vena cava occlusion and during saline injection. The data were analysed with Advantage software. After the catheter was withdrawn, the carotid artery was ligated.
Statistical analysis
Data are presented as mean ± SD unless otherwise stated. Continuous data were compared between young and aging using unpaired Student’s t-test and categorical variables using Chi-square test. Timecourse echocardiography data was analyzed within groups using ANOVA with Fisher’s post hoc test. Mortality following TAC was analyzed using Kaplan-Meier methodology after early surgical deaths were excluded. Other statistical methods are included in the figure legends. Microarrays were normalized for array-specific effects using Affymetrix’s “Robust Multi-Array” (RMA) normalization. For statistical analyses, we removed all array probesets where no experimental groups had an average log2 intensity greater than 3.0. This is a standard cutoff, below which expression is indistinguishable from background noise. Linear models were fitted for each gene using the Bioconductor “limma” package in R [14,15].
Results
Size appropriate aortic constriction results in comparable physiologic effects in young and aging mice
Aging mice had significantly (28%) larger ascending aortic cross-sectional areas (2.22±0.22 mm2 vs 1.73±0.13 mm2, P=0.001) than young mice. We therefore calculated that ligating the transverse aorta using a 27 gauge needle in the young and a 26 gauge needle in the aging mice would result in a 92.5% and 92.3% proportional reduction in aortic cross sectional area (CSA) respectively (Figure 1A). To assess the hemodynamic effects of this approach, the left ventricular and aortic pressures were measured in young and aging mice at baseline and following TAC. There was a similar pressure drop across the aortic banding in both young and aging mice (Figure 1B). Further, B-type natriuretic peptide was similarly upregulated in young and aging mice early following TAC (Figure 1C). These findings demonstrate that size appropriate TAC results in equivalent pressure gradients and induction of fetal gene expression.
Figure 1.
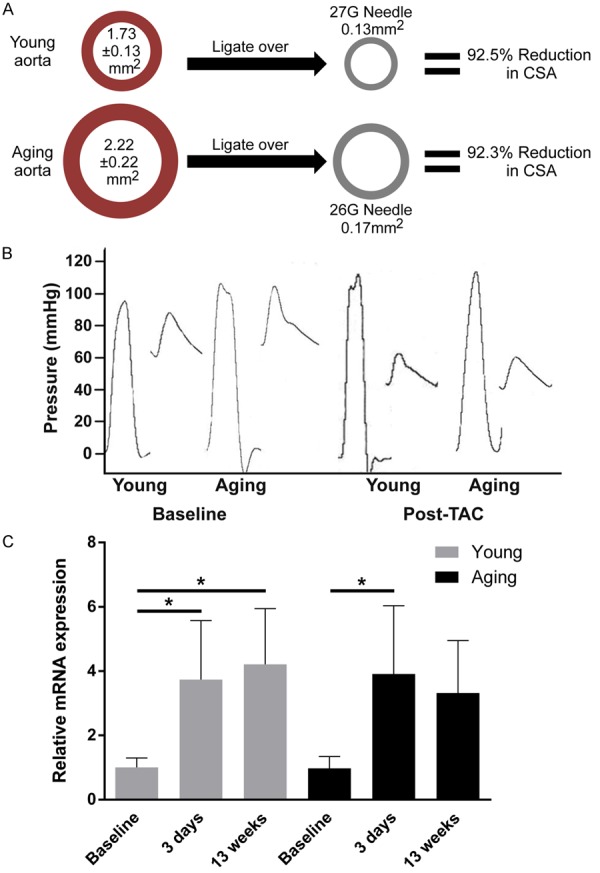
Size-appropriate aortic constriction results in similar relative reduction in aortic CSA and similar hemodynamic stress on the LV. A. Using a 26 gauge needle for aging mice and a 27 gauge for young mice results in similar reduction in CSA. B. Left ventricular and aortic pressure measurements show no difference in systolic pressure at baseline, and a similar drop in pressure is seen across the aortic constriction in young and aging mice. C. A 3.5 and 4-fold increase in BNP mRNA expression was detected in both young and aging mice respectively 3 days post-TAC. *P<0.01. Data were analyzed using ANOVA with Fisher’s post hoc test. BNP = brain natriuretic peptide (n=4 mice per group).
Increased left ventricular dilation and decreased survival in young mice following aortic constriction
Young and aging mice both undergo dilation of the left ventricle 12 weeks post-TAC with young mice demonstrating larger proportional increases in end-diastolic and end-systolic volumes compared to their baseline volumes than aging mice (P<0.01, Figure 2B). We also observed a significant reduction in left ventricular ejection fraction in young mice 12 weeks post-TAC (P<0.01, Figure 2C), which was not observed in aging mice. Taken together, these findings indicate that young mice experience a greater extent of LV dilation than aged mice following TAC despite young mice having significantly smaller LV cavities at baseline. Moreover, young mice demonstrated a 50% mortality rate following TAC, whereas no mortality was observed in the aging group (Chi square P=0.005, log rank P=0.006, Figure 2A).
Figure 2.
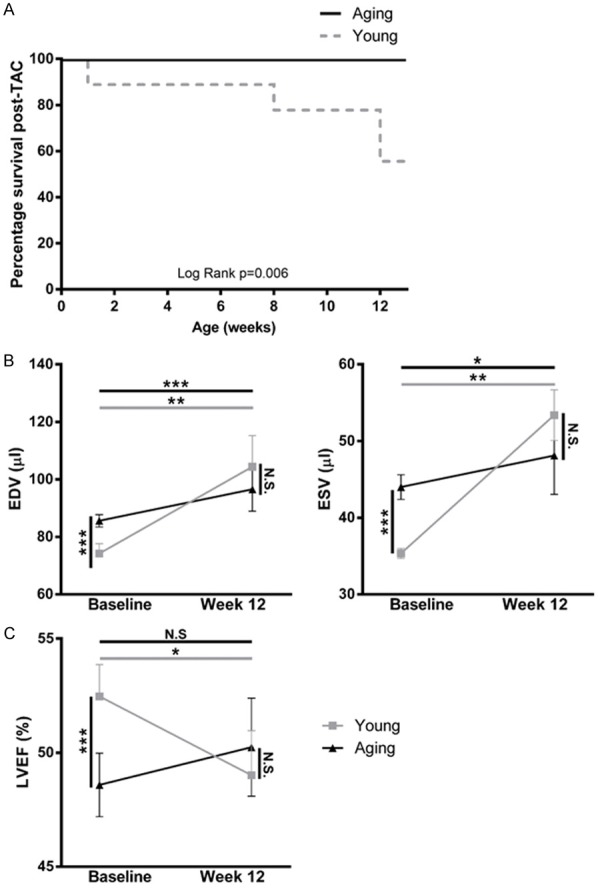
Aging mice are protected from LV remodeling and death following TAC. A. The Kaplan-Meier survival curve indicates that survival following TAC is age dependent. Mortality following TAC declined to 50% by 12 weeks after the procedure in young mice (n=14 young mice, n=18 aging mice). Survival curves were compared with log rank test. B. Echocardiography was used to determine the EDV, ESV and LVEF of young and aging mice at baseline and 12 weeks post-TAC. The EDV and ESV of young and aging mice both increased significantly following TAC (P<0.01). C. The LVEF significantly increased in aging mice by 12 weeks post-TAC, however decreased in young mice (P<0.01). Baseline and 12-week post TAC values were compared using paired t-tests, and values were compared across groups at each time-point using non-paired t-tests. LVEF = Left Ventricular Ejection Fraction; EDV = End-Diastolic Volume; ESV = End-Systolic Volume; *P<0.01 to Baseline.
Increased hypertrophy of the myocardium in young mice following aortic constriction
At baseline aging mice have significantly greater heart weight (117 mg vs 96 mg; P<0.01), LV mass (36 mg vs 33 mg; P<0.01), posterior wall thickness (0.67 vs 0.75 mm; P=0.03) and anterior wall thickness (0.73 vs 0.85 mm) compared to young mice (Figure 3A and 3B). Although both young and aging mice demonstrate myocardial hypertrophy 12 weeks post-TAC, the changes were consistently greater in young mice, which show a larger increase in anterior wall thickness (30% and 20% increase respectively, Figure 3B). In addition, young mice show a significant increase in posterior wall thickness (10% increase, P=0.02, Figure 3B), which was not observed in aging mice. In the left ventricle we see comparable, but modest 20% increase in LV mass at 12 weeks post-TAC in both young and aging mice (Figure 3A). The overall increased level of hypertrophy observed in young mice leads to indistinguishable heart weight, anterior and posterior wall thickness in young and aging mice 12 weeks post-TAC, despite higher baseline values for aging mice.
Figure 3.

Myocardial hypertrophy post-TAC. At baseline, aging mice exhibit greater heart weight, LV mass, anterior wall thickness and posterior wall thickness. Young and aging mice undergo significant hypertrophy 12 weeks post-TAC (A). Heart weight, anterior and posterior wall thickness of young and aging mice are no longer discernibly different between young and aging mice 12 weeks post-TAC (B). Baseline and 12-week post TAC values were compared using paired t-tests, and values were compared across groups at each time-point using non-paired t-tests. (C) Representative pressure volume loops from young and aging mice. *P<0.05, **P<0.01, ***P<0.001 (n=6 young mice, n=6 aging mice).
Impairment of systolic function is apparent early after TAC in young mice
Pressure-volume loops were obtained by cardiac catheterization at baseline and after TAC (Figure 3C; Table 1). At baseline, the aging mice were larger and the left ventricular end-systolic volume (ESV) was larger. However, when the ESV was indexed for body weight, there was no significant difference between young and aging mice. There was no difference in systolic or diastolic function parameters between young and aging mice. After TAC, arterial elastance (Ea) increased similarly in young and aging mice, demonstrating a similar hemodynamic increase in afterload on the left ventricles of each group. The left ventricular cavities became smaller in both age groups, and this change remained significant after indexing for body size. However, young mice appeared unable to augment their systolic function to compensate for the increased afterload. There was a significant drop in stroke work index and a reduction in the slope of the end-systolic pressure volume relationship (Ees) in the young mice, indicating reduced inotropy. This was associated with numeric reductions in dP/dTmax and LVEF, but these did not reach statistical significance. Aging mice, however, showed an increased Ees with a significant increase in developed pressure and a smaller reduction in stroke work index, demonstrating increased inotropy. This was associated with an unchanged dP/dTmax and a non-significant increase in LVEF. Taken together, these results suggest that systolic function is relatively maintained in the aging mice, but deteriorates in the young mice. There were no acute differences in parameters of diastolic function.
Table 1.
Invasive hemodynamic parameters
| Young Baseline (n=6) | Aging baseline (n=6) | Young day 3 post-TAC (n=6) | Aging day 3 post-TAC (n=7) | |
|---|---|---|---|---|
| SV (μL) | 12.3±5.0 | 14.4±4.87 | 6.27±2.85* | 8.8±5.2* |
| SVI (μL g-1) | 0.58±0.23 | 0.43±0.13 | 0.31±0.14* | 0.28±0.17 |
| ESV (μL) | 14.3±1.8 | 18.7±4.0§ | 10.8±3.8 | 8.2±3.2* |
| ESVI (μL g-1) | 0.67±0.07 | 0.57±0.11 | 0.53±0.19 | 0.27±0.10*,§ |
| EDV (μL) | 22.1±7.8 | 29.6±7.6 | 13.0±4.7* | 14.1±6.7* |
| EDVI (μL g-1) | 1.04±0.35 | 0.91±0.21 | 0.64±0.23* | 0.46±0.22* |
| Cardiac index (ml min-1 × kg) | 238±102 | 175±59 | 166±95 | 127±89 |
| HR (min-1) | 407±21 | 401±82 | 522±75* | 430±69§ |
| EF (%) | 51±5 | 47±9 | 46±10 | 57±13 |
| Pdev (mmHg) | 78.6±9.4 | 69.2±9.3 | 82.8±15.4 | 83.0±6.4* |
| Ea (mm Hg μl-1) | 7±3 | 5±2 | 16±8 | 15±14 |
| dP/dTmax (mm Hg s-1) | 6138±1175 | 5055±1277 | 4741±1527 | 5045±1449 |
| dP/dTmin (mm Hg s-1) | 5841±1886 | 4781±1540 | 5378±2242 | 5865±1751 |
| Ees (mm Hg μl-1) | 1.77±1.15 | 1.33±0.87 | 0.67±2.23 | 2.47±0.95§ |
| V0 (μL) | 54.35±9.48 | 36.26±8.19 | 67.68±30.95 | 42.46±16.11§ |
| SWI (mm Hg ml g-1) | 36.95±20.5 | 25.5±9.4 | 17.9±15.5* | 19.1±13.4 |
| Tau (ms) | 8.6±1.9 | 9.4±2.4 | 11.5±5.6 | 9.0±5.6 |
| EDPVR | 0.06±0.04 | 0.04±0.02 | 0.026±0.097 | 0.056±0.03 |
| Body weight (g) | 21.2±0.74 | 32.5±1.37§ | 20.2±0.23* | 30.8±0.92*,§ |
SV = stroke volume. SVI = stroke volume index. ESV = end-systolic volume. ESVI = end-systolic volume index. EDV = end-diastolic volume. EDVI = end-diastolic volume index. HR = heart rate. EF = ejection fraction. Pdev = developed pressure. SWI = stroke work index. EDPVR = end-diastolic pressure volume relation. V0 = intercept of the Volume axis. Ea = arterial elastance. dP/dT max = maximal rise in pressure. dP/dT min = maximal rate of pressure decline. Ees = end-systolic elastance.
P<0.05 vs respective baseline.
P<0.05 young vs aging at same time-point.
Increased myocardial expression of extracellular matrix regulators as an early adaptive response to aortic constriction in aging mice
Aging mice are known to have expanded ECM compared to young mice at baseline [6]. Following TAC, the ECM are no longer different between young and aging mice (Figure 4A), suggesting a greater expansion in the young than the aging mice in response to TAC. To determine the mechanism by which aging mice are protected from LV dilation and death, we examined mice early (day 3) post-TAC to assess changes in the ECM. The matrix metalloproteinases (MMP), MMP-3 and MMP-12 and tissue inhibitor of metalloproteinases (TIMP), TIMP-1 showed similar expression profiles in young and aging mice at baseline (Figure 4B). MMP-3 and MMP-12 gene expression levels were increased in the myocardium of aging mice early post TAC (increased 3.2 and 1.5 fold respectively, relative to baseline, P<0.001, Figure 4B), while expression levels in young mice were unchanged. TIMP-1 gene expression was found to be increased in myocardium from both young and aging mice early post-TAC (increased 4.3-fold and 8.6-fold respectively, P<0.001, Figure 4B) with the upregulation of TIMP1 being significantly greater in aging mice (P<0.01). The upregulation of MMP-3, MMP-12 and TIMP-1 returned to baseline levels in both young and aging mice by week 13 post-TAC, suggesting this is an early adaptive response to pathological changes in extracellular matrix resulting from the TAC procedure. The increased levels of TIMP1, MMP-3 and MMP-12 upregulation in the myocardium of aging mice demonstrates an enhanced adaptive capability for extracellular matrix regulation following TAC-induced cardiac stress.
Figure 4.
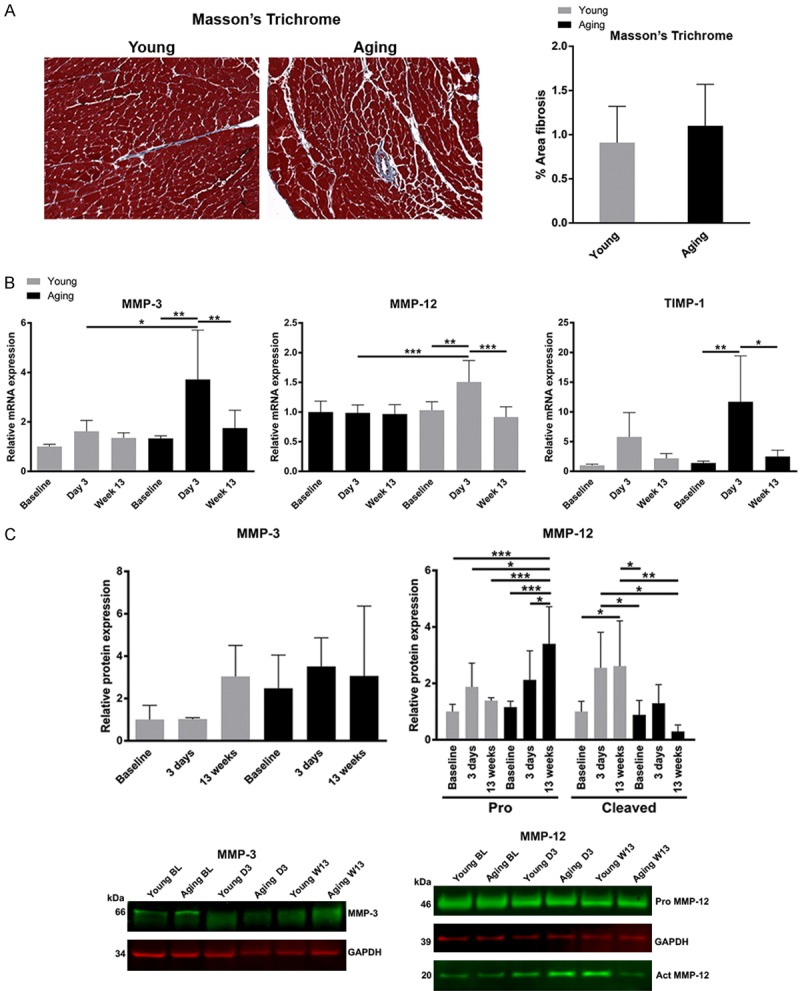
ECM remodeling post-TAC. A. Masson’s trichrome stain showing muscle as red and extracellular matrix as blue. At 13 weeks post-TAC, the proportion of extracellular matrix in the young and aging hearts becomes similar. B. The mRNA expression of MMP-3 and MMP-12 increased (3.2-fold and 1.5-fold, respectively) in aging myocardium at day 3 post-TAC while no increase was detected in young mice. An increase in TIMP-1 expression was detected in both young and aging mice (4.3-fold and 8.6-fold, respectively) at day 3 post-TAC although TIMP-1 was upregulated significantly more so in the aging mice than in the young (P<0.01). The expression of MMP-3, MMP-12 and TIMP-1 returned to baseline levels by week-13 post TAC. Data were analyzed using ANOVA with Tukey’s post hoc test. C. Protein expression of MMP-3 and MMP-12 are shown. There are no significant changes in MMP-3 protein levels. The levels of pro-MMP-12 increase in the aging mice, but active MMP12 is higher in the young mice. *P<0.05, **P<0.01, ***P<0.001 (n=3-5 mice per group).
Taken together, our findings suggest that resistance to, or protective mechanisms against, pathological remodeling following pressure overload may be more efficient in the aging heart.
Discussion
This series of experiments demonstrated a number of key findings. First, it showed that young and aging mice experience equivalent changes in pressure and cardiac stress in response to size appropriate TAC induced pressure overload. Second, TAC resulted in increased pathological remodeling, increased myocardial hypertrophy, and increased mortality in young mice compared to aging mice. Last, the aging myocardium demonstrates adaptive ECM remodeling in response to pressure overload.
Animal models of pressure overload cardiomyopathy are generally carried out with young mice, despite heart failure occurring more commonly in aging adults. Understanding the pathological mechanisms specific to the aging heart is vital in the development of targeted new treatments. We have previously shown that aging mice demonstrate increased ECM and hypertrophied cardiomyocytes compared to young mice [6]. Herein, we confirmed that left ventricular remodeling characteristics are age-dependent. Despite older mice demonstrating greater degrees of myocardial hypertrophy and larger left ventricular cavities at baseline (Figures 2 and 3), following pressure overload, they showed less remodeling than young mice. Not only did young mice display a greater increase in myocardial hypertrophy and left ventricular dilation, they demonstrated higher mortality. The cumulative survival findings are in stark contrast to previous studies that have demonstrated increased pressure overload-related mortality in aging mice [7] and rats [8]. The discrepancy between our findings and prior reports may be due to the degree of aortic constriction between different sized animals not being accounted for in the previous studies. When TAC is caused by ligation around the same size needle in small and larger aortas, leaving a similar cross sectional area of the aorta, this results in a greater proportional reduction in luminal area of the larger aortas. This in turn leads to a greater pressure gradient across the stenosis and a larger hemodynamic stress on the left ventricle. Our study is the first in the literature to define aortic size in mice of different ages and perform size-appropriate aortic constriction.
LV remodeling may have been prevented in the aging myocardium by the upregulation of MMP-3, MMP-12 and TIMP-1 detected three days following TAC (Figure 4). MMPs are proteolytic enzymes involved in the degradation and remodeling of extracellular matrix components such as fibronectin, elastin and collagen subtypes. The upregulation of MMPs and TIMPs following TAC may assist in reducing LV remodeling in the aging myocardium by maintaining integrity of the ECM and by reducing cardiomyocyte hypertrophy. The upregulation of MMP-3 in the aging LV may minimize the dilation and hypertrophy, similar to that observed in transduced vein grafts where overexpression of MMP-3 was associated with the inhibition of smooth muscle cell migration and blood vessel thickening [16]. The mechanism that underpins this process is unclear; however, existing studies suggest that ECM degradation by high levels of MMP-3 deprives cells of anchorage sites essential for migration [16-18]. Additionally, LV hypertrophy may be inhibited in aging mice due to inhibition of angiogenesis by increased expression of MMP-12, which thereby reduces the potential for cardiomyocyte hypertrophy. Overexpression of MMP-12 has been found to reduce angiogenesis by inhibiting microvascular endothelial cell proliferation, migration and capillary morphogenesis through the cleavage of urokinase-type plasminogen activator receptor, a glycoprotein essential for angiogenesis [19,20]. Consistent with these findings, prior literature has reported that the inhibition of MMP-12 leads to prolonged inflammation and worsened cardiac remodeling post-MI [21], suggesting that MMP-12 plays an important role in reducing inflammation and ameliorating post-MI remodeling. Interestingly, both MMP-3 and MMP-12 RNA was undetectable in patients with dilated cardiomyopathy undergoing transplantation [22], suggesting that lower levels are associated with worse heart function.
Furthermore, overexpression of TIMP-1 has recently been implicated in improving cardiac function and remodeling following ischemia and myocardial infarction by inhibiting the hypertrophy-inducing FAK, AKT and ERK pathways in addition to reducing apoptosis [23,24]. We observed TIMP-1 upregulation in the left ventricle of both young and aging mice following TAC with twice the level of TIMP-1 expression detected in the aging mice compared to the young (Figure 4). The lesser degree of hypertrophy in the aging mice may be due, at least in part, to increased TIMP-1 expression.
Another potential explanation for the improved outcomes in aging mice is mechanical, rather than molecular. We have previously shown that aging left ventricles have approximately twice the extracellular matrix area compared to young mice [6]. The combination of fibrosis and hypertrophy at baseline may protect the aging heart from dilation by mechanically splinting the heart. It is known that LV dilation is the largest predictor of death following myocardial infarction [25]. It is enticing to speculate that simply having some degree of LV fibrosis and hypertrophy may actually be protective against LV dilation and therefore against death, although this would need to be confirmed in future studies.
In summary, our findings demonstrate that aging is associated with protection against left ventricular dilation and death following pressure overload. This is associated with increased expression of MMP-3, MMP-12 and TIMP-1, suggesting that adaptive changes in the left ventricle are responsible for these improved outcomes in aging mice.
Acknowledgements
This project was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI Grant Number UL1 TR000004. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. This project was also supported by the Ellison Medical Foundation.
Disclosure of conflict of interest
None.
References
- 1.WHO Cause-Specific Mortality Global Health estimates 2000-2012. World Health Organisation. 2013 [Google Scholar]
- 2.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Executive summary: heart disease and stroke statistics 2013 update: a report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 3.Cheng S, Fernandes VR, Bluemke DA, McClelland RL, Kronmal RA, Lima JA. Age-related left ventricular remodeling and associated risk for cardiovascular outcomes: the multi-ethnic study of atherosclerosis. Circ Cardiovasc Imaging. 2009;2:191–198. doi: 10.1161/CIRCIMAGING.108.819938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gebhard C, Stahli BE, Gebhard CE, Tasnady H, Zihler D, Wischnewsky MB, Jenni R, Tanner FC. Age- and gender-dependent left ventricular remodeling. Echocardiography. 2013;30:1143–1150. doi: 10.1111/echo.12264. [DOI] [PubMed] [Google Scholar]
- 5.Ganau A, Saba PS, Roman MJ, de Simone G, Realdi G, Devereux RB. Ageing induces left ventricular concentric remodelling in normotensive subjects. J Hypertens. 1995;13:1818–1822. [PubMed] [Google Scholar]
- 6.Boyle AJ, Shih H, Hwang J, Ye J, Lee B, Zhang Y, Kwon D, Jun K, Zheng D, Sievers R, Angeli F, Yeghiazarians Y, Lee R. Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp Gerontol. 2011;46:549–559. doi: 10.1016/j.exger.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sopko NA, Turturice BA, Becker ME, Brown CR, Dong F, Popovic ZB, Penn MS. Bone marrow support of the heart in pressure overload is lost with aging. PLoS One. 2010;5:e15187. doi: 10.1371/journal.pone.0015187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isoyama S, Wei JY, Izumo S, Fort P, Schoen FJ, Grossman W. Effect of age on the development of cardiac hypertrophy produced by aortic constriction in the rat. Circ Res. 1987;61:337–345. doi: 10.1161/01.res.61.3.337. [DOI] [PubMed] [Google Scholar]
- 9.Stilli D, Bocchi L, Berni R, Zaniboni M, Cacciani F, Chaponnier C, Musso E, Gabbiani G, Clément S. Correlation of alpha-skeletal actin expression, ventricular fibrosis and heart function with the degree of pressure overload cardiac hypertrophy in rats. Exp Physiol. 2006;91:571–580. doi: 10.1113/expphysiol.2005.032607. [DOI] [PubMed] [Google Scholar]
- 10.Boyle AJ, Hwang J, Ye J, Shih H, Jun K, Zhang Y, Fang Q, Sievers R, Yeghiazarians Y, Lee RJ. The effects of aging on apoptosis following myocardial infarction. Cardiovasc Ther. 2013;31:e102–110. doi: 10.1111/1755-5922.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeghiazarians Y, Zhang Y, Prasad M, Shih H, Saini SA, Takagawa J, Sievers RE, Wong ML, Kapasi NK, Mirsky R, Koskenvuo J, Minasi P, Ye J, Viswanathan MN, Angeli FS, Boyle AJ, Springer ML, Grossman W. Injection of bone marrow cell extract into infarcted hearts results in functional improvement comparable to intact cell therapy. Mol Ther. 2009;17:1250–1256. doi: 10.1038/mt.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyle AJ, Kelly DJ, Zhang Y, Cox AJ, Gow RM, Way K, Itescu S, Krum H, Gilbert RE. Inhibition of protein kinase C reduces left ventricular fibrosis and dysfunction following myocardial infarction. J Mol Cell Cardiol. 2005;39:213–221. doi: 10.1016/j.yjmcc.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Boyle AJ, Schuster M, Witkowski P, Xiang G, Seki T, Way K, Itescu S. Additive effects of endothelial progenitor cells combined with ACE inhibition and beta-blockade on left ventricular function following acute myocardial infarction. J Renin Angiotensin Aldosterone Syst. 2005;6:33–37. doi: 10.3317/jraas.2005.004. [DOI] [PubMed] [Google Scholar]
- 14.Gentleman R, Carey V, Bates D, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini A, Sawitzki G, Smith C, Smyth G, Tierney L, Yang J, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article 3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 16.Kallenbach K, Salcher R, Heim A, Karck M, Mignatti P, Haverich A. Inhibition of smooth muscle cell migration and neointima formation in vein grafts by overexpression of matrix metalloproteinase-3. J Vasc Surg. 2009;49:750–758. doi: 10.1016/j.jvs.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimura T, Nakamura K, Kishioka Y, Kato-Mori Y, Wakamatsu J, Hattori A. Inhibition of matrix metalloproteinases suppresses the migration of skeletal muscle cells. J Muscle Res Cell Motil. 2008;29:37–44. doi: 10.1007/s10974-008-9140-2. [DOI] [PubMed] [Google Scholar]
- 18.Grotendorst GR, Seppa HE, Kleinman HK, Martin GR. Attachment of smooth muscle cells to collagen and their migration toward plateletderived growth factor. Proc Natl Acad Sci U S A. 1981;78:3669–3672. doi: 10.1073/pnas.78.6.3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Alessio S, Fibbi G, Cinelli M, Guiducci S, Del Rosso A, Margheri F, Serrati S, Pucci M, Kahaleh B, Fan P, Annunziato F, Cosmi L, Liotta F, Matucci-Cerinic M, Del Rosso M. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004;50:3275–3285. doi: 10.1002/art.20562. [DOI] [PubMed] [Google Scholar]
- 20.Serrati S, Cinelli M, Margheri F, Guiducci S, Del Rosso A, Pucci M, Fibbi G, Bazzichi L, Bombardieri S, Matucci-Cerinic M, Del Rosso M. Systemic sclerosis fibroblasts inhibit in vitro angiogenesis by MMP-12-dependent cleavage of the endothelial cell urokinase receptor. J Pathol. 2006;210:240–248. doi: 10.1002/path.2048. [DOI] [PubMed] [Google Scholar]
- 21.Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Brás LE, Lindsey ML. Early matrix metalloproteinase-12 inhibition worsens postmyocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol. 2015;185:198–208. doi: 10.1016/j.ijcard.2015.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Felkin LE, Birks EJ, George R, Wong S, Khaghani A, Yacoub MH, Barton PJ. A quantitative gene expression profile of matrix metalloproteinases (MMPS) and their inhibitors (TIMPS) in the myocardium of patients with deteriorating heart failure requiring left ventricular assist device support. J Heart Lung Transplant. 2006;25:1413–1419. doi: 10.1016/j.healun.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Glass C, Singla DK. Overexpression of TIMP-1 in embryonic stem cells attenuates adverse cardiac remodeling following myocardial infarction. Cell Transplant. 2012;21:1931–1944. doi: 10.3727/096368911X627561. [DOI] [PubMed] [Google Scholar]
- 24.Uchinaka A, Kawaguchi N, Mori S, Hamada Y, Miyagawa S, Saito A, Sawa Y, Matsuura N. Tissue inhibitor of metalloproteinase-1 and -3 improves cardiac function in an ischemic cardiomyopathy model rat. Tissue Eng Part A. 2014;20:3073–3084. doi: 10.1089/ten.tea.2013.0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolognese L, Neskovic AN, Parodi G, Cerisano G, Buonamici P, Santoro GM, Antoniucci D. Left ventricular remodeling after primary coronary angioplasty: patterns of left ventricular dilation and long-term prognostic implications. Circulation. 2002;106:2351–2357. doi: 10.1161/01.cir.0000036014.90197.fa. [DOI] [PubMed] [Google Scholar]