

Abstract
Background: A colorectal cancer may develop through a particular molecular genetic pathway, raising the question of whether the particular molecular changes are random, or are unique to the particular segment of colon. We wanted to determine whether molecular changes found within a colorectal cancer might also be detected in separate adenomas and polyps removed from the same area of colon at surgery. Microsatellite instability was chosen as a marker for a pathway of colon carcinogenesis. Methods: We studied a total of 46 primary colorectal cancers with microsatellite instability and 77 synchronous adenomas and polyps. All tumors were evaluated for microsatellite instability, BRAF and KRAS mutations, and methylation using standard polymerase chain reaction based methods. Results: Forty-nine benign tumors did not follow a pathway similar to that of their 31 synchronous primary cancers. For two distinct subsets of the microsatellite unstable colorectal cancers, those with acquired methylation and BRAF mutation, and those without methylation suggestive of an underlying germ line mutation, the molecular changes in the majority of their synchronous benign tumors were different from the colorectal cancer. Conclusions: These differences suggest a stochastic process within the colon regarding the particular molecular carcinogenic pathways followed by the synchronous tumors, rather than a ‘field defect’ within the colon segments. Variability in molecular findings was present for colorectal cancers arising from acquired methylation, as well as those cancers suggestive of a germ line origin.
Keywords: Adenoma, BRAF gene, colorectal cancer, field defect, hyperplastic polyp, KRAS gene, methylation, microsatellite instability, sessile polyp
Introduction
Colorectal cancer (CRC) can develop through several general pathways, resulting in cancers with particular phenotypic and molecular characteristics [1]. One pathway involves chromosomal instability, and gives rise to the majority of CRCs. A second pathway involves aberrant epigenetic DNA hypermethylation of CpG islands within gene promotor regions, referred to as the CpG island methylator phenotype (CIMP). This pathway is considered to be the major mechanism driving the serrated polyp pathway to CRC. It is closely linked with somatic BRAF p.V600E mutations. A third pathway involves defective DNA mismatch repair, leading to microsatellite instability (MSI) [2]. This pathway represents approximately 15% of all CRCs [3]. Colorectal cancers with MSI frequently demonstrate methylation of one of the mismatch repair genes, particularly the MLH1 gene, as well as frequent BRAF mutations. DNA methylation has been suggested as a mediator of ‘field defect’ within a region of colon mucosa, thereby acting as a risk factor for developing tumors [4].
Adenomas and hyperplastic polyps are frequently the precursor neoplasm for colon cancers. Various genetic alterations have been reported in adenomas, but significantly fewer than in colorectal cancers [5]. For example, early publications on the incidence of MSI in adenomas reported from 2% to 9%, but with small sample sizes [6-8]. A more recent study found CIMP-high in only 1% of 580 conventional adenomas, but in 26% of 419 serrated lesions [9]. Yamme et al found no MSI in 50 adenomas detected during colonoscopies [10]. However, the adenomatous portions of MSI-high carcinomas were reported to be also MSI-high in 9 cases [11]. Using microarrays to assess DNA methylation of one half-million CpG dinucleotides, Grady et al demonstrated a higher frequency of methylation in CRC-associated normal mucosa compared with colon mucosa from cancer-free individuals [12].
We were interested in determining whether molecular changes found within a colorectal cancer might also be detected in separate adenomas and polyps removed from the adjacent area of colon at the time of primary colon cancer resection. Finding one or more similar molecular changes within both the cancer and the synchronous benign tumor might suggest that the particular changes are an early alteration within the carcinogenesis process. It could also suggest a similar molecular response within the colon segment to a particular carcinogenic stimulus. Further, similar findings could reflect an underlying ‘field defect’ upon which a carcinogenic stimulus is acting. However, different molecular findings between the cancer and the synchronous benign tumor might suggest a more stochastic process with regard to molecular genetic changes in the colorectal carcinogenesis process.
We utilized microsatellite instability as an indication for one of the CRC carcinogenesis pathways, and we focused our analysis on colorectal cancers found to have MSI, as well as synchronous polyps and/or adenomas. In addition, we assayed both the CRC and the benign tumors for BRAF and KRAS mutations and for methylation. Additionally, we also assayed the benign portions of those MSI carcinomas with residual adenomatous tissue for the same molecular changes.
Materials and methods
Ascertainment of cases
We have completed several studies evaluating colorectal neoplasms utilizing banked tissue blocks from our Department of Pathology computerized files [13,14]. Each study was approved by the Saint Barnabas Medical Center Institutional Review Board, under a limited data certification for material de-identified of any protected health information. The current cases are those determined to be microsatellite unstable and to have accompanying adenomas found in the surgical specimens. The current analysis was also approved by the Saint Barnabas Medical Center Institutional Review Board on the same basis. Written informed consent was not obtained from the patients whose material was studied. This was a pathological study only, based upon banked tissue samples, there was no direct patient contact.
Clinical material primarily reflected a suburban community of middle economic level, with substantial representations from various minority groups (Asian, African-American). Histological slides stained with hematoxylin and eosin (H&E), and paraffin blocks containing adequate tumor material to ensure sufficient DNA for analysis, were available for all cases. Normal colonic mucosa from each patient was used as control tissue for all studies. One clinical pathologist reviewed all histological slides and identified the areas for molecular analysis. All cancers were surgically removed prior to the administration of any radiation or chemotherapy to the patient. Criteria for differentiation of adenomas followed the World Health Organization guidelines with respect to villous component: tubular adenomas, <20%; tubulovillous adenomas, 20-80%; and villous adenomas, >80%. Right side colonic segments were defined as cecum, ascending, hepatic flexure, transverse; and the left side was considered the splenic flexure, descending, sigmoid and rectum. We defined carcinomas with residual adenoma as tumors in which both a benign component and an invasive malignant component were present within the surgically removed specimen. All authors had access to the study data and reviewed and approved the final manuscript.
DNA extraction and purification
All tissue specimens were formalin-fixed and paraffin-embedded. Histological slides stained with H&E were examined and the area of relevant tissue and an area of normal mucosa were identified. Consecutive unstained slides from the blocks were prepared and the corresponding areas were isolated under a dissecting microscope by manual dissection. Micro-dissections were approximately 75% tumor cells. The paraffin wax was removed by xylene and ethanol washes. Cellular material was lysed in a proteinase K buffer solution. DNA was isolated and purified using the QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA). DNA concentration was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE).
Sequence analysis of the KRAS and BRAF genes
The codon 12/13 region in exon 2 of the KRAS gene was amplified using the primer set 5’-AAGGCCTGCTGAAAATGACTG-3’ and 5’-GGTCCTGCACCAGTAATATGCA-3’. The codon 600 region in exon 15 of the BRAF gene was amplified using the primer set 5’-CATAATGCTTGCTCTGATAGGAAA-3’ and 5’-GATCCAGACAACTGTTCAAACTG-3’. Primers were ordered through Sigma Life Sciences Custom Oligomer Service (customorders@sial.com). PCR was performed in 50 µl volumes with AmpliTaq Gold polymerase and ABI reagents (Applied Biosystems, Foster City, CA) using 100 ng of template DNA, 50 pmols of primer, and 2.0 mM MgCl2 on a GeneAmp PCR System 9700 (Applied Biosystems). PCR consisted of an initial 8 minute denaturation at 94°C, followed by 40 total cycles of a 30 second denaturation at 94°C, a 30 second annealing, and a one minute elongation at 72°C, with a final 30 minute extension at 72°C. The annealing temperature was stepped down at 62°C, 60°C, and 58°C for 5, 15, and 20 cycles, respectively.
The post-PCR products were quality checked by agarose gel and then purified using the QIAquick PCR Purification Kit (Qiagen Inc., Valencia, CA) prior to sequencing. The sequencing reactions were performed in 20 µl volumes using 0.25× BigDye Terminator Cycle Sequencing Reagents (Applied Biosystems, Foster City, CA) with 5.0 pmol of primer (reverse KRAS or forward BRAF) and 1.0 µl of the purified PCR reaction. Reactions were run on the GeneAmp PCR System 9700 for 25 cycles using a 2-minute extension time. The sequencing reaction fragments were cleaned using isopropanol precipitation. Sequencing products were separated by capillary electrophoresis with an ABI 3130 Genetic Analyzer and the data were processed with Sequencing Analysis v5.2 (Applied Biosystems, Foster City, CA) software (Figure 1).
Figure 1.

BRAF DNA sequence analysis. A: DNA from a tubular adenoma. The arrow indicates the wild-type pattern with a single peak at nucleotide number 1799, codon 600 of exon 15. B: DNA from a second tubular adenoma from the same colon segment. The arrow indicates a heterozygous mutant peak under the wild-type peak. C: DNA from a colon carcinoma from the same colon segment. The arrow indicates a heterozygous mutant peak of equal intensity to the wild-type peak.
Microsatellite instability (MSI) analysis
MSI was detected using the Bethesda panel of markers, which includes two mononucleotide markers BAT25 and BAT26, and three dinucleotide markers D2S123, D5S346, and D17S250. All microsatellite primer sets were ordered through the Life Technologies Custom Oligo Synthesis Service (genomicorders@lifetech.com). In all primer sets the forward primer contains a 5’fluorescent label while the reverse primer contains a 5’-GTGTCTT tail.
All PCR reactions were performed in 30 µl volumes using 100 ng of template with Applied Biosystems reagents and final 1.5 mM MgCl2 concentrations. Reactions were run on the GeneAmp PCR System 9700 under the following conditions: 5 minutes denaturation at 94°C, followed by 35 cycles of a 30 second denaturation at 94°C, 30 second annealing at 55°C, and a 60 second elongation at 72°C, with a final 30 minute extension at 72°C. PCR products were separated by capillary electrophoresis with an ABI 3130 Genetic Analyzer and the data was processed with GeneMapper v4.0 (Applied Biosystems, Foster City, CA) software. Microsatellite instability for a given primer set was defined as a change in the allele pattern, with the appearance of one or more new PCR products relative to those produced by the normal DNA. A tumor was defined as MSI-high if two or more of the five markers had a changed allele pattern, and referred to as “MSI” (Figure 2).
Figure 2.
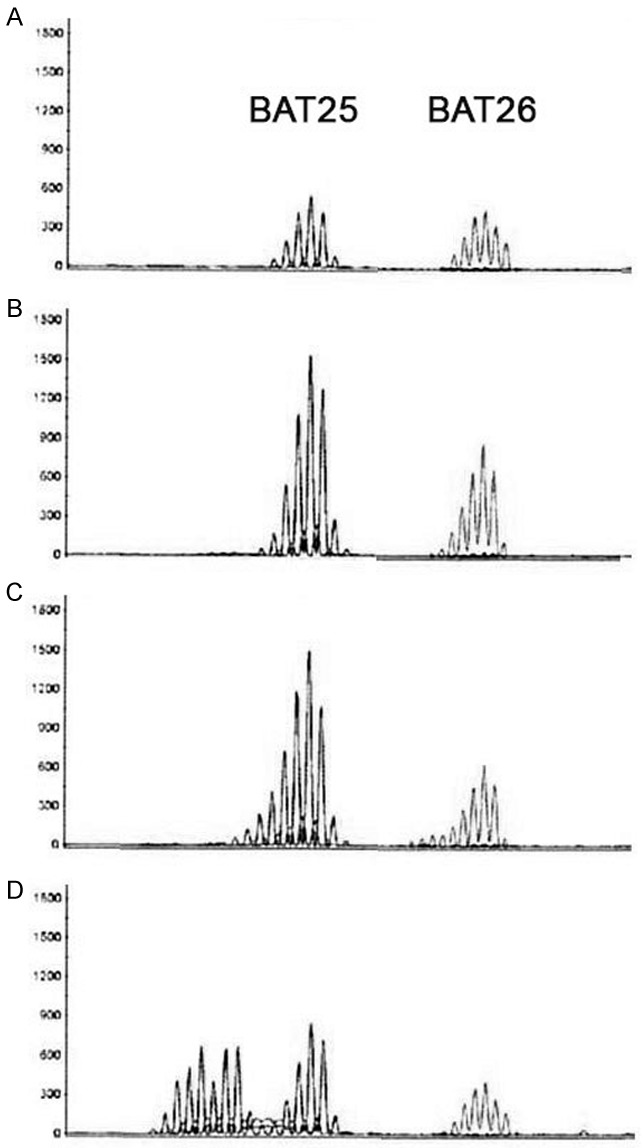
MSI analysis: Paired normal and tumor results demonstrating microsatellite instability (MSI) with the noncoding mononucleotide repeats BAT25 and BAT26. A: DNA from normal mucosa. B: DNA from a tubular adenoma showing equal numbers of peaks as the normal tissue. C: DNA from a second tubular adenoma from the same segment showing additional peaks indicating MSI. D: DNA from a colon carcinoma from the same segment showing many more additional peaks indicating MSI.
Methylation analysis
The methylation status of the mismatch repair (MMR) system was ascertained using the SALSA® MS-MLPA® Methylation-specific DNA detection Kit #ME011 (MRC-Holland, Amsterdam, The Netherlands). Briefly, 200 ng of genomic DNA was hybridized overnight with the 32-probe mix. This hybridization mixture was then split for two separate reactions, a ligation reaction, and a dual ligation and Hha1 restriction enzyme cutting reaction. Electrophoresis of PCR fragments was performed on an ABI 3130 Genetic Analyzer and the raw data were processed with Genemapper v4.0 software. The Genemapper data were subsequently exported and the methylation status was analyzed using Coffalyser v9.4 software (MRC-Holland website, www.mlpa.com). The “Direct Methylation Status” analysis option was chosen to normalize and analyze the MS-MLPA data. Methylation status for the 21 probe target sites in each sample was determined by comparing the PCR products from the normal DNA to that of the tumor. Based upon guidelines provided by the Coffalyser v9.4 software, ratios of tumor to normal peak areas for a given probe that are from 0.7 to 1.0 are assigned as methylated. Ratios of tumor to normal peak areas for a given probe that are <0.3 range are assigned unmethylated. Ratios from 0.3 to 0.7 are considered as partially methylated or hemi-methylated (Figure 3).
Figure 3.

Methylation analysis: Capillary electrophoresis patterns from the PCR products of Hha1 digestion reaction for tumors from one colon resection. A: Normal mucosa and B: Tubular adenoma. All probes that hybridize to an unmethylated Hha1 site were cut and no PCR product was observed for those probes. C: Second tubular adenoma. PCR products from methylation sensitive probes for MLH1 and MGMT are present in small amounts. D: Colon cancer. The MLH1 and MGMT probes are prominent, indicating full methylation for these targets. u = unmethyated site; m = methylated site.
Statistical analysis
Descriptive statistics using proportions was utilized throughout.
Results
Group A: colorectal cancers with synchronous polyps and adenomas
In this group, a total of 33 patients had colorectal cancer demonstrating MSI as well as a synchronous adenoma or hyperplastic polyp. There were 26 (78.8%) females and 7 (21.2%) males. The average age was 75.3 years, with a range from 53 to 94 years. The distribution of these cancers was: right colon = 5, cecum = 10, ascending = 12, transverse = 4, descending = 1, sigmoid = 1. Size was available for 31 cancers and the average was 6.51 cm in greatest dimension (S.D. = 3.15). Pathological features are shown in Table 1. These 33 patients had a total of 58 synchronous tumors. All synchronous tumors were in the same segment of the colon as the primary cancer for 30 patients. Three primary cancers were in the right colon, with a synchronous tubular adenoma in the left colon.
Table 1.
Clinical findings in patients with microsatellite unstable colorectal cancer and synchronous polyps and adenomas
| Group | Cancer No. (%) | Cancer with residual adenoma |
|---|---|---|
|
|
||
| 33 | 13 | |
| Gender | ||
| Female | 26 (78.8) | 7 (53.8) |
| Male | 7 (21.2) | 6 (46.2) |
| Age | 75.3 | 75.4 |
| Range | 53-94 | 46-91 |
| Right side | 31 (93.9) | 10 (76.9) |
| Left side | 2 (6.1) | 3 (23.1) |
| Stage | ||
| 0 | 0 (0) | 1 (7.7) |
| 1 | 6 (18.2) | 5 (38.5) |
| 2 | 15 (45.4) | 4 (30.8) |
| 3 | 9 (27.3) | 0 (0) |
| Unknown | 3 (9.1) | 3 (23.1) |
| Grade | ||
| 1 | 2 (6.1) | 6 (46.2) |
| 2 | 11 (33.3) | 2 (15.4) |
| 3 | 19 (57.6) | 3 (23.1) |
| Unknown | 1 (3.0) | 2 (15.4) |
| Mucin | ||
| Yes | 6 (18.2) | 1 (7.7) |
| No | 27 (81.8) | 8 (61.5) |
| Intermediate | 4 (30.8) | |
| No. accompanying Polyps + adenomas | 58 | 19 |
The overall comparisons between the CRCs and synchronous tumors are shown in Table 2 and in more detail through Figures 4, 5. Twenty-five of the 33 (75.8%) CRCs demonstrated MSI, methylation of MLH1 and BRAF mutation. For 18 (54.5%) of these cancers, 29 synchronous polyps and adenomas were all normal with regard to these molecular changes. Specifically, the synchronous tumors were microsatellite stable, showed no methylation and were BRAF and KRAS wild type. These 29 benign tumors included: 2 hyperplastic polyps, 25 tubular adenomas and 2 tubulovillous adenomas (Table 2 and Figure 4A).
Table 2.
Comparison of molecular changes in microsatellite unstable colorectal cancers and in their synchronous tumors
| Colorectal cancers No. (%) | No. accompanying tumors | Accompanying tumors | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| MSI | Methylation | BRAF mut | KRAS mut | |||
| Group A | 33 | CRC with synchronous polys and adenomas | ||||
| [MSI + Methylated + BRAF mutated] | 25 (75.7) | 42 | 1 | 10 | 11 | 0 |
| [MSI + Unmethylated + BRAF wild] | 6 (18.2) | 14 | 5 | 1 | 1 | 3 |
| [MSI + Methylated + BRAF wild] | 2 (6.1) | 2 | 0 | 0 | 0 | 0 |
| Group B | 13 | CRC with residual adenomatous tissue and synchronous adenomas | ||||
| [MSI + Methylated + BRAF mutated] | 7 | 9 | 1 | 1 | 1 | 0 |
| [MSI + Unmethylated + BRAF wild] | 4 | 7 | 4 | 0 | 0 | 0 |
| [MSI + Methylated + BRAF wild KRAS mutated] | 1 | 1 | 0 | 0 | 0 | 0 |
| [MSI + Methylated + BRAF wild] | 1 | 2 | 0 | 2 | 0 | 0 |
| CRC with residual adenoma; comparison of changes in the cancer and adenomatous tissue (from Group B) | ||||||
| Residual adenomatous tissue | ||||||
| [MSI + Methylated + BRAF mutated] | 5 | 5 | 4 | 5 | 4 | 0 |
| [MSI + Methylated + BRAF wild] | 1 | 1 | 1 | 1 | 0 | 0 |
| [MSI + Unmethylated + BRAF wild KRAS wild] | 3 | 3 | 3 | 0 | 0 | 0 |
Figure 4.

Molecular findings in 33 microsatellite unstable colorectal cancers and their synchronous polyps and adenomas. *aden = adenoma. tubular = tubular adenoma. villous = villous adenoma. hyperplastic = hyperplastic polyp. Serrated = sessile serrated adenoma. This figure provides a detailed description of those benign tumors that accompany carcinomas demonstrating four specific molecular profiles.
Figure 5.
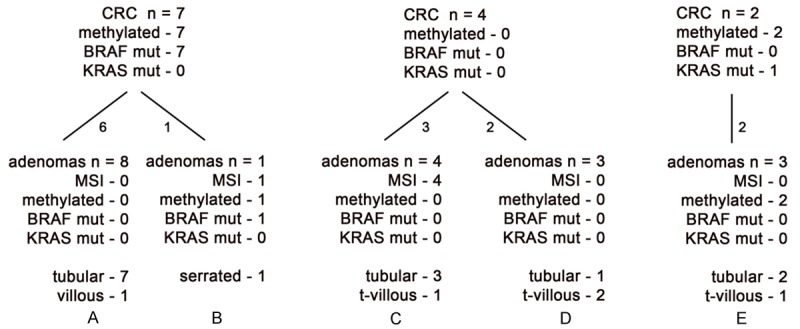
Molecular findings in 13 microsatellite unstable colorectal cancers with residual adenomatous tissue and their accompanying polyps and adenomas. tubular = tubular adenoma. villous = villous adenoma. hyperplastic = hyperplastic polyp. Serrated = sessile serrated adenoma. This figure provides a detailed description of those benign tumors that accompany carcinomas demonstrating three specific molecular profiles.
The other 7 of these 25 CRCs had 13 synchronous tumors. Twelve of the 13 were microsatellite stable, but 10 demonstrated methylation of MLH1, and 11 contained a BRAF mutation. These 13 benign tumors were: 5 hyperplastic polyps, 2 sessile serrated adenomas and 6 tubular adenomas (Figure 4B). Of these seven cancers, one carcinoma had four synchronous hyperplastic polyps, all of which were microsatellite stable, but were methylated and BRAF mutated. One carcinoma had two synchronous sessile serrated adenomas, both of which were microsatellite stable, but methylated and BRAF mutated. A third carcinoma had a synchronous hyperplastic polyp with just a BRAF mutation, but also a synchronous tubular adenoma with a BRAF mutation and methylation. A fourth carcinoma had two synchronous tubular adenomas; one had none of the molecular changes and the other had all three: BRAF mutation, methylation and MSI. The carcinoma and both tubular adenomas were all from the ascending colon. The other 3 carcinomas each had one synchronous tubular adenoma, with methylation (1), a BRAF mutation (1), or both (1). These 7 individuals were all elderly, with ages between 61 and 87 years.
Two additional MSI CRCs demonstrated methylation, but were BRAF wild. One contained a KRAS mutation. The synchronous tumors were a tubular adenoma and a hyperplastic polyp; both were microsatellite stable, showed no methylation, and were BRAF and KRAS wild (Figure 4C).
The remaining 6 (18.2%) of the 33 CRCs were variable in their molecular findings, and their synchronous tumors were dissimilar in their molecular findings from the primary cancer. All six of these CRCs were MSI, but did not have methylation and were BRAF wild, suggesting the possibility of a germ line mutation. Three demonstrated a KRAS mutation. The average age for these 6 patients was 68.3 years (54-81 years).
These 6 CRCs had a total of 14 synchronous tumors: 3 hyperplastic polyps, 10 tubular adenomas, and 1 villous adenoma. Five of the tubular adenomas were MSI; only one hyperplastic polyp demonstrated methylation and a BRAF mutation, the other 8 tumors were all microsatellite stable, unmethylated and BRAF wild; three demonstrated a KRAS mutation. (Figure 4D and 4E).
Three patients had a second primary carcinoma removed at the time of surgery. For each patient, the second cancer was in the same or adjacent colon segment as the first primary. For one patient, the molecular changes in the two primaries were identical. However, for the other two patients, there were differences between the two primaries, with one primary showing microsatellite instability and methylation, while the second primary was microsatellite stable and did not demonstrate methylation (data not shown).
Group B: colorectal carcinomas with residual adenomatous tissue and synchronous adenomas
The classical cancer model for colon carcinogenesis proposes a series of stages with stepwise accumulation of somatic mutations, and the development of a malignant tumor from a pre-existing adenoma [15]. During the malignant progression, adenoma tissue is typically destroyed. However, malignant lesions with residual adenomatous tissue present an unusual setting, one that raises the question of whether the residual benign tissue had merely ‘failed’ to be completely destroyed, or whether, in these tumors, there is parallel evolution for the benign and malignant parts of the tumor mass [16,17]. Since these cancers might, therefore arise through a different mechanism, we have elected to present separately the results for our CRCs that had residual adenomatous tissue.
A total of 13 patients had a colorectal carcinoma demonstrating MSI, with residual adenoma in the primary tumor; and, additionally, had separate, synchronous tumors. There were 7 (53.8%) females and 6 (46.2%) males. The average age was 75.4 years, with a span from 46 to 91 years. Ten carcinomas were from the right colon and 3 from the left colon. The distribution was: right = 2, cecum = 3, ascending = 4, transverse = 1, sigmoid = 1 and rectum = 2 (Table 1). The residual adenomatous tissue within the primary tumors was: tubular = 7, villous = 5, and tubulovillous = 1. Pathological features are shown in Table 1. One of these 13 primary tumors was from a patient also included in the first group of colorectal cancers; there was a span of 19 years between the patient’s two colorectal cancers. Size was available for 12 of these cancers with residual adenoma, and the average size was 4.65 cm in greatest dimension (S.D. = 2.47).
These 13 patients had a total of 19 separate, synchronous adenomas (Table 2). Adenomas were in the same segment of the colon as the primary cancer for 17 (89.5%).
Two primary cancers were from the rectum, with a synchronous adenoma in the transverse colon (one tubular adenoma and one tubulovillous adenoma). Seven of these 13 (53.8%) CRCs demonstrated methylation and a BRAF mutation. For 6 (85.7%) of these 7 CRCs, the eight synchronous adenomas were all microsatellite stable, unmethylated, and were BRAF and KRAS wild (Table 2 and Figure 5A). Only one of these seven CRCs had a synchronous adenoma with the same findings, and it was a sessile serrated adenoma (Figure 5B). Four (30.1%) MSI CRCs with residual adenomatous tissue lacked methylation and were BRAF and KRAS wild; and among their synchronous adenomas, 4 were MSI, but were not methylated and were BRAF and KRAS wild, while 3 other adenomas were microsatellite stable, were not methylated, and were BRAF and KRAS wild (Figure 5C and 5D). Two additional MSI CRCs with residual adenomatous tissue were also methylated and one contained a KRAS mutation. The three synchronous adenomas were microsatellite stable, two were methylated, and all three were BRAF and KRAS wild (Table 2 and Figure 5E).
Comparison of primary MSI colorectal carcinomas with their residual adenomatous tissue
For nine carcinomas with residual adenomatous tissue, the residual adenomatous tissue was also evaluated for the molecular changes. The molecular changes in the adenomatous tissue were identical to the changes found in the carcinomatous portion for 7 (77.8%) of these cancers. For one tumor, the adenomatous part lacked a BRAF mutation, but was otherwise identical to the carcinomatous part, demonstrating MSI and MLH1 methylation. For the other tumor, the adenomatous portion did not show microsatellite instability, as seen in the carcinomatous portion. However, both parts demonstrated methylation and a BRAF mutation (Table 2).
Discussion
Microsatellite instability (MSI) is a mutational change resulting from inactivation of DNA mismatch repair systems, first reported in colorectal cancers in the early 1990s [18]. Normal DNA contains repetitive nucleotide sequences, referred to as microsatellites. When DNA mismatch repair is defective, the microsatellites may become either shorter or longer in length due to deletion or insertion of repeating units, as compared with normal DNA from the same individual. Mismatch repair may be defective because of a germ line mutation in one of several genes that participate in the repair system. These germ line mutations account for approximately 2% of all colorectal cancers [19] and for approximately 12% of all colorectal cancers that are MSI-H [20].
However, for the majority of colorectal cancers demonstrating MSI, the instability is not caused by a germ line mutation, but is the result of an acquired loss of DNA repair function, usually resulting from biallelic methylation of the promotor region of a repair gene, frequently the gene MLH1 [21]. These cancers frequently also demonstrate a point mutation in the BRAF gene. Colorectal cancers with acquired methylation of MLH1 have an overall background of numerous genes in which sequences of at least 200 bases in length with >50% cytosine and guanine content have become methylated, referred to as CpG island methylator phenotype (CIMP) [22]. Thus, MSI-high may be considered a characteristic of the CIMP phenotype.
There are different epidemiological approaches to analyzing colorectal tissue for a possible ‘field defect’. For example, molecular data of synchronous colorectal tumors have been reported in several publications. In a study of 47 synchronous colorectal cancers, those pairs in either the proximal or the distal colon showed a significantly concordant pattern of CpG island methylation compared with pairs with one proximal and one distal tumor [23]. The authors suggested that this finding supported the concept of field effect in the colorectal carcinogenic process. However, their methylation and BRAF mutation data from cancers and paired adjacent normal tissue were inconclusive. Similar results were reported by others [24].
We previously reported that informative synchronous carcinomas from the same segment showed the same genetic findings in approximately two-thirds of pairs; but when separated by one or more colonic segments, less than half of the pairs had consistent findings [13]. Rosa et al suggested that co-localization of carcinomas and adenomas in their study favored a regional field defect in the colon [25]. Thus, the presence of a field effect in sporadic colorectal cancer has not yet been strongly established.
In a recent publication, Kim et al compared the carcinomatous and adenomatous portions of eight surgically removed colorectal cancers using whole-exome sequencing. They found differing molecular profiles between the two portions, suggesting independent evolutionary histories for the adenomatous and carcinomatous tissue within the overall cancer [16]. Their findings are consistent with the “Big Bang” model of colorectal carcinogenesis, in which there is an initial transformation, followed by tumor growth through numerous intermixed subclones, resulting in intra-tumor genomic heterogeneity [17]. However, we found the molecular changes in the residual adenomatous tissue to be identical to the changes found in the carcinomatous tissue from 7 of 9 tumors with both carcinomatous and benign features that were evaluated in this study.
A recent study of two colorectal carcinomas and their four synchronous adenomatous polyps and one hyperplastic polyp utilized whole exome sequencing to compare the mutational spectrum of the multiple samples from the two patients. No single recurrent mutation was detected in the tumors from the same patient [26].
Colorectal cancers with MSI-H are more likely to be right-sided and to occur in older individuals, particularly women [27]. Our data support this observation, with 71.7% of our primary CRCs from women, of whom only 10 were under the age of 70; while 7 were in their 70s, and 16 were above age 80 years.
Our data illustrate several key findings regarding the relationship between colorectal cancers and synchronous polyps and adenomas. First, approximately half (24 of 46, or 52.2%) of the studied CRCs with MSI, methylation and a BRAF mutation have synchronous adenomas and hyperplastic polyps (n = 37) with none of these three molecular changes. Thus, for approximately half of the CRCs with these changes, the neoplastic process appears to be stochastic and not related to a field defect.
Second, when accompanying adenomas and hyperplastic polyps follow a similar pathway as the primary CRC involving these three molecular changes, the synchronous benign tumors almost all have a BRAF mutation, are less likely to have methylation, and most likely do not have microsatellite instability. These data support the proposed sequence of these changes in sporadic CRCs with methylation. First, the development of a BRAF mutation, followed by methylation of MLH1, and finally the development of microsatellite instability as a late event occurring at the interface of serrated adenoma and invasive carcinoma [28]. However, some evidence from the literature suggests that CpG island methylation develops early in the sequence [28,29].
Third, primary CRCs with MSI that are unmethylated and BRAF wild suggest a germ line mutation in one of the mismatch repair genes (Figures 4D, 4E and 5C, 5D). For our cancers suggesting a germ line mutation, eight of 21 (38.1%) of their synchronous adenomas and hyperplastic polyps were also MSI, and also unmethylated and BRAF wild. Only one accompanying hyperplastic polyp was methylated and BRAF mutated. This suggests that approximately one-third of adenomas synchronous with CRCs in patients with a presumed germ line mutation in a mismatch repair gene may follow the same molecular pathway, but the majority of synchronous adenomas, even from the same colon segment as the CRC, may be following a different molecular pathway.
Fourth, primary CRCs with MSI and methylation may have a mutation in KRAS rather than BRAF (Figures 4C and 5E). The 5 synchronous adenomas and hyperplastic polyps of our primary CRCs with KRAS mutation did not have a KRAS mutation, and only two were methylated. Fifth, our data show that a primary carcinoma may demonstrate molecular changes that are strictly mirrored in one synchronous adenoma, but not in a second synchronous, adjacent adenoma. Similarly, as discussed about, synchronous carcinomas may, or may not, demonstrate similar molecular changes.
Certain limitations of the present study should be noted. The total number of primary CRCs studied (46) is not large. However, MSI is found in only a minority of all CRCs, and the probability of MSI CRCs found with synchronous adenomas and polyps further diminishes the sample size. Second, we did not study the primary CRCs for germ line mutations. However, it is reasonable to assume that a patient with a CRC that is MSI, but unmethylated and BRAF wild represents a carrier of a germ line mutation in one of the mismatch repair genes [30].
In summary, we have studied 46 primary colorectal cancers with microsatellite instability and 77 synchronous adenomas and hyperplastic polyps to assess whether molecular genetic changes suggest a similar pathway of carcinogenesis. For primary CRCs with acquired methylation and BRAF mutation, as well as primary CRCs suggesting an underlying germ line mutation in mismatch repair, only a minority of the synchronous tumors appear to follow a pathway similar to that of the carcinoma. The molecular changes in the majority of synchronous tumors will be different from the primary CRC, suggesting a stochastic process, rather than a field defect, within the colon regarding the molecular pathway of carcinogenesis.
Acknowledgements
The authors thank Dr. Errol Berman for review of all histological slides. Funding was provided by the H. Nussbaum Foundation of Saint Barnabas Medical Center, which had no role in the design of the study, collection of data, analysis, interpretation or writing of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Jass JR. Molecular heterogeneity of colorectal cancer: implications for cancer control. Surg Oncol. 2007;1:S7–9. doi: 10.1016/j.suronc.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 2.Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, Burt RW, Goldblum JR, Guillem JG, Kahi CJ, Kalady MF, O’Brien MJ, Odze RD, Ogino S, Parry S, Snover DC, Torlakovic EE, Wise PE, Young J, Church J. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol. 2012;107:1315–1329. doi: 10.1038/ajg.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burgart LJ. Testing for defective DNA mismatch repair in colorectal carcinoma. Arch Pathol Lab Med. 2005;129:1385–1389. doi: 10.5858/2005-129-1385-TFDDMR. [DOI] [PubMed] [Google Scholar]
- 4.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JJ. MGMT promotor methylation and field defect in sporadic colorectal cancer. J Natl Can Inst. 2005;97:1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 5.Luo Y, Wong CJ, Kaz AM, Dzieciatkowski S, Carter KT, Morris SM, Wang J, Willis JE, Makar KW, Ulrich CM, Lutterbaugh JD, Shrubsole MJ, Zheng W, Markowitz SD, Grady WM. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology. 2014;147:418–429. doi: 10.1053/j.gastro.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samowitz WS, Slattery ML. Microsatellite instability in colorectal adenomas. Gastroenterology. 1997;112:1515–1519. doi: 10.1016/s0016-5085(97)70032-5. [DOI] [PubMed] [Google Scholar]
- 7.Konishi K, Yamochi T, Makino R, Kaneko K, Yamamoto T, Nozawa H, Katagiri A, Ito H, Nakayama K, Ota H, Mitamura K, Imawari M. Molecular differences between sporadic serrated and conventional colorectal adenomas. Clin Cancer Res. 2004;10:3082–3090. doi: 10.1158/1078-0432.ccr-03-0334. [DOI] [PubMed] [Google Scholar]
- 8.Ricciardiello L, Goel A, Mantovani V, Fiorini T, Fossi S, Chang DK, Lunedei V, Pozzato P, Zagari RM, De Luca L, Fuccio L, Martinelli GN, Roda E, Boland CR, Bazzoli F. Frequent loss of hMLH1 by promoter hypermethylation leads to microsatellite instability in adenomatous polyps of patients with a single first-degree member affected by colon cancer. Cancer Res. 2003;63:787–792. [PubMed] [Google Scholar]
- 9.Burnett-Hartman AN, Newcomb PA, Potter JD, Passarelli MN, Phipps AI, Wurscher MA, Grady WM, Zhu LC, Upton MP, Makar KW. Genomic aberrations occurring in subsets of serrated colorectal lesions but not conventional adenomas. Cancer Res. 2013;73:2863–2872. doi: 10.1158/0008-5472.CAN-12-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamane LS, Scapulatempo-Neto C, Alvarenga L, Oliveira CZ, Berardinelli GN, Almodova E, Cunha TR, Fava G, Colaiacovo W, Melani A, Fregnani JH, Reis RM, Guimaraes DP. KRAS and BRAF mutations and MSI status in precursor lesions of colorectal cancer detected by colonoscopy. Oncology Reports. 2014;32:1419–1426. doi: 10.3892/or.2014.3338. [DOI] [PubMed] [Google Scholar]
- 11.de Maat MF, Umetani N, Sunami E, Turner RR, Hoon DS. Assessment of methylation events during colorectal tumor progression by absolute quantitative analysis of methylated alleles. Mol Cancer Res. 2007;5:461–471. doi: 10.1158/1541-7786.MCR-06-0358. [DOI] [PubMed] [Google Scholar]
- 12.Grady WM, Parkin RK, Mitchell PS, Lee JH, Kim YH, Tsuchiya KD, Washington MK, Paraskeva C, Willson JK, Kaz AM, Kroh EM, Allen A, Fritz BR, Markowitz SD, Tewari M. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene. 2008;27:3880–3888. doi: 10.1038/onc.2008.10. [DOI] [PubMed] [Google Scholar]
- 13.Zauber P, Huang J, Sabbath-Solitare M, Marotta S. Similarities of molecular genetic changes in synchronous and metachronous colorectal cancers are limited and related to the cancers’ proximities to each other. J Mol Diagn. 2013;15:652–660. doi: 10.1016/j.jmoldx.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 14.Zauber P, Marotta S, Sabbath-Solitare M. Colorectal cancers with the uncommon findings of KRAS mutation and microsatellite instability. Cytogenet Genome Res. 2015;146:2617–2623. doi: 10.1159/000441086. [DOI] [PubMed] [Google Scholar]
- 15.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 16.Kim TM, An CH, Rhee JK, Jung SH, Lee SH, Baek IP, Kim MS, Lee SH, Chung YJ. Clonal origins and parallel evolution of regionally synchronous colorectal adenoma and carcinoma. Oncotarget. 2015;6:27725–27735. doi: 10.18632/oncotarget.4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, Curtis C. A big bang model of human colorectal tumor growth. Nat Genet. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thibodeau SN, Schaid BG. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 19.Cunningham JM, Kim CY, Christensen ER, Parc Y, Burgart LJ, Halling KC, McDonnell SK, Schaid DJ, Vockley CW, Kubly V, Nelson H, Michels VV, Thibodeau SN. The frequency of hereditary defective mismatch repair in prospective series of unselected colorectal carcinomas. Am J Hum Genet. 2001;69:780–790. doi: 10.1086/323658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poynter JN, Siegmund KD, Weisenberger DJ, Long TI, Thibodeau SN, Lindor N, Young J, Jenkins MA, Hopper JL, Baron JA, Buchanan D, Casey G, Levine AJ, Le Marchand L, Gallinger S, Bapat B, Potter JD, Newcomb PA, Haile RW, Laird PW Colon Cancer Family Registry Investigators. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol Biomarkers Prev. 2008;17:3208–3215. doi: 10.1158/1055-9965.EPI-08-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 23.Nosho K, Kure S, Irahara N, Shima K, Baba Y, Spiegelman D, Meyerhardt JA, Giovannucci EL, Fuchs CS, Ogino S. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology. 2009;137:1609–1620. doi: 10.1053/j.gastro.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konishi K, Shen L, Jelinek J, Watanabe Y, Ahmed S, Kaneko K, Kogo M, Takano T, Imawari M, Hamilton S, Issa JJ. Concordant DNA methylation in synchronous colorectal carcinomas. Cancer Prev Res. 2009;2:814–822. doi: 10.1158/1940-6207.CAPR-09-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosa I, Fidalgo P, Chaves P, Pereira AD. The colocalization of carcinomas and adenomas favors a regional field defect in the colon: an observational study. Int J Colorectal Dis. 2015;30:323–327. doi: 10.1007/s00384-014-2087-4. [DOI] [PubMed] [Google Scholar]
- 26.Vaque JP, Martinez N, Varela I, Fernandez F, Mayorga M, Derdak S, Beltran S, Moreno T, Almaraz C, De las Heras G, Bayes M, Gut I, Crespo J, Piris MA. Colorectal adenomas contain multiple somatic mutations that do not coincide with synchronous adenocarcinoma spectrum. PLoS One. 2015;10:e0119946. doi: 10.1371/journal.pone.0119946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward R, Meagher A, Tomlinson I, O’Connor T, Norrie M, Wu R, Hawkins N. Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut. 2001;48:821–829. doi: 10.1136/gut.48.6.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Brien MJ, Yang S, Mack C, Xu H, Huang CS, Mucahy E, Amorosino M, Farraye FA. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol. 2006;30:491–501. doi: 10.1097/01.pas.0000213313.36306.85. [DOI] [PubMed] [Google Scholar]
- 29.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–550. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 30.Goel A, Boland R. Epigenetics of colorectal cancer. Gastroenterology. 2012;143:1442–1460. doi: 10.1053/j.gastro.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]