

Abstract
Background
Forage species of Urochloa are planted in millions of hectares of tropical and subtropical pastures in South America. Most of the planted area is covered with four species (U. ruziziensis, U. brizantha, U. decumbens and U. humidicola). Breeding programs rely on interspecific hybridizations to increase genetic diversity and introgress traits of agronomic importance. Knowledge of phylogenetic relationships is important to optimize compatible hybridizations in Urochloa, where phylogeny has been subject of some controversy. We used next-generation sequencing to assemble the chloroplast genomes of four Urochloa species to investigate their phylogenetic relationships, compute their times of divergence and identify chloroplast DNA markers (microsatellites, SNPs and InDels).
Results
Whole plastid genome sizes were 138,765 bp in U. ruziziensis, 138,945 bp in U. decumbens, 138,946 bp in U. brizantha and 138,976 bp in U. humidicola. Each Urochloa chloroplast genome contained 130 predicted coding regions and structural features that are typical of Panicoid grasses. U. brizantha and U. decumbens chloroplast sequences are highly similar and show reduced SNP, InDel and SSR polymorphism as compared to U. ruziziensis and U. humidicola. Most of the structural and sequence polymorphisms were located in intergenic regions, and reflected phylogenetic distances between species. Divergence of U. humidicola from a common ancestor with the three other Urochloa species was estimated at 9.46 mya. U. ruziziensis, U. decumbens, and U. brizantha formed a clade where the U. ruziziensis lineage would have diverged by 5.67 mya, followed by a recent divergence event between U. decumbens and U. brizantha around 1.6 mya.
Conclusion
Low-coverage Illumina sequencing allowed the successful sequence analysis of plastid genomes in four species of Urochloa used as forages in the tropics. Pairwise sequence comparisons detected multiple microsatellite, SNP and InDel sites prone to be used as molecular markers in genetic analysis of Urochloa. Our results placed the origin of U. humidicola and U. ruziziensis divergence in the Miocene-Pliocene boundary, and the split between U. brizantha and U. decumbens in the Pleistocene.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3904-2) contains supplementary material, which is available to authorized users.
Keywords: cpDNA, Brachiaria, Plastid, Phylogeny, Chloroplast markers
Background
Forage grasses belonging to four species of Urochloa (previously included in Brachiaria) represent 85% of planted pastures in Brazil [1], extending over 115 Mha [2]. These pastures feed 90% of the commercial cattle herd raised in the country, which added up to 209 million heads in 2010 [3]. While U. brizantha, U. decumbens, and U. humidicola are the main species used as forages, interest in U. ruziziensis has grown due to its recent use in crop-livestock integrated systems, which could restore 18 Mha of degraded pastures in the next few years [2]. The four species are native to Africa and distributed in the humid and sub-humid tropics in South-East Asia, the Pacific Islands, Northern Australia and South America [4]. While U. brizantha, U. decumbens, and U. humidicola are predominantly polyploid and apomictic, U. ruziziensis is a sexual diploid species. The genus includes 110 species, and it is the largest of subtribe Melinidinae [5]. Inclusion of species in either Brachiaria or Urochloa has changed over time [6], and many research groups - forage breeders in particular - still refer to them as Brachiaria.
Phylogenetic relationships between the four main species of Urochloa have been subject of some controversy. Phylogenetic analysis of Urochloa species based on morphological traits included U. ruziziensis, U. brizantha, and U. decumbens in a group with U. eminii, U. dura, and U. oligobrachiata [7], a pattern that in part has not been confirmed in phylogenies based on molecular data. Analysis of ITS nuclear rDNA [6] clustered U. brizantha and U. ruziziensis in a clade with U. comata and U. dura, while U. decumbens was included in a group with U. subulifolia, Melinis repens, and U. eruciformis. U. humidicola joined another group, which included U. dictyoneura and B. leersioides. These relationships were maintained when molecular and morphological data were combined. Analysis of chloroplast DNA (cpDNA) regions, however, grouped U. ruziziensis, U. brizantha, and U. decumbens in a strongly supported clade [5], while U. humidicola was included in a separate clade with U. dictyoneura and U. dura.
Analyses of plastid genomes traditionally involve laborious isolation of chloroplasts, extraction and purification of plastid DNA, followed by sequencing and assembly [8–10]. New sequencing technologies have allowed investigation of plastid genomes in a more cost-effective, time-saving manner, with huge increases in sequence throughput [11–14]. This resulted in the publication of a growing number of plastid genomes of species for which genomic information was scarce or even absent. Examples in recent literature include species of bamboo [12], coconut palm [15] and the Lolium-Festuca complex [16]. Chloroplast genome sequencing and assembly also benefit research groups focused on chloroplast transformation for crop improvement [17–19]. Studies on structural and sequence variation in chloroplast genomes have contributed to plant phylogeny, ecology, comparative genomics, population genetics and evolution, particularly in angiosperms [20–22].
Chloroplast DNA ranges between 120 and 160 kb in size in most plants, with each chloroplast containing multiple copies of a circular chromosome composed of four regions: Large Single Copy (LSC), Small Single Copy (SSC), and two copies of an Inverted Repeat (IRa and IRb) [23]. Chromosomal organization, as well as the linear order of genes in cpDNA, vary little in angiosperms [23, 24]. Nucleotide substitution rates are low in coding regions of cpDNA, due to strong selection on the photosynthetic machinery, which restricts nucleotide mutation rates [24, 25]. It is possible, however, to detect structural and sequence variations that can be useful for phylogenetic analysis [26–28]. Use of cpDNA in phylogenetic analyses is also favored by the abundance of cpDNA after DNA extraction from leaf tissue [29, 30], by its usually maternal inheritance [31], and by the absence of recombination [32]. Structural and sequence variations in chloroplasts include single-nucleotide polymorphisms (SNPs), insertion-deletions (InDels), as well as microsatellites. Chloroplast microsatellites or SSRs (Simple Sequence Repeats) can be useful tools for the conservation and use of plant genetic resources. Chloroplast SSRs usually show polymorphism, and are generally composed of poly-A or poly-T sequences of approximately 20 bp [33–35]. Mononucleotide chloroplast SSRs have been used in studies of plant population structure and diversity, as well as in maternity tests [17, 34, 35]. Chloroplast SNPs and InDels have been recently applied to cultivar and food product differentiation in ginseng [36], germplasm identification in cacao [37], and species differentiation and identification in grasses [38, 39], and Populus [40].
Although sequence data generated by phylogenetic studies in Urochloa-Brachiaria is available on public databases, complete chloroplast genome sequences for members of these genera have not been published to date. Whole cpDNA sequence analysis could be used to investigate the phylogenetic relationships of the four cultivated species, identify chloroplast DNA markers (SSRs, SNPs and InDels), estimate their time of divergence and contribute to the current understanding of the Urochloa genus. This information is important for breeding programs which rely on interspecific hybridizations to increase genetic diversity and to introgress traits of agronomic importance in Urochloa. Therefore, the objectives of this study were to sequence, assemble, annotate, and compare complete chloroplast genomes of four species of Urochloa used as tropical grass forages. This data was then used to investigate phylogenetic relationships between these species based on complete cpDNA sequences, to estimate their times of divergence and to describe polymorphic cpDNA regions which will be useful for genetic analysis of Urochloa.
Methods
Plant material and DNA sequencing
Genomic DNA from samples of the four Urochloa species was extracted using a standard CTAB protocol [41] with modifications [42]. Samples included a selfed clone of U. ruziziensis cv. Kennedy (FSS-1) [43]; U. brizantha BRS cv. Marandu; U. decumbens cv. Basilisk; and U. humidicola cv. Tupi. Genomic libraries were prepared according to manufacturer’s instructions (Illumina, San Diego, CA, USA). In summary, DNA fragments were obtained by nebulization, and their 3′ ends were added an adenine, to which adapter fragments were ligated. Ligation products were run on an 1% agarose gel, and fragments of ~200 bp were excised and purified. Sequencing of each paired-end genomic DNA fragment library was performed on the Illumina GAII sequencer, with six sequencing lanes for U. ruziziensis and one lane each for the other three species.
Chloroplast genome assembly
FASTQ formatted files containing DNA sequencing reads were submitted to the short-read correction tool of SOAPdenovo (Release 1.05), designed to correct Illumina GA reads [14]. The KmerFreq and ErrorCorrection routines were run with default parameters (seed length = 17, quality cutoff = 5). Illumina sequencing adapters and low quality reads were eliminated using the CLC trimmer function (default limit = 0.05) (CLC Genomics Workbench 4.1 software, CLC Bio, Aarhus, Denmark). Error corrected FASTQ files were then submitted to assembly routines performed on CLC Genomics. Reads from the four Urochloa species were initially assembled using the Panicum virgatum cv. Summer chloroplast genome (NC_015990) as a reference, with assembly routines of the CLC Genomics Workbench. High quality and matching reads (e-value = −10) were initially selected for assembly. Additionally, four de novo assemblies of the cpDNA molecules of each Urochloa species were also performed and compared to results of assemblies using P. virgatum as reference. In this case, sequence reads BLASTed against P. virgatum were submitted to assembly routines performed on CLC Genomics with de novo assembly using paired-end reads. Bubble size was automatically defined by the software as 50 bp. Assembly Length Fraction and Similarity parameters were set to 0.5 and 0.8, respectively. Mismatch, deletion and insertion cost parameters were set to 2, 3 and 3, respectively. The k-mer size on CLC Bio assembler was set to 25 bp and the coverage cutoff to 1000X.
Chloroplast genome sequence analysis
Annotation was performed using DOGMA [44] with default parameters. Predicted coding regions were manually adjusted for their start and stop codons, after inspection and comparison with available chloroplast genomes in tribe Paniceae. Corrections were made using Sequin (http://www.ncbi.nlm.nih.gov/Sequin/) and Artemis [45]. Graphic representations of the annotated plastid genomes were obtained with OGDraw [46]. Complete chloroplast genomes from nine species in tribe Paniceae were compared regarding their levels of sequence conservation, using the Multi-LAGAN alignment program [47] included in mVISTA [48, 49], with default parameters. The chloroplast genome sequence of U. humidicola was used as a reference for these alignments. In addition to the sequences of four Urochloa species from this study, complete plastid sequences from switchgrass (Panicum virgatum, NC_015990), pearl millet (Cenchrus americanus, NC_024171), foxtail millet (Setaria italica, NC_022850), late barnyard grass (Echinochloa oryzicola, NC_024643), and white fonio (Digitaria exilis, NC_024176) were included in this analysis.
Assembled chloroplast sequences were analyzed with the perl script MISA [50] for the detection of microsatellite regions. Parameters included searches for regions with repeat units ranging from one to six. Thresholds for a minimum number of repeat units were established as follows: at least 10 repeat units for mononucleotide regions; five repeat units for dinucleotides; three repeat units for tri- and tetranucletides; and five repeat units for penta- and hexanucleotides.
SNPs and InDels were detected from pairwise alignments of complete plastid sequences using the NUCmer program included in MUMmer v 3.23 [51]. A perl script was used to parse output files from NUCmer and produce VCF files containing SNP and InDel information from each pairwise comparison (mummer2Vcf.pl script available at https://github.com/marcopessoa/bioinfo-scripts). The programs snpEff and SnpSift v. 4.2 [52, 53] were used to annotate genomic regions and types of effects of the detected SNPs and InDels. For each pairwise comparison, effects were annotated using the most ancestral species as a reference, based on results of phylogenetic analyses.
Phylogenetic analyses
Plastid nucleotide sequences from the four Urochloa species and from 26 other Poaceae species were aligned using parallel MAFFT v. 7.187 [54] on XSEDE [55] via the CIPRES Science Gateway [56]. Alignments included the LSC region, only one copy of the IR, and the SSC region of each plastid. The FFT-NS-i [57] executable was used, with 1000 cycles of iterative refinement. The list of species included in this analysis is presented on Additional file 1. This dataset was analyzed using Maximum Parsimony (MP), Maximum Likelihood (ML, [58]), and Bayesian Markov Chain Monte Carlo inference (BI, [59]).
MP analyses were performed with PAUP*4.0a144 [60], using an initial Heuristic Search with 1000 random-taxon-addition replicates, the tree-bisection-reconnection (TBR) branch-swapping algorithm, and the “MulTrees” option in effect. Non-parametric bootstrap [61] was applied with 10,000 replicates, each with 10 random-taxon-addition replicates. ML analyses was performed on RAxML-HPC2 on XSEDE [62] using the GTRGAMMA model for the rapid bootstrapping phase, and Puelia olyriformis as an outgroup. The search for the best-scoring ML tree and the rapid bootstrap were performed in a single run. Bootstrapping was stopped automatically with the autoMRE Majority Rule Criterion, and was followed by ML optimization steps. BI was conducted using MrBayes v. 3.2.3 [63], with two independent runs and four chains. Each run was performed until completion, and included 20,000,000 generations, with sampling every 100 generations. The first 3000 generations were discarded as burn-in. Trees were visualized with FigTree v1.4.2 [64].
Divergence time estimation
The ML tree obtained with RAxML was used as input to generate an ultrametric tree with the chronos function of the R package ape [65], (lambda = 0, a relaxed model, and a calibration for the most recent common ancestor [MRCA] of rice and maize with a minimum age of 32 and a maximum age of 66 million years). These constraints were based on phytolith data from the North American Great Plains [66], which suggest that BEP and PACMAD clades had diverged by 35 mya [67]. The resulting ultrametric tree was used as the starting tree for BEAST v2.3.1 [68] runs, which was used for molecular dating. BEAST parameters were set under an uncorrelated log-normal clock model [69], a Calibrated Yule tree prior, and the GTR substitution model. Site model parameters included four gamma categories, a value of 1.0 for the gamma shape distribution, a proportion of invariant sites set to 0.5, and an empirical frequency model. A prior for the calibration of the MRCA node for the BEP-PACMAD group was included following an exponential distribution with mean 20.0 and offset 35.0. The BEP-PACMAD group was constrained as monophyletic during MCMC analysis. Each MCMC run had a chain length of 100,000,000 iterations, with sampling every 10,000 steps. An input file with all analyses parameters was prepared using BEAUTi v2.3.1, and is included as Additional file 2. Convergence between two MCMC runs, as well as their respective means and ESS values for logged statistics were assessed using Tracer v1.6.0 [70]. Tree and log files were combined with Logcombiner v2.3.1.
Results and discussion
Chloroplast genome sequencing, assembly, and annotation
Input data from U. ruziziensis was 3X greater than that from the other species, but coverage and proportion of mapped reads on the reference P. virgatum chloroplast genome did not increase proportionally, since the cpDNA molecule is small. In U. brizantha, for instance, a higher proportion (4%) of reads were mapped on the P. virgatum chloroplast genome sequence, while for the other species this value ranged between 1 and 2% of all sequence reads (Table 1). This resulted in an average coverage in U. brizantha (2791X) that was higher than that observed for U. ruziziensis (2011X). The high average coverage values for the assembled contigs, all of which were greater than 1000X (Table 1), did not seem to be related to the initial number of reads obtained for each species, given that U. brizantha had a higher mean coverage. It seems, though, that U. brizantha whole DNA extraction had proportionally more cpDNA sequences than the other species. A possible explanation for this observation should still be pursued.
Table 1.
Assembly metrics of four chloroplast genomes of Urochloa forage species, using the P. virgatum plastid sequence as a reference
| Species | Total reads (bp) | Reads mapped to P. virgatum cpDNA (bp) | % of mapped reads | cpDNA assembly size (bp) | LSC size (bp) | SSC size (bp) | IRa size (bp) | IRb size (bp) | Mean coverage |
|---|---|---|---|---|---|---|---|---|---|
| U. ruziziensis | 20,211,010,448 | 279,025,488 | 1% | 138,765 | 80,798 | 12,537 | 2715 | 22,715 | 2011 |
| U. brizantha | 8,643,705,720 | 387,850,876 | 4% | 138,946 | 81,008 | 12,535 | 22,699 | 22,704 | 2791 |
| U. decumbens | 9,018,811,776 | 168,717,644 | 2% | 138,945 | 81,005 | 12,537 | 22,699 | 22,704 | 1321 |
| U. humidicola | 8,476,910,040 | 183,602,548 | 2% | 138,976 | 81,017 | 12,535 | 22,711 | 22,713 | 1214 |
| P. virgatum | - | - | - | 139,619 |
Annotated chloroplast genome sequences were submitted to GenBank and are available under accession numbers NC_030066-NC030069. The four chloroplast genomes showed a typical circular chromosome including the LSC region (ranging from 80,798 bp in U. ruzizienis to 81,017 bp in U. humidicola), the SSC region (12,535 in U. brizantha and U. humidicola; 12,537 bp in U. ruziziensis and U. decumbens), and the two IR regions (ranging from 22,699 in U. brizantha and U. decumbens to 22,715 bp in U. ruziziensis) (Table 1). The two IR regions had the same size in U. ruziziensis, but differed by 2 bp in U. humidicola, and by 5 bp in U. brizantha and U. decumbens. Whole plastid genome sizes ranged between 138,765 bp in U. ruziziensis to 138,976 bp in U. humidicola, all genomes sequenced being smaller than the reference chloroplast genome (139,619 bp in Panicum virgatum) (Table 1). Size differences between assemblies for U. brizantha and U. decumbens reached only 1 bp, going up to 211 bp between U. humidicola and U. ruziziensis. De novo assemblies of each cpDNA molecule represented 92.89 to 99.45% of chloroplast genomes assembled using P. virgatum cpDNA as reference (Additional file 3). Sequence alignment corroborated the results obtained using P. virgatum as reference.
Each of the four Urochloa chloroplast genomes contained 130 predicted coding regions, 112 of which were unique, and 18 of which were duplicated in the two IRs. These regions included 77 protein-coding genes, 31 tRNAs, and 4 rRNAs. Eleven protein-coding genes and seven tRNAs contained introns. Coding regions ranged between 51.6% and 51.8%, and GC contents ranged between 38.5 and 38.63%. Figure 1 shows the genome structure and mapping of these genes on the U. ruziziensis chloroplast genome. Typical features of chloroplast genome organization of Panicoid grasses were found, such as the loss of genes accD, ycf1, and ycf2 [71]. The IR regions also contained trnH-GUG and rps19 near the IR-LSC junction [71, 72]. The IRb/SSC boundary included 29-bp of the ndhF gene in IRb, a feature that is unique to subfamily Panicoideae [71]. Recent reports have shown the feasibility of fully assembling chloroplast genomes de novo using either Illumina short reads [73] or PacBio single-molecule real-time (SMRT) sequences. This will certainly increase the number of large scale phylogenomic studies using either complete chloroplast sequences [74] or single nucleotide variants (SNVs) [75].
Fig. 1.
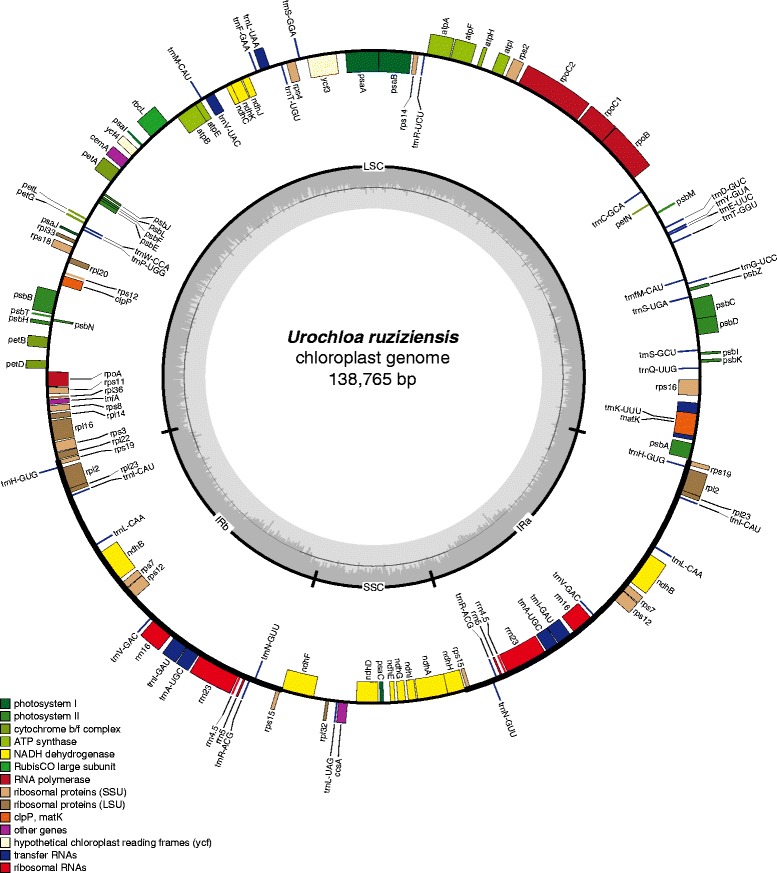
Genome structure and mapping of genes in the U. ruziziensis chloroplast genome. The LSC, SSC, and IR regions are labeled accordingly. The inner circle depicts GC content. Genes on the outside of the map are in the forward strand, while genes on the inside are in the reverse strand. Genes are colored according to their functions as shown in the legend
Comparative analysis between the four Urochloa chloroplast genomes showed high values of sequence conservation between U. ruziziensis, U. brizantha and U. decumbens when compared to U. humidicola (Fig. 2). Values are close to 100% in coding regions, with very few regions of lower similarity (between 50% and 60%) in non-coding regions. On average, U. humidicola had 98.3% sequence similarity with U. decumbens and U. brizantha, and 98.2% with U. ruziziensis. As expected, sequence conservation decreases when U. humidicola is compared to more distantly related species in tribe Paniceae (Fig. 2). However, average similarity still ranged between 94.9% between U. humidicola and Digitaria exilis and 97.2% between U. humidicola and P. virgatum.
Fig. 2.
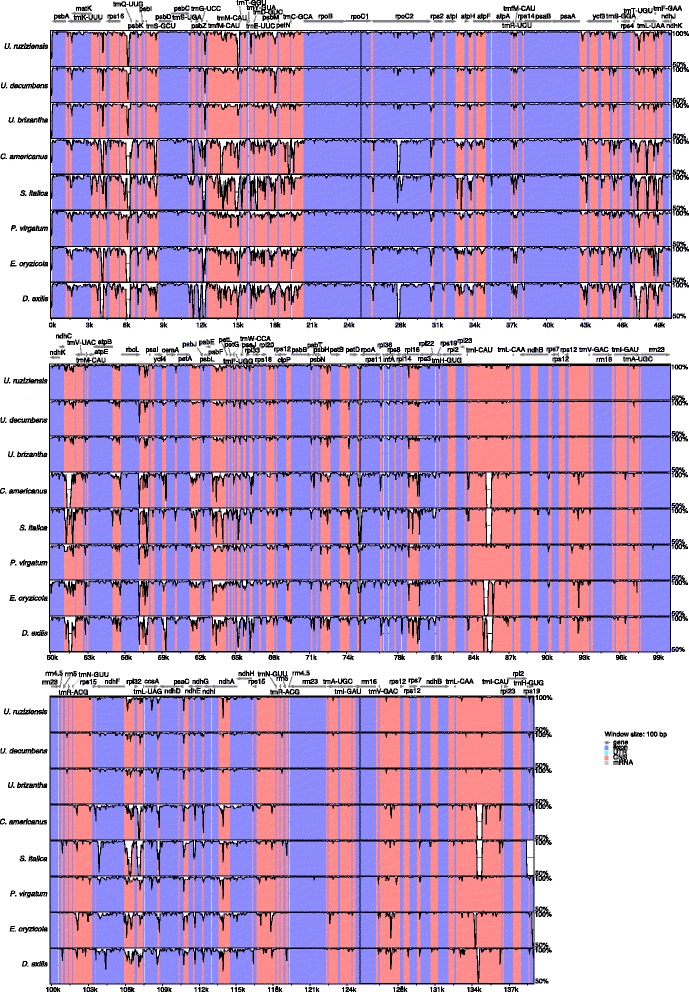
Alignment and percentage identity of complete chloroplast sequences of Urochloa and five species of Paniceae using mVISTA. Measures of similarity are relative to U. humidicola, which was used as a reference. Coding regions are shaded in blue, while noncoding sequences are shaded in pink. Arrows indicate positions of annotated genes in U. humidicola
Chloroplast molecular markers
Microsatellites
The number of cpSSRs detected in the four Urochloa species ranged between 80 in U. decumbens and 84 in U. brizantha (Table 2). Loci with tri-nucleotide repeat motifs were the most abundant, followed by those with mono-nucleotide repeats. U. ruziziensis presented the largest mono-nucleotide locus, with 24 bp, located at position 44,778 bp of intron 1 of ycf3. The number of mono-nucleotide repeat loci ranged between 24 in U. decumbens and U. humidicola and 29 loci in U. ruziziensis. These values are in agreement with what was described for P. virgatum [17], with 25 mono-nucleotide microsatellite loci of length 10 bp or greater in that species. In common wheat, 24 loci having more than ten mononucleotide repeats have been detected [76].
Table 2.
Number of microsatellites with mono-, di-, tri-, and tetra-nucleotide repeat motifs in chloroplast genomes of four Urochloa species
| Unit size | Species | |||
|---|---|---|---|---|
| U. brizantha | U. decumbens | U. ruziziensis | U. humidicola | |
| 1 | 28 | 24 | 29 | 24 |
| 2 | 4 | 5 | 5 | 5 |
| 3 | 42 | 41 | 39 | 42 |
| 4 | 10 | 10 | 9 | 10 |
| Total | 84 | 80 | 82 | 81 |
| Largest mononucleotide repeat (bp) / Location (bp) | 15 / 50,621 | 21 / 30,776 | 24 / 44,778 | 17 / 45,087 |
We looked for inter-specific cpSSR polymorphisms between the four Urochloa chloroplast genomes using in silico analysis (Additional file 4). Out of 84 cpSSRs detected in U. brizantha, for instance, 32 sites were potentially polymorphic in at least one of the other three Urochloa species. Forty-five cpSSRs were located in genic regions, 38 of which were in exons (Additional file 4). The gene with the largest number of cpSSRs was rpoC2, with seven loci. One trinucleotide locus detected in U. brizantha and U. decumbens (trnfM-CAU-trnT-GGU intergenic region, position 15,138 bp in U. brizantha) was absent in U. ruziziensis and U. humidicola (Additional file 4). Three other loci presented changes in their repeat motifs, which modified their status from perfect to imperfect cpSSRs. The usefulness of these loci for intra- and inter-specific analyses remains to be experimentally validated in a future study. Potential applications include studies of intra-specific plant population structure and diversity, as well as maternity tests which could be useful in breeding programs based on the generation of intra- and inter-specific hybrids. The first set of nuclear microsatellite markers for U. ruziziensis, using a draft assembly of its nuclear genome from Illumina sequence data, has been recently published and applied to genetic analysis of U. ruziziensis [43, 77].
Single nucleotide polymorphisms (SNPs) and insertion/deletion (InDel) sites
Pairwise sequence comparisons allowed the identification of SNPs in the Urochloa chloroplast genomes, with numbers ranging from 170 SNPs between U. brizantha and U. decumbens, up to 1338 SNPs between U. decumbens and U. humidicola (Table 3). Most of the detected SNPs in all pairwise comparisons were located in intergenic regions. The number of chloroplast SNPs detected between U. brizantha and U. decumbens is 4.5× smaller than between U. brizantha and U. ruziziensis, and almost 8× smaller than between U. brizantha x U. humidicola. A similar numbers were observed in comparisons between U. decumbens and U. ruziziensis, as well as between U. decumbens and U. humidicola (Table 3). Approximately 28% of the SNP effects, i.e., mutations with potential effect on gene expression, are located in genic regions in the U. brizantha x U. decumbens comparison, while this number increases up to 41% in other comparisons. Thirty-nine SNPs located in exons were detected between U. brizantha x U. decumbens (19 of which were missense mutations using U. brizantha as a reference). This number increases in other pairwise comparisons and it was found to be as high as 439 SNPs between U. decumbens x U. humidicola (144 of which were missense mutations using U. humidicola as reference). Similar pattern was observed in tRNA loci mutations, which were about 2% of the total number of SNP effects detected between U. brizantha x U. decumbens, and up to 7.1% between U. ruziziensis x U. humidicola. Additional file 5 includes SNPs detected in genes for all pairwise comparisons and their respective positions in base-pairs. The three genes with the largest numbers of SNPs in exons were rpoC2, ndhF, and matK, for all pairwise comparisons but one: rpoC2 was followed by ccsA and rps18 when U. brizantha and U. decumbens were compared.
Table 3.
Total number of SNPs, their locations (inter or intragenic), and effects in pairwise comparisons of four Urochloa chloroplast genomes
| Pairwise comparison |
U. brizantha
x U. decumbens |
U. ruziziensis
x U. decumbens |
U. ruziziensis
x U. brizantha |
U. humidicola
x U. ruziziensis |
U. humidicola
x U. brizantha |
U. humidicola
x U. decumbens |
|---|---|---|---|---|---|---|
| Total number of SNPs | 170 | 752 | 788 | 1319 | 1307 | 1338 |
| Number of Effects | 172 | 767 | 805 | 1356 | 1343 | 1371 |
| Intergenic | 119 | 418 | 440 | 734 | 749 | 772 |
| Intragenic | 49 | 315 | 328 | 526 | 502 | 511 |
| Intron | 10 | 52 | 61 | 82 | 66 | 72 |
| Exon | 39 | 263 | 267 | 444 | 436 | 439 |
| synonymous coding mutation | 20 | 174 | 175 | 305 | 293 | 296 |
| non-synonymous coding mutation | 19 | 89 | 92 | 140 | 144 | 144 |
| Missense | 19 | 89 | 92 | 139 | 144 | 144 |
| Nonsense | - | - | - | 1 | - | - |
| Other (tRNAs) | 4 | 34 | 37 | 96 | 92 | 88 |
Pairwise cpDNA sequence comparisons also allowed the identification of InDels between the four Urochloa species. The lowest number of InDels (91) was found between U. brizantha and U. decumbens, while the largest number (259) was found between U. brizantha and U. humidicola. Results for all pairwise comparisons are shown in Additional file 6. A high correlation between the number of identified InDels and SNPs between species was detected (0.996). The number of InDels located in chloroplast genic regions ranged from 12 between U. decumbens and U. brizantha, to 30 between U. humidicola and U. ruziziensis. When all pairwise comparisons are considered, these InDels were mapped on genes rpoC2, ccsA, rbcL, rps18, and ndhK.
The chloroplast gene rpoC2 presented the largest numbers of cpSSRs, SNPs and InDels. This gene codes for the β” subunit of RNA polymerase, and is well known as a hotspot of structural and sequence variation in chloroplasts of grass species [78]. Recently, PCR markers based on rpoC2 structural variation were developed for differentiation and identification of species used in commercial food products [38]. InDels detected in Urochloa chloroplasts could be easily deployed as markers for species differentiation and identification. These would be useful, for instance, for identification of accessions in germplasm collections, and confirmation of hybridizations in breeding programs.
Phylogenetic analyses
Phylogenetic analysis using plastid nucleotide sequences of 30 species of Poaceae resulted in trees with well supported clades. The MP tree was built using 16,322 parsimony-informative characters, and the score of the best MP tree was 57,653. The best scoring ML tree had a Likelihood of - 526,037.12. The BI analysis showed that runs reached stationarity, and convergence diagnostics showed that parameters were properly sampled, with most of the Estimated Sample Size (ESS) values above 200. Tree topologies for MP, ML, and BI analyses were identical, and results are presented and discussed using the best scoring ML tree (Fig. 3) including support values for each method when they were below 100%.
Fig. 3.
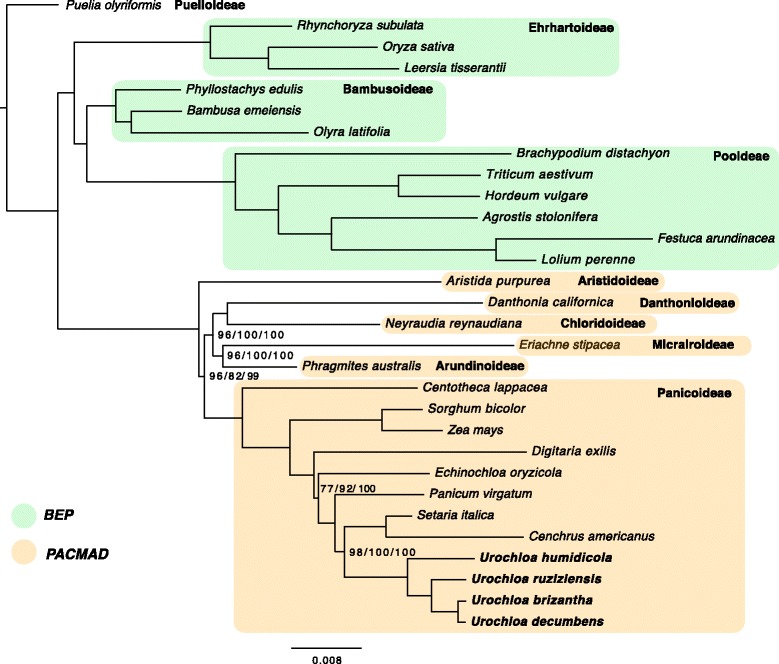
Maximum-likelihood tree based on complete chloroplast sequences of 30 species of Poaceae. Unlabeled nodes had supporting values of 100%. Where noted, supporting values are shown in the following order: MP/ML/BI
Using Puelia olyriformis as an outgroup, the split between the BEP and PACMAD clades was clearly observed with 100% support. Puelia is one of two genera belonging to the Puelioideae, a deeply diverging grass subfamily endemic to tropical forests of West Africa [79]. Nodes were strongly supported with bootstrap values above 90%, with the following exceptions: (i) the MP tree showed a bootstrap value of 72% for divergence of Echinochloa oryzicola from Panicum, Setaria, Cenchrus and Urochloa; (ii) support for the relationship of Panicoideae to other members of the PACMAD clade was 82% in the ML tree. Relationships in the PACMAD clade were in agreement with those found by the Grass Phylogeny Working Group II [80]. In summary, Aristidoideae species were shown as the first diverging clade in PACMAD, sister to other subfamilies, and followed by Panicoideae. In a recent study using fully assembled plastome sequences [73], Panicoideae species were grouped as a sister clade to all other PACMAD grasses (with Aristidoideae being sister to the CMAD clade), but results were not statistically different from what was described by GPWG II. Taxon sampling in our study is small and may lead to artifactual groups [73]. However, increase in character sampling from the use of full chloroplast genome sequences results in strong support of phylogenetic relationships [73]. This is evident from the agreement with topologies of larger studies such as the one performed by GPWG II, in addition to strong support values and consistency between different phylogenetic analysis methods.
Inside Paniceae, Digitaria exilis (subtribe Anthephorinae) was the earliest diverging species, followed by Echinochloa oryzicola (subtribe Boivinellinae), and Panicum virgatum (subtribe Panicinae). Finally, Setaria italica and Cenchrus americanus (both members of subtribe Cenchrinae) appeared in a sister group to the four Urochloa species (subtribe Melinidinae). These results are also in agreement with those found by recent phylogenies [67, 80, 81], and confirm that the use of full plastome sequences can lead to well supported and consistent phylogenetic reconstructions.
Grouping of the four Urochloa species was consistent with what was found in a previous study [5] using rpl16/trnL intron/trnL-F spacer/ndhF sequences: U. ruziziensis, U. brizantha, and U. decumbens were grouped in a strongly supported clade, while U. humidicola was shown as a sister taxon. In another study using rpl16 and ndhF, U. brizantha and U. decumbens were also found to be closely related, and separated from U. humidicola and U. dictyoneura [82]. Interestingly, the first published molecular phylogeny for Urochloa and Brachiaria, using ITS nuclear ribosomal DNA analysis [6], showed different results: U. brizantha and U. ruziziensis were included in a clade with U. comata and U. dura, while U. decumbens was included in a group with U. subulifolia, Melinis repens, and U. eruciformis. These relationships were maintained when molecular and morphological data were combined [6]. Contrasting these findings, morphological analysis alone had included U. ruziziensis, U. brizantha, and U. decumbens in a group with U. eminii, U. dura, and U. oligobrachiata [7].
The grouping of U. ruziziensis, U. decumbens, and U. brizantha is also consistent with their belonging to an agamic complex-a group of species considered as being distinct, and reproductively isolated by ploidy levels and apomixis [7, 83, 84] (see next section). This reproductive barrier, however, can be overcome by polyploidization of sexual diploids, allowing inter-specific hybrid production, a strategy that is currently applied in Urochloa breeding programs in Brazil and at the International Center for Tropical Agriculture (CIAT, Colombia) [7, 85, 86]. Results presented in the previous section regarding the presence of SNPs and InDels between these species pointed in the same direction. The number of inter-specific structural and sequence polymorphisms also indicated that U. ruziziensis is more closely related to U. brizantha than to U. humidicola, and that chloroplast sequence similarity between U. brizantha and U. decumbens is high, given the lower number of chloroplast SNPs and InDels found between these two species.
Divergence estimates
Divergence time estimates were based on a single calibration point at the BEP-PACMAD node using phytolith data, which suggests that all major grass subfamilies had diverged by 35 mya [66]. Results of divergence dates for some of the observed clades as well as the upper and lower bounds of the 95% highest posterior density (HPD) intervals are shown on Table 4. A complete chronogram is shown in Fig. 4. The estimated divergence date for Puelioideae was 54.1 [35.22, 92.11] mya, and the BEP-PACMAD divergence date was 51.3 [35, 86.8] mya. These results are in agreement with those found using the same phytolith calibration for BEP-PACMAD, and a combination of plastid ndhF and nuclear phyB sequence alignments [67]. Their divergence estimates for Puelioideae and for the BEP-PACMAD divergence were 52.6 mya, and 49 mya, respectively. Another recent study estimated the age of BEP-PACMAD to be 54.9 mya [87]. The estimated time of divergence for the crown PACMAD node was 38.1 [21.65, 65.71] mya, and is also in agreement with recent studies [67, 73, 87].
Table 4.
Times of divergence of Poaceae clades based on BEAST analysis of complete chloroplast sequences, with a calibration for BEP-PACMAD divergence (exponential distribution, mean 20.0, offset 35.0)
| Clade | Age (mya) | 95% HPD Lower Bound | 95% HPD Upper Bound |
|---|---|---|---|
| Puelioideae | 54.1 | 35.22 | 92.11 |
| BEP-PACMAD divergence | 51.3 | 35 | 86.8 |
| Aristida - Crown PACMAD | 38.01 | 21.65 | 65.71 |
| CMAD | 33.71 | 18.36 | 58.48 |
| Panicoideae | 31.42 | 17.26 | 54.8 |
| Urochloa – Setaria | 15.37 | 6.66 | 27.82 |
| Urochloa humidicola | 9.46 | 3.94 | 17.35 |
| Urochloa ruziziensis | 5.67 | 2.04 | 10.79 |
| U. decumbens – U. brizantha | 1.6 | 0.36 | 3.43 |
Fig. 4.
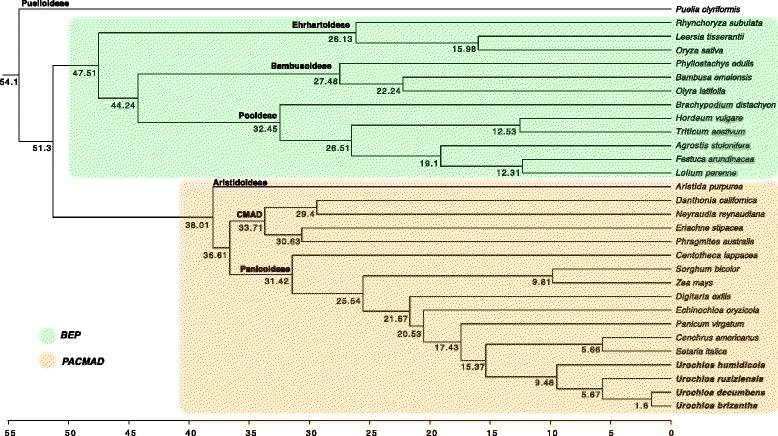
Divergence times of 30 species of Poaceae based on complete chloroplast sequences. Values at nodes indicate divergence dates in millions of years. The four Urochloa species are shown in bold
With our taxon sampling, the date of the Setaria-Urochloa divergence was estimated at 15.37 [6.66, 27.82] mya. Divergence of U. humidicola from a common ancestor with the three other Urochloa species was estimated at 9.46 [3.94, 17.35] mya. In the clade composed by U. ruziziensis, U. decumbens, and U. brizantha, U. ruziziensis would have diverged by 5.67 [2.04, 10.79] mya, followed by a recent divergence event between U. brizantha and U. decumbens around 1.6 [0.36, 3.43] mya. The Urochloa clade had an estimated date of origin at 7.2 ± 2.2 mya in a previous study [67], which overlaps with the results found in our analysis. Our results place the origins of U. humidicola and U. ruziziensis separation in the Miocene-Pliocene boundary, and the split between U. brizantha and U. decumbens in the Pleistocene.
U. ruziziensis, U. decumbens and U. brizantha have traditionally been considered members of a single agamic complex [7, 83, 84] - a group of species that includes sexual diploids and polyploids among facultative or obligate apomicts, originated from hybridizations among sexual diploid and polyploid members [88]. These hybridizations would initially take place among sexual diploids, generating hybrids at different ploidy levels [88]. Gene exchange would still be possible for sexual triploids and tetraploids, but asexual reproduction in higher ploidy levels would lead to reproductive isolation and the occurrence of microspecies with discontinuous morphological variation. Indeed, while most accessions of U. brizantha and U. decumbens are tetraploid and predominantly apomict, a few diploid sexual accessions of U. decumbens can be found in germplasm collections [89–91] and at least one sexual diploid accession of U. brizantha is available [92, 93]. One hexaploid sexual accession of U. humidicola has also been reported [94].
The similarity between chloroplast genomes of U. brizantha and U. decumbens is striking. Their cpDNA sizes differ by just 1 bp, and the cpDNA polymorphism (SNPs, InDels, SSRs) detected on coding and intergenic regions is smaller than in pairwise comparisons with U. ruziziensis and U. humidicola. The average divergence time between U. brizantha and U. decumbens is estimated to be recent (1.6 mya). Given the high sequence similarity between their cpDNA genomes, these combined data would indicate that a single polyploidization event took place to establish the U. brizantha and U. decumbens lineages. However, complementary analysis of cpDNA sequence data of germplasm accessions of both species would be necessary to confirm this hypothesis.
The taxonomic complexity of the Urochloa genus is also characteristic of forage grass species in general [95]. Hybridization and allopolyploidization are probably common processes in Urochloa, leading to reticulate evolution events and to potential incongruences between nuclear and chloroplast phylogenies [5, 96, 97]. A recent paper on the phylogeny of photosynthesis in Paniceae using a combination of chloroplast, mitochondrial and nuclear rDNA found that phylogenies from different types of markers did differ in certain areas of the trees [81]. In order to further investigate taxonomic relationships between the species described here, the inclusion of accessions of U. brizantha and U. decumbens with different ploidy levels, especially the diploids, would be important. In addition to larger taxon sampling, a robust nuclear phylogeny [5] would be necessary to properly identify the most likely parent species of Urochloa polyploids used as forages.
Conclusions
Use of low-coverage Illumina sequencing allowed the successful assembly and annotation of plastid genomes in four species of Urochloa extensively used as forages in the tropics (U. ruziziensis, U. brizantha, U. decumbens and U. humidicola). Comparative analyses of these chloroplast genomes allowed the identification of sequence and structural polymorphisms that will be useful for future genetic studies in Urochloa species. Results were consistent with previous phylogenies that group U. ruziziensis, U. brizantha and U. decumbens in a well-supported clade. U. brizantha and U. decumbens chloroplast sequences are highly similar and show reduced SNP, InDel and SSR polymorphism as compared to U. ruziziensis and U. humidicola. Future phylogenetic studies based on complete plastid sequences should include diploid samples of U. decumbens and U. brizantha, in addition to nuclear markers that could provide a better understanding of relationships between these species. The increased throughput and reduced costs of next-generation sequencing technologies bring the opportunity for the execution of phylogenetic studies based on either complete or large fragments of plastids, including a high number of taxa.
Additional files
List of Poaceae species with available plastid sequences used in phylogenetic analyses in this study, their respective tribes, and NCBI accession codes. (PDF 72 kb)
XML input file used for analysis in BEAST. (TXT 3981 kb)
Sequencing and de novo assembly metrics of chloroplast genomes of four Urochloa species. (PDF 13 kb)
Annotation of chloroplast microsatellites in four Urochloa species. Genome locations are based on the plastid genome of U. brizantha. The table includes locations of cpSSRs relative to annotated genes, types of regions (exon, intron, intergenic), repeat motifs, physical position in bp, polymorphism information, and number of repeats in U. decumbens, U. ruziziensis, and U. humidicola. (PDF 44 kb)
Annotation of SNPs located in genes in six pairwise comparisons between Urochloa species. The table includes genes, mutations, and SNP positions in bp. (PDF 46 kb)
Total number of InDels, their effects, and genome locations in six pairwise comparisons between Urochloa species. InDels located in genes are listed with their respective variations (insertion/deletion) and genome positions. (PDF 23 kb)
Acknowledgements
We thank Ediene G. Gouvea, Ana Luisa S. Azevedo, Claudio Takao Karia, Marcelo Ayres Carvalho and Fausto de Souza Sobrinho for providing support to this work.
Funding
This research was sponsored by EMBRAPA Macroprograma 2 - Grant # 02.12.02.002.00.00.
Availability of data and materials
The resulting MAFFT alignment matrix and consensus trees for MP, ML, BI, and divergence time estimation analyses are available at TreeBASE (http://treebase.org) under study ID 21243. The XML input file for BEAST is available as Additional file 2. Annotated plastid genomes generated in this study are available at NCBI under accession numbers NC_030066, NC_030067, NC_030068, and NC_030069. Scripts in R and perl are available on GitHub (https://github.com/marcopessoa/bioinfo-scripts). Accession codes for plastid sequences of Poaceae species used in this study are available on Additional file 1.
Abbreviations
- BI
Bayesian inference
- CIAT
Centro Internacional de Agricultura Tropical (International Center of Tropical Agriculture)
- cpDNA
chloroplast DNA
- cpSSR
chloroplast simple sequence repeat
- CTAB
cetyl trimethylammonium bromide
- ESS
estimated sample size
- GPWG
Grass phylogeny working group
- InDel
Insertion-Deletion
- IRa
Inverted repeat A
- IRb
Inverted repeat B
- ITS
internal transcribed spacer
- LSC
large single copy
- MCMC
Markov chain Monte Carlo
- Mha
million hectares
- ML
maximum likelihood
- MP
maximum parsimony
- MRCA
most recent common ancestor
- Mya
million years ago
- PCR
polymerase chain reaction
- rDNA
ribosomal DNA
- SMRT
single-molecule real-time
- SNP
single-nucleotide polymorphism
- SNV
single nucleotide variant
- SSC
small single copy
- SSR
simple sequence repeat
- TBR
tree-bisection-reconnection
Authors’ contributions
MPF and AMM performed DNA extractions, assembly and annotation of plastid genomes, sequence alignments, comparative analysis of plastid genomes, phylogenetic analysis, estimation of times of divergence, and drafted the manuscript. MEF allocated resources, designed the experiments, obtained the sequencing data, supervised the study and drafted the manuscript. All authors read and contributed written sections of the final manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study has been conducted in accordance with the Brazilian legislation (Law 13.123–5/20/2015) and the “Convention on the Trade in Endangered Species of Wild Fauna and Flora”. The selfed clone of U. ruziziensis cv. Kennedy (FSS-1) was selected from commercial seeds and belongs to Embrapa’s forage breeding program, Embrapa Gado de Leite (Dairy Cattle), Juiz de Fora, MG. All other plant accessions used in the study are commercial varieties of U. brizantha, U. decumbens and U. humidicola. They belong to the Brachiaria germplasm collection and have been deposited at the long-term Germplasm Collection of Embrapa (BGEN), Embrapa Recursos Genéticos e Biotecnologia (Genetic Resources and Biotechnology), Brasília, DF, Brazil. U. brizantha cv. Marandu is deposited under code BRA-000591, U. decumbens cv. Basilisk under code BRA-001058, and U. humidicola cv. Tupi under BRA-005118. BGEN is authorized by resolution n° 073, DOU 13/09/2004 section 1, page 54, Process n° 02000.001348/2004–81, Genetic Heritage Management Council (CGEN). The experimental research described here complies with institutional, national, and international guidelines. Seeds and cuttings can be available by request. A voucher specimen of U. humidicola cv. Tupi is available at Embrapa Genetic Resources and Biotechnology Herbarium (CEN) under accession number CEN 11796. This specimen was identified by Dr. José Francisco Montenegro Valls.
Consent for publication
Not applicable. This publication does not include details, images, or videos relating to an individual person.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3904-2) contains supplementary material, which is available to authorized users.
Contributor Information
Marco Pessoa-Filho, Email: marco.pessoa@embrapa.br.
Alexandre Magalhães Martins, Email: mm.alexandre@gmail.com.
Márcio Elias Ferreira, Phone: 01-301-504-5070, Email: marcio.ferreira@embrapa.br, Email: marcio.ferreira@ars.usda.gov.
References
- 1.Barcellos AO, Vilela L, Lupinacci AV. Produção animal a pasto: desafios e oportunidades. Encontro nacional do boi verde: a pecuaria sustentável. 2001;
- 2.Strassburg BBN, Latawiec AE, Barioni LG, Nobre CA, da Silva VP, Valentim JF, Vianna M, Assad ED. When enough should be enough: improving the use of current agricultural lands could meet production demands and spare natural habitats in Brazil. Glob Environ Chang. 2014;28:84–97. doi: 10.1016/j.gloenvcha.2014.06.001. [DOI] [Google Scholar]
- 3.IBGE . Pesquisa Pecuária Municipal (1974–2010) 2010. [Google Scholar]
- 4.Miles JW, Maass BL. Valle CB. Biology, Agronomy and improvement. CIAT: Brachiaria; 1996. [Google Scholar]
- 5.Salariato DL, Zuloaga FO, Giussani LM, Morrone O. Molecular phylogeny of the subtribe Melinidinae (Poaceae: Panicoideae: Paniceae) and evolutionary trends in the homogenization of inflorescences. Mol Phylogenet Evol. 2010;56:355–369. doi: 10.1016/j.ympev.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Torres González AM, Morton CM. Molecular and morphological phylogenetic analysis of Brachiaria and Urochloa (Poaceae) Mol Phylogenet Evol. 2005;37:36–44. doi: 10.1016/j.ympev.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Valle CB, Savidan YH. Genetics, cytogenetics and reproductive biology of Brachiaria. In: Miles JW, Maass BL, Valle CB. Brachiaria: Biology, Agronomy and improvement Cali: CIAT; 1996. p. 147–163.
- 8.Asano T, Tsudzuki T, Takahashi S, Shimada H, Kadowaki K. Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res. 2004;11:93–99. doi: 10.1093/dnares/11.2.93. [DOI] [PubMed] [Google Scholar]
- 9.Diekmann K, Hodkinson TR, Wolfe KH, van den Bekerom R, Dix PJ, Barth S. Complete chloroplast genome sequence of a major Allogamous forage species, perennial ryegrass (Lolium perenne L.) DNA Res. 2009;16:165–176. doi: 10.1093/dnares/dsp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shahid Masood M, Nishikawa T, Fukuoka S, Njenga PK, Tsudzuki T, Kadowaki K. The complete nucleotide sequence of wild rice (Oryza nivara) chloroplast genome: first genome wide comparative sequence analysis of wild and cultivated rice. Gene. 2004;340:133–139. doi: 10.1016/j.gene.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Moore MJ, Dhingra A, Soltis PS, Shaw R, Farmerie WG, Folta KM, Soltis DE. Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol. 2006;6:17. doi: 10.1186/1471-2229-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y-J, Ma P-F, Li D-Z. High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae) PLoS One. 2011;6 doi: 10.1371/journal.pone.0020596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cronn R, Liston A, Parks M, Gernandt DS, Shen R, Mockler T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkn502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atherton RA, McComish BJ, Shepherd LD, Berry LA, Albert NW, Lockhart PJ. Whole genome sequencing of enriched chloroplast DNA using the Illumina GAII platform. Plant Methods. 2010;6:22. doi: 10.1186/1746-4811-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Y-Y, Matzke AJM, Matzke M. Complete sequence and comparative analysis of the chloroplast genome of coconut palm (Cocos nucifera) PLoS One. 2013;8 doi: 10.1371/journal.pone.0074736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hand ML, Spangenberg GC, Forster JW, Cogan NOI. Plastome Sequence Determination and Comparative Analysis for Members of the Lolium-Festuca Grass Species Complex. G3. 2013;3(4):607–616. [DOI] [PMC free article] [PubMed]
- 17.Young HA, Lanzatella CL, Sarath G, Tobias CM. Chloroplast genome variation in upland and lowland switchgrass. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniell H, Kumar S, Dufourmantel N. Breakthrough in chloroplast genetic engineering of agronomically important crops. Trends Biotechnol. 2005;23:238–245. doi: 10.1016/j.tibtech.2005.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan I-C, Liao D-C, Wu F-H, Daniell H, Singh ND, Chang C, Shih M-C, Chan M-T, Lin C-S. Complete chloroplast genome sequence of an orchid model plant candidate: Erycina pusilla apply in tropical Oncidium breeding. PLoS One. 2012;7 doi: 10.1371/journal.pone.0034738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. P Natl Acad Sci USA. 2010;107:4623–4628. doi: 10.1073/pnas.0907801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Givnish TJ, Ames M, JR MN, MR MK, Steele PR, de Pamphilis CW, Graham SW, Pires JC, Stevenson DW, Zomlefer WB, Briggs BG, Duvall MR, Moore MJ, Heaney JM, Soltis DE, Soltis PS, Thiele K, Leebens-Mack JH. Assembling the tree of the monocotyledons: Plastome sequence phylogeny and evolution of Poales. Ann Missouri Bot Gard. 2010;97:584–616. doi: 10.3417/2010023. [DOI] [Google Scholar]
- 22.Shimada H, Sugiura M. Fine structural features of the chloroplast genome: comparison of the sequenced chloroplast genomes. Nucleic Acids Res. 1991;19:983–995. doi: 10.1093/nar/19.5.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer JD. Comparative organization of chloroplast genomes. Ann Rev Genet. 1985;19:325–354. doi: 10.1146/annurev.ge.19.120185.001545. [DOI] [PubMed] [Google Scholar]
- 24.Palmer JD, Stein DB. Conservation of chloroplast genome structure among vascular plants. Curr Genet. 1986;10:823–833. doi: 10.1007/BF00418529. [DOI] [Google Scholar]
- 25.Kapralov MV, Filatov DA. Widespread positive selection in the photosynthetic Rubisco enzyme. BMC Evol Biol. 2007;7:73. doi: 10.1186/1471-2148-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham SW, Olmstead RG. Utility of 17 chloroplast genes for inferring the phylogeny of the basal angiosperms. Am J Bot. 2000;87:1712–1730. doi: 10.2307/2656749. [DOI] [PubMed] [Google Scholar]
- 27.Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J, Siripun KC, Winder CT, Schilling EE, Small RL. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot. 2005;92:142–166. doi: 10.3732/ajb.92.1.142. [DOI] [PubMed] [Google Scholar]
- 28.Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94:275–288. doi: 10.3732/ajb.94.3.275. [DOI] [PubMed] [Google Scholar]
- 29.Lutz KA, Wang W, Zdepski A, Michael TP. Isolation and analysis of high quality nuclear DNA with reduced organellar DNA for plant genome sequencing and resequencing. BMC Biotechnol. 2011;11:54. doi: 10.1186/1472-6750-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaver JM, Oldenburg DJ, Bendich AJ. Changes in chloroplast DNA during development in tobacco, Medicago truncatula, pea, and maize. Planta. 2006;224:72–82. doi: 10.1007/s00425-005-0195-7. [DOI] [PubMed] [Google Scholar]
- 31.Birky CW. The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms, and models. Ann Rev Genet. 2001;35:125–148. doi: 10.1146/annurev.genet.35.102401.090231. [DOI] [PubMed] [Google Scholar]
- 32.Ravi V, Khurana JP, Tyagi AK, Khurana P. An update on chloroplast genomes. Plant Syst Evol. 2007;271:101–122. doi: 10.1007/s00606-007-0608-0. [DOI] [Google Scholar]
- 33.Vendramin GG, Lelli L, Rossi P, Morgante M. A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol Ecol. 1996;5:595–598. doi: 10.1111/j.1365-294X.1996.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 34.Angioi SA, Desiderio F, Rau D, Bitocchi E, Attene G, Papa R. Development and use of chloroplast microsatellites in Phaseolus spp. and other legumes. Plant Biol. 2009;11:598–612. doi: 10.1111/j.1438-8677.2008.00143.x. [DOI] [PubMed] [Google Scholar]
- 35.Powell W, Morgante M, McDevitt R, Vendramin GG, Rafalski JA. Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. P Natl Acad Sci USA. 1995;92:7759–7763. doi: 10.1073/pnas.92.17.7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung J, Kim KH, Yang K, Bang K-H, Yang T-J. Practical application of DNA markers for high-throughput authentication of Panax ginseng and Panax quinquefolius from commercial ginseng products. J Ginseng Res. 2014;38:123–129. doi: 10.1016/j.jgr.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kane N, Sveinsson S, Dempewolf H, Yang JY, Zhang D, Engels JMM, Cronk Q. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am J Bot. 2012;99:320–329. doi: 10.3732/ajb.1100570. [DOI] [PubMed] [Google Scholar]
- 38.Moon J-C, Kim J-H, Jang CS. Development of multiplex PCR for species-specific identification of the Poaceae. Appl Biol Chem. 2016;59:201–207. doi: 10.1007/s13765-016-0155-x. [DOI] [Google Scholar]
- 39.Nock CJ, Waters DLE, Edwards MA, Bowen SG, Rice N, Cordeiro GM, Henry RJ. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol J. 2011;9:328–333. doi: 10.1111/j.1467-7652.2010.00558.x. [DOI] [PubMed] [Google Scholar]
- 40.Kersten B, Rampant PF, Mader M, Le Paslier M-C, Bounon R, Bérard A, Vettori C, Schroeder H, Leplé J-C, Fladung M. Genome Sequences of Populus tremula Chloroplast and Mitochondrion: Implications for Holistic Poplar Breeding. bioRxiv. 2016;035899. [DOI] [PMC free article] [PubMed]
- 41.Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- 42.Ferreira M, Grattapaglia D. Introdução ao uso de marcadores moleculares em análise genética. 3. Embrapa: Brasília; 1998. [Google Scholar]
- 43.Silva PI, Martins AM, Gouvea EG, Pessoa-Filho M, Ferreira ME. Development and validation of microsatellite markers for Brachiaria ruziziensis obtained by partial genome assembly of Illumina single-end reads. BMC Genomics. 2013;14:17. doi: 10.1186/1471-2164-14-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- 45.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 46.Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52:267–274. doi: 10.1007/s00294-007-0161-y. [DOI] [PubMed] [Google Scholar]
- 47.Brudno M, Do CB, Cooper GM, Kim MF, Davydov E. NISC comparative sequencing program, green ED, Sidow a. Batzoglou S LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA Genome Res. 2003;13:721–731. doi: 10.1101/gr.926603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W237–W239. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–1047. doi: 10.1093/bioinformatics/16.11.1046. [DOI] [PubMed] [Google Scholar]
- 50.MISA - MIcroSAtellite identification tool. http://pgrc.ipk-gatersleben.de/misa/. Accessed 1 July 2016.
- 51.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, Lu X. Using Drosophila melanogaster as a model for Genotoxic chemical mutational studies with a new program. SnpSift Front Genet. 2012;3 [DOI] [PMC free article] [PubMed]
- 54.Katoh K, Toh H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. 2010;26:1899–1900. doi: 10.1093/bioinformatics/btq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Towns J, Peterson GD, Roskies R, Scott JR, Wilkens-Diehr N, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, Hazlewood V, Lathrop S, Lifka D. XSEDE: accelerating scientific discovery. Comput Sci Eng. 2014;16:62–74. doi: 10.1109/MCSE.2014.80. [DOI] [Google Scholar]
- 56.Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science Gateway for inference of large phylogenetic trees. Gateway Computing Environments Workshop (GCE) 2010;2010:1–8. [Google Scholar]
- 57.Katoh K, Misawa K. Kuma K ichi. Miyata T MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 59.Yang Z, Rannala B. Bayesian phylogenetic inference using DNA sequences: a Markov chain Monte Carlo method. Mol Biol Evol. 1997;14:717–724. doi: 10.1093/oxfordjournals.molbev.a025811. [DOI] [PubMed] [Google Scholar]
- 60.Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). 2002.
- 61.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 62.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.FigTree. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 1 July 2016.
- 65.Paradis E, Claude J, Strimmer K. APE: analyses of Phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 66.Strömberg CAE. Decoupled taxonomic radiation and ecological expansion of open-habitat grasses in the Cenozoic of North America. P Natl Acad Sci USA. 2005;102:11980–11984. doi: 10.1073/pnas.0505700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vicentini A, Barber JC, Aliscioni SS, Giussani LM, Kellogg EA. The age of the grasses and clusters of origins of C4 photosynthesis. Glob Chang Biol. 2008;14:2963–2977. doi: 10.1111/j.1365-2486.2008.01688.x. [DOI] [Google Scholar]
- 68.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10 doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed Phylogenetics and dating with confidence. PLoS Biol. 2006;4 doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tracer v1.6. http://beast.bio.ed.ac.uk/Tracer. Accessed 1 July 2016.
- 71.Guisinger MM, Chumley TW, Kuehl JV, Boore JL, Jansen RK. Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J Mol Evol. 2010;70:149–166. doi: 10.1007/s00239-009-9317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang R-J, Cheng C-L, Chang C-C, Wu C-L, Su T-M, Chaw S-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol Biol. 2008;8:36. [DOI] [PMC free article] [PubMed]
- 73.Cotton JL, Wysocki WP, Clark LG, Kelchner SA, Pires JC, Edger PP, Mayfield-Jones D, Duvall MR. Resolving deep relationships of PACMAD grasses: a phylogenomic approach. BMC Plant Biol. 2015;15:178. doi: 10.1186/s12870-015-0563-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu Q, Xiong G, Li P, He F, Huang Y, Wang K, Li Z, Hua J. Analysis of complete nucleotide sequences of 12 Gossypium chloroplast genomes: origin and evolution of Allotetraploids. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carbonell-Caballero J, Alonso R, Ibañez V, Terol J, Talon M, Dopazo J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol Biol Evol. 2015;32(8):2015–2035. doi: 10.1093/molbev/msv082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ishii T, Mori N, Ogihara Y. Evaluation of allelic diversity at chloroplast microsatellite loci among common wheat and its ancestral species. Theor Appl Genet. 2001;103:896–904. doi: 10.1007/s001220100715. [DOI] [Google Scholar]
- 77.Pessoa-Filho M, Azevedo ALS, Sobrinho FS, Gouvea EG, Martins AM, Ferreira ME. Genetic diversity and structure of Ruzigrass Germplasm collected in Africa and Brazil. Crop Sci. 2015;55:2736. doi: 10.2135/cropsci2015.02.0096. [DOI] [Google Scholar]
- 78.Cummings MP, King LM, Kellogg EA. Slipped-strand mispairing in a plastid gene: rpoC2 in grasses (Poaceae) Mol Biol Evol. 1994;11:1–8. doi: 10.1093/oxfordjournals.molbev.a040084. [DOI] [PubMed] [Google Scholar]
- 79.Jones SS, Burke SV, Duvall MR. Phylogenomics, molecular evolution, and estimated ages of lineages from the deep phylogeny of Poaceae. Plant Syst Evol. 2014;300:1421–1436. doi: 10.1007/s00606-013-0971-y. [DOI] [Google Scholar]
- 80.Grass Phylogeny Working Group II New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol. 2012;193:304–312. doi: 10.1111/j.1469-8137.2011.03972.x. [DOI] [PubMed] [Google Scholar]
- 81.Washburn JD, Schnable JC, Davidse G, Pires JC. Phylogeny and photosynthesis of the grass tribe Paniceae. Am J Bot. 2015;102(9):1493–1505. doi: 10.3732/ajb.1500222. [DOI] [PubMed] [Google Scholar]
- 82.Salariato DL, Giussani LM, Morrone O, Zuloaga FO. Rupichloa, a new genus segregated from Urochloa (Poaceae) based on morphological and molecular data. Taxon. 2009;58(2):381–391. [Google Scholar]
- 83.Quero Carrillo AR, Enriquez Quiroz JF, Morales Nieto CR, Miranda JL. Apomixis importance for tropical forage grass selection and breeding. Review Rev Mex Cienc Pecu. 2010;1:25–42. [Google Scholar]
- 84.Lutts S, Ndikumana J, Louant BP. Fertility of Brachiaria ruziziensis in interspecific crosses with Brachiaria decumbens and Brachiaria brizantha: meiotic behaviour, pollen viability and seed set. Euphytica. 1991;57:267–274. doi: 10.1007/BF00039673. [DOI] [Google Scholar]
- 85.Jank L, Valle CB, Resende RMS. Breeding tropical forages. Crop Breed Appl Biot. 2011;11:27–34. doi: 10.1590/S1984-70332011000500005. [DOI] [Google Scholar]
- 86.Miles JW. Apomixis for Cultivar Development in Tropical Forage Grasses. Crop Sci. 2007;47 Suppl 3;S-238-S-249.
- 87.Christin P-A, Besnard G, Samaritani E, Duvall MR, Hodkinson TR, Savolainen V, Salamin N. Oligocene CO2 decline promoted C4 photosynthesis in grasses. Curr Biol. 2008;18:37–43. doi: 10.1016/j.cub.2007.11.058. [DOI] [PubMed] [Google Scholar]
- 88.Bayer RJ. Evolution and phylogenetic relationships of the Antennaria (Asteraceae: Inuleae) polyploid agamic complexes. Biol Zent. 1987;106:683–698. [Google Scholar]
- 89.Do Valle CB, Savidan YH. Apomixis and sexuality in Brachiaria decumbens Stapf. XVI International Grassland Congress. 1989:407–8.
- 90.Penteado MI de O, Sandos ACM dos, Rodrigues IF, do Valle CB, Seixas MAC, Esteves A. Determinação de ploidia e avaliação da quantidade de DNA total em diferentes espécies do gênero Brachiaria. Boletim de Pesquisa, 11. Embrapa Gado de Corte. 2000.
- 91.Naumova TN, Hayward MD, Wagenvoort M. Apomixis and sexuality in diploid and tetraploid accessions of Brachiaria decumbens. Sex Plant Reprod. 1999;12:43–52. doi: 10.1007/s004970050170. [DOI] [Google Scholar]
- 92.Araujo ACG, Falcão R, Carneiro VT de C. Seed abortion in the sexual counterpart of Brachiaria brizantha apomicts (Poaceae) Sex Plant Reprod. 2007;20:109–121. doi: 10.1007/s00497-007-0048-6. [DOI] [Google Scholar]
- 93.Do Valle CB, Glienke C. New sexual accessions in Brachiaria. Apomixis Newsl. 1991;3:11–13. [Google Scholar]
- 94.Jungmann L, Vigna BBZ, Paiva J, Sousa ACB. Do Valle CB, Laborda PR, Zucchi MI, DE Souza AP. Development of microsatellite markers for Brachiaria humidicola (Rendle) Schweick. Conservation Genet Resour. 2009;1:475–479. doi: 10.1007/s12686-009-9111-y. [DOI] [Google Scholar]
- 95.Clayton WD. Tropical grasses. In: McIvor JG, editor. Bray RA. Genetic Resources of Forage Plants Melbourne: CSIRO; 1983. pp. 38–46. [Google Scholar]
- 96.McBreen K, Lockhart PJ. Reconstructing reticulate evolutionary histories of plants. Trends Plant Sci. 2006;11:398–404. doi: 10.1016/j.tplants.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 97.Linder CR, Rieseberg LH. Reconstructing patterns of reticulate evolution in plants. Am J Bot. 2004;91:1700–1708. doi: 10.3732/ajb.91.10.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of Poaceae species with available plastid sequences used in phylogenetic analyses in this study, their respective tribes, and NCBI accession codes. (PDF 72 kb)
XML input file used for analysis in BEAST. (TXT 3981 kb)
Sequencing and de novo assembly metrics of chloroplast genomes of four Urochloa species. (PDF 13 kb)
Annotation of chloroplast microsatellites in four Urochloa species. Genome locations are based on the plastid genome of U. brizantha. The table includes locations of cpSSRs relative to annotated genes, types of regions (exon, intron, intergenic), repeat motifs, physical position in bp, polymorphism information, and number of repeats in U. decumbens, U. ruziziensis, and U. humidicola. (PDF 44 kb)
Annotation of SNPs located in genes in six pairwise comparisons between Urochloa species. The table includes genes, mutations, and SNP positions in bp. (PDF 46 kb)
Total number of InDels, their effects, and genome locations in six pairwise comparisons between Urochloa species. InDels located in genes are listed with their respective variations (insertion/deletion) and genome positions. (PDF 23 kb)
Data Availability Statement
The resulting MAFFT alignment matrix and consensus trees for MP, ML, BI, and divergence time estimation analyses are available at TreeBASE (http://treebase.org) under study ID 21243. The XML input file for BEAST is available as Additional file 2. Annotated plastid genomes generated in this study are available at NCBI under accession numbers NC_030066, NC_030067, NC_030068, and NC_030069. Scripts in R and perl are available on GitHub (https://github.com/marcopessoa/bioinfo-scripts). Accession codes for plastid sequences of Poaceae species used in this study are available on Additional file 1.