

Abstract
Although ample evidence links hepatic lipid accumulation with hepatic insulin resistance, the mechanistic basis of this association is incompletely understood and controversial. Diacylglycerols and ceramides have emerged as the two best-studied putative mediators of lipid-induced hepatic insulin resistance. Both lipids were first associated with insulin resistance in skeletal muscle, and subsequently hypothesized to mediate insulin resistance in liver. However, the putative roles for diacylglycerols and ceramides in hepatic insulin resistance have proved more complex than originally imagined, with various genetic and pharmacologic manipulations yielding a vast and occasionally contradictory trove of data to sort through. In this review, we examine the state of this field, turning a critical eye toward both diacylglycerols and ceramides as putative mediators of lipid-induced hepatic insulin resistance.
Keywords: Insulin resistance, ectopic lipid, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), insulin receptor kinase, protein kinase C epsilon, ceramide
Lipid-Induced Hepatic Insulin Resistance
The World Health Organization estimated the 2016 global prevalence of type 2 diabetes (T2D) at a staggering 8.5%: more than 1 in 12 people worldwide [1]. The liver is the source of ~90% of endogenous glucose production, and increased hepatic gluconeogenesis is the proximate cause of the fasting hyperglycemia that defines T2D [2]. In addition to the increase in fasting hepatic glucose production (HGP), insulin suppression of HGP is impaired in T2D [3]. This is generally considered to reflect hepatic insulin resistance (see Glossary), although indirect (i.e., extrahepatic) insulin action also contributes substantially to acute insulin suppression of HGP [4]. The major direct acute effect of insulin on hepatocellular glucose metabolism is the stimulation of glycogen synthesis, and hepatic insulin resistance to glycogen metabolism appears to manifest as a damping of the normal oscillations produced by fasting and feeding: hepatic glycogen stores during fasting are decreased and both glycogen synthesis and glycogenolysis are diminished in T2D [2,3,5–7]. Hepatic insulin resistance is reversible: in one study, insulin resistance to suppression of HGP was reversed by modest (~8 kg) weight loss, and this was associated with normalization of fasting HGP and fasting glycemia [8]. Yet for many obese insulin resistant nondiabetic subjects, sustained weight loss is elusive and the eventual decompensation of the endocrine pancreas in the face of progressive insulin resistance results in overt T2D. The contribution of hepatic insulin resistance to the overall pathophysiology of T2D is thus significant, and so the question of why hepatic insulin resistance develops is of great interest.
The strong association of hepatic insulin resistance and hepatic steatosis is highly reproducible. Hepatic steatosis, or nonalcoholic fatty liver disease (NAFLD), is present in about 70% of type 2 diabetics [9,10] and nearly all obese type 2 diabetics [11] and is a particularly strong predictor of insulin resistance [12–15]. For example, IHTG is better correlated with insulin resistance than visceral adipose tissue volume [16], and increased body mass index is not associated with increased insulin resistance unless a parallel increase in IHTG is present [17]. Near-normalization of intrahepatic triglyceride (IHTG) content in type 2 diabetic subjects by modest weight loss restored sensitivity of HGP to insulin without significant improvements in skeletal muscle insulin resistance [8]. The presence of NAFLD predicts future T2D risk, and improvement of NAFLD decreases future risk of T2D [18]. Recent human genetic data indicate that impaired adipose tissue storage capacity is an important contributor to whole-body insulin resistance as measured by fasting insulin level, consistent with the ‘ectopic lipid’ model of hepatic insulin resistance [19]. These clinical observations have invited sustained investigation into whether hepatic insulin resistance and hepatic steatosis are causally related.
Does hepatic steatosis cause hepatic insulin resistance? Human and rodent clinical studies are broadly consistent with this hypothesis in that IHTG and hepatic insulin resistance are correlated and nearly always move in the same direction upon clinical, dietary, or pharmacologic interventions [8,18,20,21]. For example, specific reversal of hepatosteatosis — by liver-targeted mitochondrial protonophore treatment in high-fat fed rats, by niclosamide-induced mitochondrial uncoupling in fat-fed mice, by modest weight loss in type 2 diabetic humans, by adipose transplantation in lipodystrophic mice, or by leptin treatment in lipodystrophic humans — dramatically reverses hepatic insulin resistance [8,20,22–26].
Triglyceride is not a signaling lipid, however, and the search for lipid moieties with a potential mechanistic role in hepatic insulin resistance has centered on two major lipid classes: diacylglycerol (DAG) and ceramide. In this review, we critically examine the substantial literature investigating DAGs and ceramides as putative mediators of lipid-induced hepatic insulin resistance. We attempt a synthesis of available research examining: 1) levels of DAGs and ceramides in human and rodent models of lipid-induced hepatic insulin resistance, 2) proposed molecular mechanisms for DAG- and ceramide-mediated hepatic insulin resistance, and 3) rodent models interrogating DAG- and ceramide-mediated hepatic insulin resistance. We conclude with a formal evaluation of DAG- and ceramide-induced hepatic insulin resistance using the Bradford Hill criteria for causality in biological processes.
Diacylglycerol in hepatic insulin resistance
Early clues to potential anti-insulin actions of diacylglycerol derived from studies of the DAG analog phorbol 12-myristate 13-acetate (PMA). Cultured hepatocytes treated with PMA displayed impairments in insulin receptor tyrosine kinase (IRK) activity and insulin-stimulated glycogen synthase activity [27,28]. Soon after, livers from Zucker obese rats were found to contain ~1.8-fold higher levels of sn-1,2-DAG content than lean control rats [29]. Because sn-1,2-DAG was a known bioactive signaling lipid, a simple hypothesis — impairment of insulin signaling by DAG activation of protein kinase C (PKC) isoforms was proposed, although the source of the DAGs remained unknown [30].
Association of DAG and hepatic insulin resistance
Evidence for increased hepatic diacylglycerol in models of lipid-induced hepatic insulin resistance is abundant. Because DAG is the synthetic precursor to triglyceride, IHTG correlates well with intrahepatic DAG if lipid handling pathways are genetically intact [31–36]. In one study, multiple putative mediators and pathways were systematically assessed in liver biopsies from obese non-diabetic subjects across a wide range of homeostatic model assessment of insulin resistance (HOMA-IR) levels [31]. Total hepatic DAG content was more strongly correlated with HOMA-IR than any other variable, including body mass index, long-chain fatty acyl CoA content, ceramide content, multiple markers of endoplasmic reticulum stress, c-Jun N-terminal kinase (JNK) phosphorylation, and multiple plasma inflammatory cytokine concentrations — though this study was limited by the availability of biopsy samples and was underpowered to definitively rule out effects of these other variables on HOMA-IR [31]. In another study that used suppression of HGP during hyperinsulinemic-euglycemic clamps as the measure of hepatic insulin resistance rather than HOMA-IR, intrahepatic DAG was again significantly correlated with hepatic insulin resistance [35]. In the third human study to measure intrahepatic DAG and HOMA-IR, four of five DAG species measured were positively correlated with HOMA-IR [37]. Furthermore, reversal of high-fat diet-induced hepatic insulin resistance is accompanied by decreased intrahepatic DAG in multiple rodent models including pharmacologic FGF-21 treatment, niclosamide (mitochondrial uncoupler) treatment, apolipoprotein A5 knockdown, low-dose 2,4-dinitrophenol treatment in multiple rodent models, acetyl CoA carboxylase inhibition, and estradiol treatment in ovariectomized female mice among others [20,23,24,26,33,34,38–42]. Measurements of intrahepatic DAG in human subjects after an intervention that reverses hepatic insulin resistance have not yet been reported, likely owing to the difficulty of obtaining liver biopsy samples outside the setting of surgery.
Mechanism of DAG-mediated hepatic insulin resistance
An attractive feature of the hypothesis that DAG mediates lipid-induced hepatic insulin resistance is the existence of a plausible mechanism for inhibition of insulin signaling — the aforementioned activation of PKC by sn-1,2-DAG. Systematic examination of all PKC isoforms expressed in liver revealed that activation of the ε isoform was particularly prominent in rats fed a 3-day high fat diet [20]. ASO knockdown of PKCε prevented hepatic insulin resistance in this model, and PKCε knockout mice are protected from glucose intolerance after 7 days of high fat feeding [43,44]. The protection from hepatic insulin resistance observed in PKCε ASO-treated mice appeared to be caused by protection from IRK inhibition, and recently insulin receptor (INSR) Thr1160 was identified as a PKCε substrate that may be responsible for this effect [43,45]. INSR Thr1160 phosphorylation destabilizes the active configuration of the IRK, impairing its tyrosine kinase activity and all downstream insulin action [45] (Figure 1). InsrT1150A mice genetically insusceptible to inhibition by PKCε were protected from high-fat diet-induced hepatic insulin resistance [45].
Figure 1. The DAG-PKCε-INSR axis in lipid-induced hepatic insulin resistance.
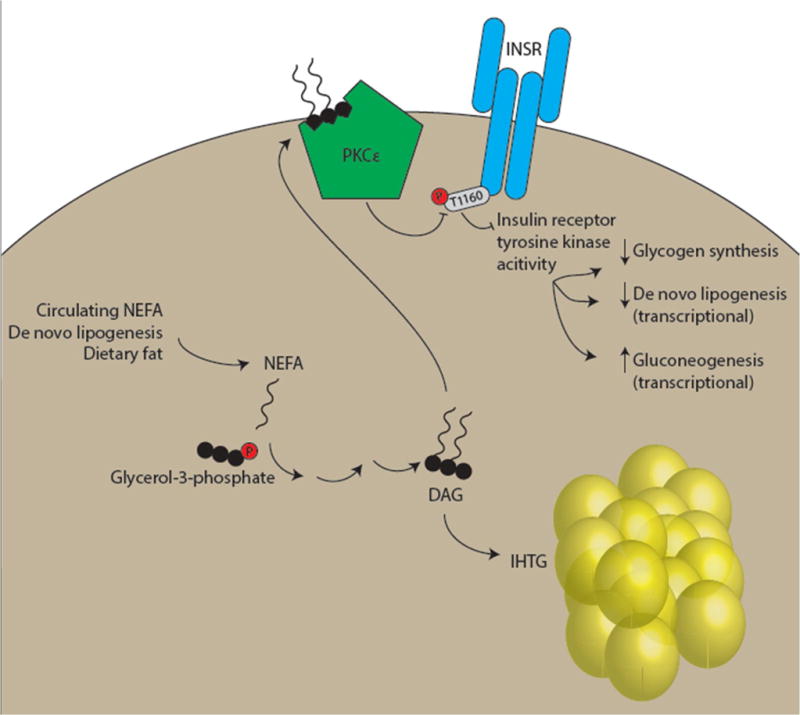
In subjects with increased intrahepatic triglyceride (IHTG), sn-1,2-diacylglycerol (DAG) accumulates. DAG activates protein kinase C (PKC) isoforms, and activation of the ε isoform (PKCε) is most consistently observed in insulin-resistant liver. PKCε phosphorylates insulin receptor (INSR) Thr1160, resulting in inhibition of INSR tyrosine kinase activity. All downstream arms of hepatocellular insulin signaling, including stimulation of net glycogen synthesis, transcriptional upregulation of de novo lipogenic genes, and transcriptional downregulation of gluconeogenic genes, are predicted to be affected by this mechanism.
Rodent models of DAG-mediated hepatic insulin resistance
Several genetically modified mouse models have been generated to examine the DAG hypothesis of lipid-induced hepatic insulin resistance. Mice with loss-of-function of mtGPAT, a major hepatic glycerol-3-phosphate acyltransferase, accumulated less intrahepatic DAG and triacylglycerol (TAG) than controls upon high-fat feeding, associated with protection from hepatic insulin resistance [46]. In mice overexpressing mtGPAT, the converse was true: increased hepatic DAG and hepatic insulin resistance on regular chow diet were observed [47]. There are two phosphatidic acid phosphatases, or lipins, which generate DAG. Knockdown of either lipin-1 or lipin-2 by shRNA in fat-fed mice decreased hepatic DAG and TAG and improved glucose tolerance, and adenoviral overexpression of lipin-2 resulted in the opposite phenotype: increased hepatic DAG and TAG accompanied by glucose intolerance [48,49]. Diacylglycerol acyltransferase (DGAT) synthesizes TAG from DAG. Knockdown of DGAT1 in rats does not alter liver triglyceride levels [50]. However, liver-specific overexpression of DGAT2 in mice leads to increased hepatic TAG and, unexpectedly, increased total hepatic DAG content [51,52]. This is associated with increased PKCε translocation and impaired suppression of HGP during hyperinsulinemic-euglycemic clamps [52]. Similarly, ASO knockdown of DGAT2 protected rats from high fat diet-induced increases in intrahepatic TAG, DAG, and PKCε translocation, and prevented hepatic insulin resistance [50].
As discussed above, intrahepatic DAG has been dissociated from hepatic insulin resistance in a few genetically modified mouse models of altered lipid handling, casting doubt on the DAG-PKCε-INSR hypothesis of lipid-induced hepatic insulin resistance [53]. There are several possible interpretations of these data. One possibility is that DAG is an excellent biomarker of hepatic insulin resistance but not a causal factor. Another reasonable possibility is that DAG-mediated hepatic insulin resistance can be overcome by one or more insulin-sensitizing factors in these models. A third possibility that is beginning to be explored is that only some DAG — specific chemical species, or in specific subcellular localizations, or as a product of specific lipid handling pathways — is capable of activating PKCε and inhibiting IRK activity. The possibility that specific DAG species trigger hepatic insulin resistance has not been systematically evaluated in humans, but advances in lipidomics may enable such studies. In one recent human study, four DAG species (32:1, 34:1, 36:2, 36:3) were significantly increased in a high HOMA-IR versus a low HOMA-IR group [37]. In kinase assays using DAG vesicles, PKCε does not appear to display a strong preference for specific DAG species [54]. However, in COS-7 cells, PKCε translocation was observed in response to treatment with tridecanoate, pentadecanoate, linoleate, arachidonate, and docosahexaenoate, but not other fatty acids including palmitate, stearate, oleate, and others [55]. The hypothesis that subcellular DAG compartmentation might modulate the relationship between hepatic DAG content and hepatic insulin resistance emerged from studies of mice with targeted knockdown of the adipose triglyceride lipase (ATGL) cofactor CGI-58 [56,57]. CGI-58 ASO-treated mice, with increased hepatic DAG content but preserved hepatic insulin sensitivity, displayed an unusual tendency to recruit PKCε to the lipid droplet [57]. It is intuitive that the critical subcellular compartment for the DAG-PKCε-INSR axis would be a membrane-associated compartment such as the plasma membrane, ER, or Golgi (though this has not been directly demonstrated), so this ‘sequestration’ of DAG and PKCε in the lipid droplet may account for the preserved hepatic insulin sensitivity in the CGI-58 ASO-treated mice as well as in other mice with defects in triglyceride mobilization (e.g., microsomal triglyceride transfer protein (MTTP) knockout mice [58]). A similar mechanism may also account for the improved glucose tolerance but increased intrahepatic DAG of mice treated with a monoacylglycerol acyltransferase 1 (MGAT1) ASO; these mice displayed decreased membrane-associated PKCε content [59]. A small percentage of humans with NAFLD may exhibit preserved hepatic insulin action through such a ‘sequestration’ mechanism. However, in this regard, it is interesting to note that in human liver biopsy samples, lipid droplet DAG content correlated better with HOMA-IR than membrane DAG content [31]. It is not obvious how to reconcile these results with the hypothesis that the membrane-associated compartment contains the ‘active’ DAG pool; however, it is possible that handling and storage of the human liver biopsy samples following biopsy may have contributed to this distribution. Measurement of DAG from both membrane-associated and cytosolic/lipid droplet compartments has become more common [23,31,45,57,59], but is not yet standard in the field [37,60,61]. Although the technique commonly employed is relatively crude (the membrane fraction includes all membranous organelles and the plasma membrane), it may be further refined in the future. It is interesting to note that cell-based studies have implicated the Golgi, mitochondrion, and plasma membrane as sites of PKCε translocation [62]. A final important consideration for the DAG-PKCε hypothesis is that triglyceride hydrolysis by ATGL in the hepatocyte preferentially produces sn-1,3-DAG, which is incapable of activating PKC [63,64]. Thus it is unlikely that DAG generated through this flux participates in the DAG-PKCε-INSR axis. Overall, the phenotypes of mice with genetically altered hepatocellular triglyceride synthesis have revealed important caveats to, but are generally consistent with, the DAG-PKCε-INSR hypothesis of lipid-induced hepatic insulin resistance. Such models have also highlighted the diversity of DAG within the hepatocyte. Although the most pathophysiologically relevant approach to measuring and reporting DAG content remains uncertain, it is unlikely to be the simple sum of all DAG species from all subcellular localizations.
Ceramides in hepatic insulin resistance
The 1990 study reporting increased sn-1,2-DAG concentrations in obese rat livers also observed modest, but significant, increases in hepatic ceramides [29]. Early investigations of ceramide-induced insulin resistance were performed primarily in skeletal muscle [65–67]. More recently, ceramides have been explored as putative mediators of hepatic insulin resistance as well [68–70]. We now examine available data concerning the role of ceramides in lipid-induced hepatic insulin resistance.
Association of ceramides and hepatic insulin resistance
Studies differ with respect to whether hepatic ceramide content is associated with hepatic insulin resistance in humans. In two studies of obese nondiabetic humans, hepatic ceramide content was not significantly associated with HOMA-IR [31] or insulin suppression of HGP [35]. However, a recent lipidomic study of obese humans observed strong associations between hepatic ceramides with HOMA-IR score [37]. This latter study is notable for its analysis of specific species of ceramide, rather than reporting the sum of several abundant species.
Are hepatic ceramides increased in rodent models of lipid-induced hepatic insulin resistance? In many cases, hepatic insulin resistance is not accompanied by increases in hepatic ceramides. In C57BL/6 mice, high fat feeding for 7 or 21 days induces hepatic insulin resistance, hepatic steatosis, and hepatic DAG accumulation [71]. At 7 days, no hepatic ceramide species were altered; at 21 days, 16:0 and 18:0 ceramides were unchanged, 20:0 and 22:0 ceramides were increased, and 24:1 and 24:0 ceramides were decreased [71]. Indeed, even chronic high-fat diet protocols of 8 to 12 weeks fail to increase total hepatic ceramides, a finding consistent across multiple mouse strains in most [72–74], though not all [61] studies. In Sprague-Dawley rats, high fat feeding (with either saturated fat- or unsaturated fat-based diets) for just 3 days is sufficient to induce hepatic insulin signaling defects but not to increase total hepatic ceramides [75]. Genetically obese ZDF rats did not display increased total hepatic ceramides compared to their lean littermates [76]. Mice with increased hepatocellular lipid uptake due to overexpression of human APOC3 displayed hepatic insulin resistance associated with increased hepatic DAG and TAG but unchanged total hepatic ceramide content [77]. In many rodent studies in which reversal of hepatosteatosis is accompanied by decreased hepatic DAG and improvements in hepatic insulin action, total hepatic ceramides are not decreased [24,26,33,34,38]. Additionally, several of the mouse models of altered hepatic triglyceride handling notable for dissociating hepatic steatosis and DAG accumulation from hepatic insulin resistance, such as CGI-58 ASO treated mice, microsomal triglyceride transfer protein (MTTP) knockout mice, and perilipin 5-overexpressing mice also display increased total hepatic ceramides but do not develop hepatic insulin resistance [56–58,78]. In these rodent models, hepatic ceramides are dissociated from lipid-induced hepatic insulin resistance.
Conversely, hepatic ceramides have been reported to correlate with hepatic insulin resistance in several rodent models. Mice with liver-specific overexpression of DGAT2 displayed hepatic insulin resistance associated with increased total hepatic ceramides, DAG, and TAG [52]. Wistar rats fed a five-week high-fat diet were reported to display increased total hepatic ceramides (and hepatic DAG) in concert with increased HOMA-IR [79]. Longer durations of high-fat feeding appear to be required to detect increased hepatic ceramides in mice. In one study, C57BL/6N mice fed HFD for 14 weeks displayed increased 14:0, 16:0, 18:0, 20:0, and 24:1 ceramides [68]. In another study, C57BL/6 mice fed HFD for 16–17 weeks displayed increased hepatic ceramides (primarily driven by increases in 16:0, 20:0, and 22:0 species) and glucose intolerance; fenretinide or salicylate treatment abrogated the increase in hepatic ceramides and modestly improved glucose tolerance — though both of these treatments have ceramide-independent metabolic effects: RBP4 blockade for fenretinide and IKKβ inhibition for salicylates) [80–83]. Leptin-deficient ob/ob mice were also reported to display increased total hepatic ceramides [84]. In rats, acute dexamethasone treatment increased total hepatic ceramides, impaired hepatocellular insulin signaling, and impaired insulin suppression of HGP during hyperinsulinemic-euglycemic clamps [76]. These effects were prevented by pre-treatment with the SPT inhibitor myriocin [76].
Myriocin has been described as the “workhorse” of the field of ceramide metabolism [85]. Yet, as with all pharmacologic inhibitors, interpretation of in vivo experiments employing myriocin requires caution. In an experiment designed to differentiate the effects of the ceramide precursor palmitate from other fatty acids, rats were infused with either saturated fat-rich lard oil or unsaturated fat-rich soy oil for six hours, and observed an increase in hepatic ceramides with only lard oil, as predicted [76]. Interestingly, both lipid infusions impaired insulin suppression of HGP, and myriocin prevented this impairment in both lard oil and soy oil-infused rats, even though hepatic ceramides were totally unaffected by myriocin treatment in the soy oil-infused group [76]. This finding suggests that myriocin may exert some ceramide-independent effects. Indeed, myriocin has dramatic effects on energy expenditure and weight gain in mice, confounding attempts to identify the proximate cause of myriocin-induced improvements in hepatic insulin action [74,86]. For example, myriocin reduced hepatic steatosis in ob/ob and high-fat fed mice, so decreased DAG/PKCε/INSR axis activation cannot be ruled out as a mechanism for improved hepatic insulin sensitivity in this model [86]. Although the insulin-sensitizing effects of myriocin have been dissociated from body weight changes in some reports [74,76], it is worth bearing in mind that even minor increases in energy expenditure, too subtle to manifest as weight loss, can have dramatic effects on hepatic and skeletal muscle diacylglycerol content and insulin sensitivity [23,24].
Mechanism of ceramide-mediated hepatic insulin resistance
Several mechanisms have been proposed to explain how ceramides might induce cellular insulin resistance (Figure 2). Early work in cultured myocytes and adipocytes suggested that ceramides impair insulin activation of AKT through two mechanisms: increased protein phosphatase-2A activity and impaired AKT translocation resulting from activation of atypical protein kinase C-ζ [87–90]. Of note, these mechanistic studies relied on cell-permeable short-chain ceramides with cytotoxic properties and often used at unphysiologic concentrations [85]. Interestingly, pharmacologic inhibition of PP2A in rats paradoxically worsened hepatic insulin resistance despite activation of AKT, suggesting that activation of PP2A would not be sufficient to induce hepatocellular insulin resistance [91]. Additionally, proximal insulin signaling is impaired in insulin resistant liver, which is difficult to reconcile with AKT as the main site for insulin resistance [43,92,93]. Alternatively, PKCζ activation of the fatty acid transporter CD36 has been proposed as a mechanism for ceramide-induced hepatic steatosis [61]. Such a mechanism has the advantage of linking hepatic ceramides to other putative mediators of lipid-induced hepatic insulin resistance. The metabolically active hormones FGF-21 and adiponectin have been proposed to mediate their beneficial effects through ceramides, adding intriguing layers of complexity to ceramide physiology [84]. The adiponectin receptor in particular has been shown to contain ceramidase activity [84]. Furthermore, intriguing links between ceramides and adipose inflammation, possibly through direct activation of the NLRP3 inflammasome [94,95], could provide another mechanism for ceramide-induced insulin resistance. Indeed, two of the mouse models genetically perturbing hepatic ceramide metabolism described later display decreased hepatic ceramides in concert with decreased adipose inflammation [61,68]. But none of these studies have updated our understanding of the molecular mechanism by which ceramides might themselves directly impair insulin signaling in a hepatocyte-autonomous manner. A complete mechanism for ceramide-induced hepatocellular insulin resistance is therefore still awaited.
Figure 2. Ceramides in lipid-induced hepatic insulin resistance.
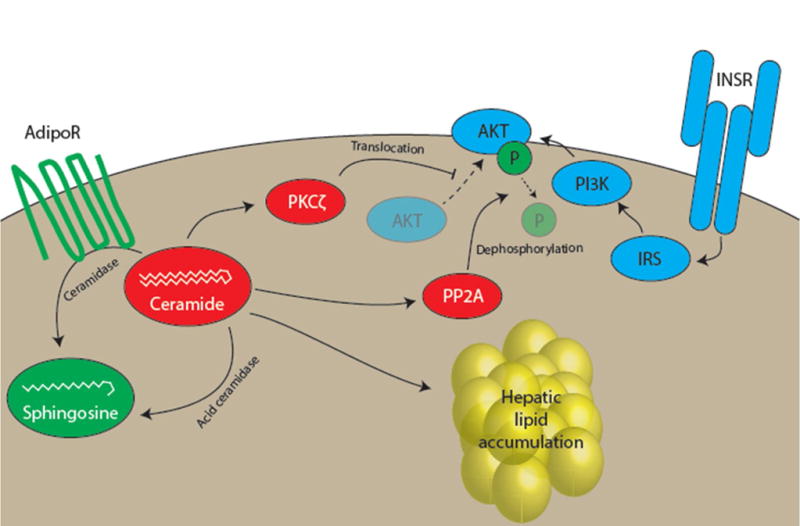
Studies in cultured cells have identified two putative direct mechanisms for ceramide-induced insulin resistance. In one, ceramide activation of protein kinase C-ζ (PKCζ) impairs translocation of AKT to the plasma membrane, preventing AKT from participating in insulin action. In the other, ceramide activation of protein phosphatase 2A leads to dephosphorylation and inactivation of AKT. The relevance of these mechanisms to hepatocellular insulin resistance has not been thoroughly investigated. Long-chain ceramides may also promote hepatic lipid accumulation. Promoting ceramide degradation, either through forced expression of acid ceramidase or through harnessing the intrinsic ceramidase activity of the adiponectin receptor (AdipoR) has been shown to improve multiple metabolic parameters in mice.
Rodent models of ceramide-mediated hepatic insulin resistance
Various mouse models genetically perturbing ceramide synthesis have been generated to examine the role of ceramides in hepatic insulin resistance. The earliest such study used mice heterozygous for deletion of the ceramide synthesis gene dihydroceramide desaturase 1 (Des1+/− mice) which displayed decreased total ceramides in several tissues, including liver, and decreased fasting HOMA-IR (although glucose tolerance was normal) [76]. In 2014, two groups reported on mice deficient in one of the six ceramide synthase (CerS) enzymes and advanced the hypothesis that C16:0 ceramides are particularly harmful [70]. CerS6 knockout mice were protected from the development of obesity when fed a high-fat diet, owing in part to increased BAT energy expenditure [68]. Interestingly, liver-specific CerS6 knockout mice displayed a selective decrease in hepatic C16:0 ceramides but also were protected from high-fat diet-induced obesity [68]. This body weight phenotype was predictably associated with improved glucose tolerance (though insulin tolerance was unchanged) [68]. Conversely, mice haploinsufficient for CerS2 displayed a compensatory increase in hepatic C16:0 ceramides [69]. The CerS2+/− mice gained weight normally on high-fat diet, but were extraordinarily susceptible to steatohepatitis and associated glucose intolerance [69]. This susceptibility to hepatic lipid accumulation was linked to impaired fatty acid oxidation, consistent with the increased fatty acid oxidation observed in CerS6 knockout mice [68,69]. Finally, an elegant mouse model of tissue-specific acid ceramidase overexpression was recently developed [61]. Liver-specific ceramidase overexpression resulted in decreased hepatic 16:0, 18:0 and 20:0 ceramides, protected high-fat fed mice from hepatic steatosis, and was associated with improvements in insulin-mediated suppression of HGP during hyperinsulinemic-euglycemic clamps and enhanced hepatic insulin signaling [61]. This protection from steatosis was attributed to increased VLDL secretion in the transgenic mice; fatty acid oxidation was unchanged [61]. Despite decreased hepatic triglycerides, hepatic DAG concentrations were reported to be increased in the transgenic mice — an unusual dissociation [61]. Similar findings of protection from hepatosteatosis and hepatic insulin resistance in association with decreased hepatic ceramides were recently reported in mice with adipose- or liver-specific inducible adiponectin receptor overexpression [96]. In summary, mouse models of altered ceramide synthesis appear to develop complex and sometimes dramatic changes in hepatic lipids. These phenotypes point to critical metabolic functions of hepatic ceramides, but, because of these potentially confounding changes in hepatic lipids, cannot address the question of whether hepatic ceramides directly impair insulin action. A related unresolved question is whether improvements in hepatosteatosis are necessary for the beneficial effects of reducing hepatic ceramides on hepatic insulin action.
Concluding Remarks and Future Perspectives
The Bradford Hill criteria provide a conceptual framework for assessing putative cause-effect relationships in biology [97]. We now attempt a brief evaluation of DAG and ceramide as causal factors in lipid-induced hepatic insulin resistance using these criteria.
The first and second criteria are strength of association and consistency. How well do hepatic DAG and ceramide levels correlate with hepatic insulin resistance, and is the correlation reproducible across investigators? Three studies have addressed this question directly in humans [31,35,37]. Hepatic DAG was associated with hepatic insulin resistance in all three studies. In contrast, hepatic ceramide content was unrelated to hepatic insulin resistance in two of the three available human studies [31,35,37]. Here, a caveat is the paucity of data on specific ceramide species (e.g., C16:0) hypothesized to mediate hepatic insulin resistance, though high-fat feeding has been reported not to increase C16:0 ceramides in five strains of mice [73].
The third criterion is specificity. Are intrahepatic DAG and ceramide accumulation associated specifically with hepatic insulin resistance, or do they have other pathophysiological consequences? Bioactive lipids, including DAG and ceramides, may also contribute to the inflammation and oxidative stress that enable progression of steatosis to steatohepatitis, and these fibrotic and inflammatory changes likely exert metabolic effects of their own [13,98]. Further, the observation that increasing hepatic ceramide content drives hepatic lipid accumulation confounds attempts to ascribe hepatic insulin resistance to direct ceramide action. In general, the profound alterations in liver and plasma lipids comorbid with hepatic insulin resistance have engendered reluctance to the hypothesis that hepatic lipids drive hepatic insulin resistance [99]. Overall, this criterion poses challenges for both DAG and ceramides.
The fourth criterion is temporality. If DAG or ceramides cause hepatic insulin resistance, they must accumulate prior to the onset of insulin resistance. Longitudinal human studies addressing this question are not available, but rodent studies are. In C57BL/6 mice, insulin resistance to suppression of HGP is present after just one week of high fat feeding [71]. At this time point, IHTG and intrahepatic DAG were increased, but ceramides (including C16:0 ceramides), were unchanged [71]. Similar results were observed in rats fed a three-day high fat diet, and held whether the diet primarily contained saturated or unsaturated fats [75]. Rodent studies which have observed increased hepatic ceramides have used much longer high fat diet protocols, such as 8 weeks [61] or 16 weeks [80]. While these data do not formally meet the criterion of temporality for DAG (because both DAG elevations and insulin resistance were present at these early time points), it may not be experimentally feasible to capture a time point in which DAG is elevated but the DAG-PKCε-INSR axis has not yet induced hepatic insulin resistance.
The fifth criterion is the presence of a biological gradient or dose-response relationship. As discussed above, the association between intrahepatic DAG and hepatic insulin resistance, whether measured by HOMA-IR or suppression of HGP, spans a wide dynamic range. In one study, a fourfold increase in intrahepatic DAG was associated with an approximately twofold increase in HOMA-IR [31]; in another, a fourfold increase in intrahepatic DAG was associated with an approximately twofold decrease in insulin-mediated suppression of HGP [35]. Intrahepatic ceramides are not associated with hepatic insulin resistance in the human or rodent studies for which dose-response data are available.
The sixth criterion is biological plausibility. Is there a viable, experimentally supported mechanism by which DAG or ceramides may mediate hepatic insulin resistance? For DAG, PKCε phosphorylation of INSR Thr1160 and consequent inhibition of INSR tyrosine kinase activity provides a direct mechanism linking intrahepatic DAG to impaired insulin signaling [43,45]. The mechanistic picture for ceramides is somewhat more blurred. An AKT-centric mechanism has long been favored; newer mechanisms involving hepatocellular lipid oxidation, VLDL export, CD36 activation, and mitochondrial dysfunction are intriguing but are indirect [61,68,69,100]. One commonality of these latter mechanisms is the conclusion that ceramides drive hepatosteatosis. If true, the deleterious effects of hepatic ceramides may be indirect: ceramides promote hepatosteatosis, and hepatosteatosis, through the DAG-INSR-PKCε axis, drives insulin resistance.
The seventh criterion is coherence. All available data, ideally, should fit the model. Table 1 provides a comprehensive overview of directional changes in hepatic insulin action, IHTG, hepatic DAG, PKCε translocation, and hepatic ceramides in studies assessing these parameters in humans and rodents. This criterion has been a major sticking point for the DAG-PKCε-INSR hypothesis of hepatic insulin resistance. Occasionally, conflicting reports arise concerning whether a given model displays hepatic insulin sensitivity or hepatic insulin resistance [51,52]. Additionally, the existence of several rodent models of genetically perturbed lipid handling with preserved hepatic insulin sensitivity despite increased hepatic DAG has led some investigators to conclude, perhaps justifiably, that increased intrahepatic DAG per se is insufficient for hepatic insulin resistance. As discussed above, however, this may reflect an oversimplified view of the sources and sites of hepatocellular DAG. There is also significant underappreciation in the field of the potentially confounding contribution of extrahepatic insulin action to readouts of “hepatic insulin action” such as the suppression of HGP [4,101,102]. An emerging paradigm emphasizes the importance of direct hepatic insulin action (and thus the DAG-PKCε-INSR axis) in the glycogen-replete state, where modulation of glycogen metabolism is a major controller of HGP, and the importance of indirect hepatic insulin action (e.g., lipolytic control of gluconeogenesis) in the glycogen-depleted state, where gluconeogenesis is the most critical component of HGP [4,103]. We further speculate that the DAG-PKCε-INSR axis is particularly relevant in the early stages of NAFLD (modeled by short-term high fat diets in rodents), and that the progression of whole-body insulin resistance is accompanied by the development of extrahepatic factors that increase HGP, such as inflammation and dysregulated lipolysis, that “dilute” the quantitative significance of direct lipid-induced hepatocellular insulin resistance. The forest can be difficult to appreciate amidst these trees. Yet ceramides also struggle with coherence. The consistent inability of multiple groups to observe increased hepatic ceramides in humans or rodents with lipid-induced hepatic insulin resistance, and the lack of a plausible mechanism directly linking ceramide action to impaired insulin signaling, are major challenges for the ceramide hypothesis of hepatic insulin resistance.
Table 1.
Comparison of human and rodent studies measuring intrahepatic DAG, hepatic PKCε translocation, intrahepatic ceramides, and hepatic insulin action. For rodent studies, hepatic insulin action was assessed as suppression of HGP during hyperinsulinemic-euglycemic clamp studies unless otherwise noted. Studies are listed in chronological order except when multiple studies describe the same model. n/a, not assessed. Rx, treatment. HFF, high-fat diet fed. ASO, antisense oligonucleotide. ZDF, Zucker diabetic fatty. STZ, streptozotocin.
| Human studies | |||||||
|---|---|---|---|---|---|---|---|
| Variable correlated to hepatic insulin resistance? | |||||||
| Reference | Readout of hepatic insulin resistance | IHTG | DAG | PKCε translocation | Ceramide | Notes | |
| Kumashiro et al. (2011) [31] | HOMA-IR | Yes | Yes (both fractions) | Yes | No | ||
| Magkos et al. (2012) [35] | Clamp EGP suppression | n/a | Yes | n/a | No | ||
| Luukonen et al. (2016) [37] | HOMA-IR | Yes | Yes | n/a | Yes | Individual lipid species reported | |
| Rodent studies | |||||||
| Reference | Model | Hepatic insulin action | IHTG | DAG | PKCε translocation | Ceramide | Notes |
| Models of perturbed hepatic triglyceride metabolism | |||||||
| Neschen et al. (2005) [46] | mtGPAT1 KO HFF mice | ↑ | ↓ | ↓ | ↓ | n/a | Individual DAG species reported |
| Nagle et al. (2007) [47] | mtGPAT1-overexpressing mice | ↓ | ↑ | ↑ | ↑ | n/a | |
| Choi et al. (2007) [50] | DGAT2 ASO HFF rats | ↑ | ↓ | ↓ | ↓ | n/a | |
| Zhang et al. (2007) [106] | LCAD KO mice | ↓ | ↑ | ↑ | ↑ | ↔ | |
| Monetti et al. (2007) [51] | DGAT2-overexpressing mice | ↔ | ↑ | ↑ | n/a | ↑ | |
| Jornayvaz et al. (2011) [52] | DGAT2-overexpressing mice | ↓ | ↑ | ↑ | ↑ | ↑ | |
| Zhang et al. (2010) | VLCAD KO HFF mice | ↑ | ↓ | ↓ | ↓ | ↔ | |
| Ryu et al. (2009) [49] | Lipin-1 knockdown in db/db mice | ↑ | ↓ | ↓ | n/a | n/a | |
| Ryu et al. (2011) [48] | Lipin-2 overexpression in HFF mice | ↓ | ↑ | ↑ | n/a | n/a | ipGTT, ITT, HOMA-IR Individual DAG species reported |
| Ryu et al. (2011) [48] | Lipin-2 knockdown in HFF mice | ↑ | ↓ | ↓ | n/a | n/a | ipGTT, ITT, HOMA-IR Individual DAG species reported |
| Hoy et al. (2011) [107] | ATGL KO HFF mice | ↑ | ↑ | ↓ (strong trend) | n/a | ↓ (strong trend) | ipGTT, ITT |
| Turpin et al. (2011) [108] | ATGL hepatic overexpression in HFF mice | ↑ | ↓ | ↓ | ↔ | ↓ | Insulin signaling |
| Cantley et al. (2013) [57] | CGI-58 ASO HFF mice | ↑ | ↑ | ↑ (lipid droplet) ↓ (membrane) |
↓ (membrane) | ↑ (all fractions) | |
| Kumashiro et al. (2013) [109] | PNPLA3 ASO HFF rats | ↑ | ↓ | ↓ | ↓ | n/a | |
| Hall et al. (2014) [59] | MGAT1 ASO HFF mice | ↑ | ↔ | ↑ (all fractions) | ↓ (membrane, total) | ↔ | Fasting insulin, ipGTT, insulin signaling |
| Soufi et al. (2014) [110] | MGAT1 ASO High trans-fat, fructose, cholesterol-fed mice | ↑ | ↓ | ↔ | n/a | n/a | ipGTT, insulin signaling |
| Trevino et al. (2015) [78] | Perilipin 5 hepatic overexpression in HFF mice | ↔ | ↑ | ↑ | n/a | ↑ | ipGTT, ITT |
| Models specifically testing the DAG-PKCε-INSR axis | |||||||
| Samuel et al. (2007) [43] | PKCε ASO 3d HFF rats | ↑ | ↔ | ↔ | ↓ | n/a | |
| Raddatz et al. (2011) [44] | PKCε KO 7d HFF mice | ↑ | ↑ | ↑ | ↓ | n/a | ipGTT, HOMA-IR |
| Petersen et al. (2016) [45] | InsrT1150A 10d HFF mice | ↑ | ↔ | ↔ | ↓ | n/a | |
| Models specifically testing the role of ceramides In hepatic Insulin resistance | |||||||
| Holland et al. (2007) [76] | Des1+/− mice | ↑ | n/a | n/a | n/a | ↓ | ITT, HOMA-IR |
| Holland et al. (2007) [76] | Myriocin Rx in ZDF rats | ↑ | n/a | ↔ | n/a | ↓ | oGTT, HOMA-IR |
| Yang et al. (2009) [86] | Myriocin Rx in HFF mice | ↑ | ↓ | n/a | n/a | n/a | ipGTT, ITT, insulin signaling |
| Holland et al. (2011) [84] | Adiponectin Rx in ob/ob mice | ↑ | n/a | ↔ | n/a | ↓ | Individual ceramide species reported |
| Holland et al. (2011) [81] | Lard oil infusion in mice | ↓ | n/a | ↔ | n/a | ↑ | |
| Holland et al. (2011) [81] | Salicylate Rx in 17 wk HFF mice | ↑ | n/a | ↔ | n/a | ↓ | ITT, ipGTT |
| Raichur et al. (2014) [69] | CerS2+/− HFF mice | ↓ | ↑ | n/a | n/a | ↑ (16:0) ↔ (total) |
Fasting insulin, ipGTT, ITT |
| Turpin et al. (2014) [68] | CerS6 KO HFF mice | ↑ | n/a (but ↓ body weight and % fat) | n/a | n/a | ↓ (16:0) | Fasting insulin, ipGTT, ITT, insulin signaling |
| Turpin et al. (2014) [68] | CerS6 liver-specific KO HFF mice | ↑ | n/a (but ↓ weight gain on HFD) | n/a | n/a | ↓ (16:0) | ipGTT |
| Xia et al. (2015) [61] | Liver-specific overexpression of acid ceramidase in HFF mice | ↑ | ↓ | ↑ | n/a | ↓ (16:0, 18:0, 20:0) | |
| Xia et al. (2015) [61] | Adipose-specific overexpression of acid ceramidase in HFF mice | ↑ | ↓ | n/a | n/a | ↓ (16:0, 18:0) | |
| Holland et al. (2017) [96] | Adipose-specific overexpression of AdipoR1/2 | ↑ | ↓ | n/a | n/a | ↓ (16:0, 18:0) | Individual ceramide species reported |
| Holland et al. (2017) [96] | Liver-specific overexpression of AdipoR1/2 | ↑ | ↓ | n/a | n/a | ↓ (16:0, 18:0) | Individual ceramide species reported |
| Miscellaneous studies examining relationships between hepatic lipids and hepatic insulin action | |||||||
| Turinsky et al. (1990) [29] | ZDF rats | n/a | n/a | ↑ | n/a | ↑ | Insulin resistance assumed |
| Savage et al. (2006) [40] | ACC1/2 ASO HFF rats | ↑ | ↓ | ↓ | ↓ | n/a | |
| Samuel et al. (2006) [111] | FOXO1 ASO HFF mice | ↑ | ↓ | ↓ | n/a | n/a | Individual DAG species reported |
| Choi et al. (2007) [112] | ACC2 KO HFF mice | ↑ | ↓ | ↓ | ↓ | n/a | |
| Matsuzaka et al. (2007) [113] | ELOVL6 KO HFF mice | ↑ | ↔ | ↓ | ↔ | ↔ | ipGTT, fasting insulin insulin signaling |
| Jaworski et al. (2009) [114] | Adipocyte phospholipase A2 KO mice | ↓ | ↑ | ↑ | n/a | n/a | |
| Erion et al. (2009) [42] | CREB ASO HFF+STZ rats | ↑ | ↓ | ↓ | ↓ | n/a | |
| Ussher et al. (2010) [74] | 12 wk HFF mice | ↓ | ↑ | n/a | n/a | ↔ | ipGTT, ITT |
| Jornayvaz et al. (2010) [115] | Ketogenic diet fed mice | ↓ | ↑ | ↑ | ↑ | ↑ | |
| Birkenfeld et al. (2011) [41] | mINDY KO HFF mice | ↑ | ↓ | ↓ | ↓ | ↔ | |
| Lee et al. (2011) [77] | ApoC3 overexpressing HFF mice | ↓ | ↑ | ↑ | ↑ | ↔ | |
| Jornayvaz et al. (2012) [116] | Thyroid hormone receptor-α KO HFF mice | ↑ | ↓ | ↓ | ↓ | n/a | |
| Jurczak et al. (2012) [117] | XBP1 conditional KO fructose-fed mice | ↑ | ↓ | ↓ | ↓ | ↑ | |
| Brown et al. (2012) [118] | FAAH KO HFF mice | ↓ | ↑ | ↑ | ↑ | ↔ | |
| Sun et al. (2012) [119] | Liver-specific HDAC3 KO mice | ↑ | ↑ | ↑ | ↔ | ↔ | |
| Camporez et al. (2013) [33] | FGF-21 Rx HFF mice | ↑ | ↓ | ↓ | ↓ | ↔ | |
| Camporez et al. (2013) [38] | Estradiol Rx ovariectomize d HFF female mice | ↑ | ↓ | ↓ | ↓ | ↔ | |
| Galbo et al. (2013) [75] | 3d HFF rats (saturated or unsaturated) | ↓ | ↑ | ↑ | ↑ | ↔ | |
| Perry et al. (2013) [24] | DNP-ME Rx HFF rats | ↑ | ↓ | ↓ | ↓ | ↔ | |
| Montgomer y et al. (2013) [72] | 8 wk HFF mice (BL/6, 129X1, DBA/2, FVB/N) | ↓ | ↑ | ↑ | n/a | ↔ | ipGTT, HOMA-IR |
| Turner et al. (2013) [71] | 1 wk HFF BL/6 mice | ↓ | ↑ | ↑ | n/a | ↔ | Individual lipid species reported |
| Turner et al. (2013) [71] | 3 wk HFF BL/6 mice | ↓ | ↑ | ↑ | n/a | ↔ (16:0, 18:0) ↑ (20:0, 22:0) ↓ (24:0, 24:1) |
Individual lipid species reported |
| Turner et al. (2013) [71] | 16 wk HFF BL/6 mice | ↓ | ↑ | ↑ | n/a | ↔ (16:0, 22:0) ↑ (18:0, 20:0) ↓ (24:0, 24:1) |
Individual lipid species reported |
| Zhu et al. (2014) [120] | Liver-specific estrogen receptor KO male HFF mice | ↓ | ↑ | ↑ | n/a | n/a | |
| Camporez et al. (2015) [34] | ApoA5 ASO HFF mice | ↑ | ↓ | ↓ | ↓ | ↑ | |
| Perry et al. (2015) [23] | CRMP Rx HFF rats | ↑ | ↓ | ↓ | ↓ | ↔ | Individual DAG species reported |
| Chan et al. (2015) [121] | Fenofibrate Rx HFF mice | ↑ | ↔ | ↔ (total) ↑ (lipid droplet) ↓ (membrane) |
n/a | ↔ (lipid droplet, membrane) | ipGTT, insulin signaling |
| Jordy et al. (2015) [122] | Exercise in HFF mice | ↑ | ↓ | ↓ | n/a | ↔ | Fasting insulin, oGTT Individual lipid species reported |
| Aroor et al. (2015) [123] | DPP-4 inhibitor Rx in Western diet-fed mice | ↑ | ↓ | ↓ | n/a | ↔ | Individual lipid species reported |
| Shang et al. (2016) [124] | Duodenal-jejunal bypass surgery in HFF mice | ↑ | ↓ | ↓ | n/a | ↓ (20:0, 22:0) ↔(16:0, 18:0) |
Individual lipid species reported |
| Wilson et al. (2016) [125] | Liver-specific CD36 KO in HFF mice | ↑ | ↓ | ↓ | n/a | ↔ | ipGTT, ITT |
| Popov et al. (2016) [126] | β-catenin ASO in HFF mice | ↑ | ↓ | ↓ | ↓ | ↑ | |
| Abulizi et al. (2017) [39] | CRMP Rx A/ZIP fatless mice | ↑ | ↓ | ↓ (all fractions) | ↓ | ↔ | |
The eighth criterion is experiment. When DAG or ceramide levels are experimentally manipulated, does hepatic insulin resistance change accordingly? For DAG, models where hepatosteatosis is specifically and acutely reversed and hepatic insulin sensitivity is improved provide inferential evidence that reducing DAG reverses hepatic insulin resistance. Liver-targeted mitochondrial uncoupling in various formulations achieves this outcome, without altering hepatic ceramides [20,23,24]. Mouse models perturbing the pathway of triacylglycerol biosynthesis have been remarkably consistent with the DAG-PKCε hypothesis, including mtGPAT−/− mice [46], mtGPAT-overexpressing mice [47], mice with Lipin1 [49] or Lipin2 [48] knockdown, Lipin2-overexpressing mice [48], Dgat2-overexpressing mice [52], and rats with Dgat2 knockdown [50]. The ceramide hypothesis also performs well on this criterion. Animal models in which ceramides are altered and hepatic insulin action moves in the predicted direction include: myriocin-treated rats[76], lard oil-infused rats [76,81], Des1+/− mice [76], adiponectin-treated mice [84], CerS2+/− mice [69], liver-specific CerS6−/− mice [68], and liver-specific acid-ceramidase overexpressing mice [61]. However as noted above these perturbations also lead to parallel alterations in hepatic lipogenesis, which impact any conclusions regarding the causal role of ceramides per se in mediating hepatic insulin resistance.
The final Bradford Hill criterion is analog — the principle that if causality has been established for one cause-effect pair, the burden of proof for related cause-effect pairs in the future should not be as strong. Both DAG and ceramide emerged as putative mediators of lipid-induced hepatic insulin resistance after first being established as putative mediators of lipid-induced skeletal muscle insulin resistance, and both hypotheses likely benefited from this familiarity among investigators. Yet the mechanisms now proposed for DAG- and ceramide-induced hepatic insulin resistance are somewhat different than those identified in skeletal muscle. In muscle, DAG is hypothesized to activate a different novel PKC isoform, PKCθ, with unclear targets in proximal insulin signaling [104]. The hepatic DAG-PKCε-INSR axis, despite emerging more recently, is now better understood than the analogous DAG-PKCθ axis in skeletal muscle. By contrast, ceramide-induced insulin resistance remains better studied in skeletal muscle than liver [65,66]. Additionally, many key mechanistic studies describing ceramide inhibition of AKT were carried out in myotubes, and have not been replicated in hepatocytes. As with DAG, however, recent progress in ceramide-induced insulin resistance has primarily concerned the liver [37,61,68,69].
Lipid-induced hepatic insulin resistance is no longer a new stage, and neither DAG nor ceramides are new players. Our current assessment of the available data is that the DAG-PKCε-INSR axis is the strongest candidate mechanism identified to account for the epidemiological link between hepatic steatosis and hepatic insulin resistance. It is unlikely to be the only such mechanism, and there is undoubtedly still ample room for development and testing of new hypotheses, especially in humans, investigating how lipids impair hepatocellular insulin action (see Outstanding Questions). Given the critical role for lipid-induced hepatic insulin resistance in the pathogenesis of type 2 diabetes, understanding this process has important implications for the development of novel drugs to treat T2D — which the USA Centers for Disease Control (CDC) predicts will impact one in three Americans by 2050 [105].
Outstanding Questions Box.
Are there specific intracellular pools of DAG, or DAG generated by specific fluxes, that are more or less capable of activating the DAG-PKCε-INSR axis in the hepatocyte?
What is the subcellular localization of the DAG-PKCε-INSR interaction? How stable is INSR inhibition by Thr1160 phosphorylation?
How might ceramides directly or indirectly impair hepatocellular insulin action? Are mechanisms previously identified in other cell types, such as AKT inhibition, functional in hepatocytes? How might specific ceramide species, such as C16:0, fit into these mechanisms?
Is protection from hepatosteatosis necessary for the insulin-sensitizing effects of decreased hepatic ceramides?
Are intrahepatic DAG and/or ceramide accumulation safely druggable targets in humans?
Trends Box.
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in industrialized nations, and is strongly associated with hepatic insulin resistance, a key driver of type 2 diabetes.
Though stored hepatic triglyceride is not thought to directly impair insulin action, two lipid classes proposed to mediate lipid-induced hepatic insulin resistance are ceramides and diacylglycerols (DAG).
A causal role for DAG in hepatic insulin resistance is supported by human correlative studies and a direct pathophysiological mechanism in rodents, but challenged by a few rodent models with increased hepatic DAG but preserved hepatic insulin sensitivity.
A causal role for ceramides in hepatic insulin resistance is supported by several rodent models in which decreasing ceramides improves hepatic insulin action, but challenged by an inconsistent relationship between hepatic ceramide content and hepatic insulin resistance.
Acknowledgments
We apologize to the many colleagues whose valuable contributions could not be discussed owing to space limitations. M.C.P. is supported by NIH Medical Scientist Training Program grant T32 GM-007205 and the NIH National Research Service Award (NRSA) grant F30 DK-104596. G.I.S. acknowledges grant support from the National Institutes of Health: R01 DK-40936, P30 DK-045735.
Abbreviations
- Insulin resistance
A condition in which the cellular response to a given ambient insulin concentration is decreased relative to a normal control. Insulin resistance as generally understood incorporates both decreased insulin sensitivity (a right shift in the insulin dose-response curve) and decreased insulin responsiveness (an impaired maximal response to high insulin concentrations). Insulin resistance has diverse manifestations in different tissues, and is a component of the ‘metabolic syndrome’ that predicts incident type 2 diabetes
- Nonalcoholic fatty liver disease (NAFLD)
Increased liver triglyceride content without an alternative etiology (e.g., alcohol use, starvation, medications). NAFLD is a risk factor for nonalcoholic steatohepatitis (NASH) and hepatocellular carcinoma. NAFLD may or may not be accompanied by biochemical signs of hepatocellular injury such as elevated serum transaminase activity, and is clinically silent in many patients
- Diacylglycerol (DAG)
A class of lipids consisting of a three-carbon glycerol backbone, two carbons of which are linked to fatty acyl chains of varying lengths. DAG exists in three stereoisomers (sn-1,2; sn-1,3 and sn-2,3); only sn-1,2-DAG is capable of activating protein kinase C (PKC) isoforms. DAG is generated through several metabolic fluxes, including triglyceride hydrolysis, triglyceride synthesis, and phosphoinositide hydrolysis
- Ceramide
A large class of lipids, many of which derive from the condensation of serine and palmitoyl CoA by serine palmitoyltransferase (SPT). Many ceramide species are bioactive and participate in diverse cellular signaling pathways
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare that there are no known conflicts of interest associated with this publication.
References
- 1.World Health Organization. Global report on diabetes. 2016. [Google Scholar]
- 2.Magnusson I, et al. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–1327. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basu R, et al. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes. 2005;54:1942–1948. doi: 10.2337/diabetes.54.7.1942. [DOI] [PubMed] [Google Scholar]
- 4.Perry RJ, et al. Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boden G, et al. Gluconeogenesis in moderately and severely hyperglycemic patients with type 2 diabetes mellitus. Am J Physiol Endocrinol Metab. 2001;280:E23–30. doi: 10.1152/ajpendo.2001.280.1.E23. [DOI] [PubMed] [Google Scholar]
- 6.Krssak M, et al. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes. 2004;53:3048–3056. doi: 10.2337/diabetes.53.12.3048. [DOI] [PubMed] [Google Scholar]
- 7.Hundal RS, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen KF, et al. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–608. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leite NC, et al. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009;29:113–119. doi: 10.1111/j.1478-3231.2008.01718.x. [DOI] [PubMed] [Google Scholar]
- 10.Targher G, et al. Prevalence of Nonalcoholic Fatty Liver Disease and Its Association With Cardiovascular Disease Among Type 2 Diabetic Patients. Diabetes Care. 2007;30:1212–1218. doi: 10.2337/dc06-2247. [DOI] [PubMed] [Google Scholar]
- 11.Silverman JF, et al. Liver pathology in morbidly obese patients with and without diabetes. Am J Gastroenterol. 1990;85:1349–1355. [PubMed] [Google Scholar]
- 12.Marchesini G, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107:450–455. doi: 10.1016/s0002-9343(99)00271-5. [DOI] [PubMed] [Google Scholar]
- 13.Bugianesi E, et al. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatol Baltim Md. 2005;42:987–1000. doi: 10.1002/hep.20920. [DOI] [PubMed] [Google Scholar]
- 14.Korenblat KM, et al. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology. 2008;134:1369–1375. doi: 10.1053/j.gastro.2008.01.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiikkainen M, et al. Liver-fat accumulation and insulin resistance in obese women with previous gestational diabetes. Obes Res. 2002;10:859–867. doi: 10.1038/oby.2002.118. [DOI] [PubMed] [Google Scholar]
- 16.Fabbrini E, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A. 2009;106:15430–15435. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magkos F, et al. Increased whole-body adiposity without a concomitant increase in liver fat is not associated with augmented metabolic dysfunction. Obes Silver Spring Md. 2010;18:1510–1515. doi: 10.1038/oby.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamazaki H, et al. Independent Association Between Improvement of Nonalcoholic Fatty Liver Disease and Reduced Incidence of Type 2 Diabetes. Diabetes Care. 2015;38:1673–1679. doi: 10.2337/dc15-0140. [DOI] [PubMed] [Google Scholar]
- 19.Lotta LA, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49:17–26. doi: 10.1038/ng.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samuel VT, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 21.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 22.Petersen KF, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perry RJ, et al. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015;347:1253–1256. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry RJ, et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 2013;18:740–748. doi: 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JK, et al. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 26.Tao H, et al. Niclosamide ethanolamine improves blood glycemic control and reduces hepatic steatosis in mice. Nat Med. 2014;20:1263–1269. doi: 10.1038/nm.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takayama S, et al. Phorbol ester-induced serine phosphorylation of the insulin receptor decreases its tyrosine kinase activity. J Biol Chem. 1988;263:3440–3447. [PubMed] [Google Scholar]
- 28.Takayama S, et al. Phorbol esters modulate insulin receptor phosphorylation and insulin action in cultured hepatoma cells. Proc Natl Acad Sci U S A. 1984;81:7797–7801. doi: 10.1073/pnas.81.24.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turinsky J, et al. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem. 1990;265:16880–16885. [PubMed] [Google Scholar]
- 30.Shmueli E, et al. Diacylglycerol/protein kinase C signalling: a mechanism for insulin resistance? J Intern Med. 1993;234:397–400. doi: 10.1111/j.1365-2796.1993.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 31.Kumashiro N, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A. 2011;108:16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry RJ, et al. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camporez JPG, et al. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology. 2013;154:3099–3109. doi: 10.1210/en.2013-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camporez JPG, et al. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat-fed mice by reducing ectopic lipid content. J Lipid Res. 2015;56:526–536. doi: 10.1194/jlr.M054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magkos F, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology. 2012;142:1444–1446.e2. doi: 10.1053/j.gastro.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jornayvaz FR, Shulman GI. Diacylglycerol activation of protein kinase Cε and hepatic insulin resistance. Cell Metab. 2012;15:574–584. doi: 10.1016/j.cmet.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luukkonen PK, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64:1167–1175. doi: 10.1016/j.jhep.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Camporez JPG, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology. 2013;154:1021–1028. doi: 10.1210/en.2012-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abulizi A, et al. A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB J Off Publ Fed Am Soc Exp Biol. 2017 doi: 10.1096/fj.201700001R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savage DB, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birkenfeld AL, et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011;14:184–195. doi: 10.1016/j.cmet.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erion DM, et al. Prevention of hepatic steatosis and hepatic insulin resistance by knockdown of cAMP response element-binding protein. Cell Metab. 2009;10:499–506. doi: 10.1016/j.cmet.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samuel VT, et al. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raddatz K, et al. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia. 2011;54:1447–1456. doi: 10.1007/s00125-011-2073-0. [DOI] [PubMed] [Google Scholar]
- 45.Petersen MC, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest. 2016;126:4361–71. doi: 10.1172/JCI86013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neschen S, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 47.Nagle CA, et al. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J Biol Chem. 2007;282:14807–14815. doi: 10.1074/jbc.M611550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ryu D, et al. Endoplasmic reticulum stress promotes LIPIN2-dependent hepatic insulin resistance. Diabetes. 2011;60:1072–1081. doi: 10.2337/db10-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryu D, et al. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metab. 2009;9:240–251. doi: 10.1016/j.cmet.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 50.Choi CS, et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem. 2007;282:22678–22688. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 51.Monetti M, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007;6:69–78. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 52.Jornayvaz FR, et al. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc Natl Acad Sci U S A. 2011;108:5748–5752. doi: 10.1073/pnas.1103451108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finck BN, Hall AM. Does Diacylglycerol Accumulation in Fatty Liver Disease Cause Hepatic Insulin Resistance? BioMed Res Int. 2015;2015:104132. doi: 10.1155/2015/104132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamiya Y, et al. Activation of conventional and novel protein kinase C isozymes by different diacylglycerol molecular species. Biochem Biophys Rep. 2016;7:361–366. doi: 10.1016/j.bbrep.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shirai Y, et al. Distinct effects of fatty acids on translocation of gamma- and epsilon-subspecies of protein kinase C. J Cell Biol. 1998;143:511–521. doi: 10.1083/jcb.143.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown JM, et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res. 2010;51:3306–3315. doi: 10.1194/jlr.M010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cantley JL, et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci U S A. 2013;110:1869–1874. doi: 10.1073/pnas.1219456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minehira K, et al. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res. 2008;49:2038–2044. doi: 10.1194/jlr.M800248-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hall AM, et al. Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes. 2014;63:2284–2296. doi: 10.2337/db13-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fullerton MD, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia JY, et al. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015;22:266–278. doi: 10.1016/j.cmet.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peterson TA, Stamnes M. ARF1-regulated coatomer directs the steady-state localization of protein kinase C epsilon at the Golgi apparatus. Biochim Biophys Acta. 2013;1833:487–493. doi: 10.1016/j.bbamcr.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eichmann TO, et al. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J Biol Chem. 2012;287:41446–41457. doi: 10.1074/jbc.M112.400416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rando RR, Young N. The stereospecific activation of protein kinase C. Biochem Biophys Res Commun. 1984;122:818–823. doi: 10.1016/s0006-291x(84)80107-2. [DOI] [PubMed] [Google Scholar]
- 65.Petersen MC, Jurczak MJ. CrossTalk opposing view: Intramyocellular ceramide accumulation does not modulate insulin resistance. J Physiol. 2016;594:3171–3174. doi: 10.1113/JP271677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Summers SA, Goodpaster BH. CrossTalk proposal: Intramyocellular ceramide accumulation does modulate insulin resistance. J Physiol. 2016;594:3167–3170. doi: 10.1113/JP271676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmitz-Peiffer C, et al. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem. 1999;274:24202–24210. doi: 10.1074/jbc.274.34.24202. [DOI] [PubMed] [Google Scholar]
- 68.Turpin SM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014;20:678–686. doi: 10.1016/j.cmet.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 69.Raichur S, et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014;20:687–695. doi: 10.1016/j.cmet.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 70.Hla T, Kolesnick R. C16:0-ceramide signals insulin resistance. Cell Metab. 2014;20:703–705. doi: 10.1016/j.cmet.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Turner N, et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia. 2013;56:1638–1648. doi: 10.1007/s00125-013-2913-1. [DOI] [PubMed] [Google Scholar]
- 72.Montgomery MK, et al. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia. 2013;56:1129–1139. doi: 10.1007/s00125-013-2846-8. [DOI] [PubMed] [Google Scholar]
- 73.Montgomery MK, et al. Regulation of glucose homeostasis and insulin action by ceramide acyl-chain length: A beneficial role for very long-chain sphingolipid species. Biochim Biophys Acta. 2016;1861:1828–1839. doi: 10.1016/j.bbalip.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 74.Ussher JR, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes. 2010;59:2453–2464. doi: 10.2337/db09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Galbo T, et al. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc Natl Acad Sci U S A. 2013;110:12780–12785. doi: 10.1073/pnas.1311176110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holland WL, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 77.Lee HY, et al. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatol Baltim Md. 2011;54:1650–1660. doi: 10.1002/hep.24571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Trevino MB, et al. Liver Perilipin 5 Expression Worsens Hepatosteatosis But Not Insulin Resistance in High Fat-Fed Mice. Mol Endocrinol Baltim Md. 2015;29:1414–1425. doi: 10.1210/me.2015-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kurek K, et al. Inhibition of ceramide de novo synthesis reduces liver lipid accumulation in rats with nonalcoholic fatty liver disease. Liver Int Off J Int Assoc Study Liver. 2014;34:1074–1083. doi: 10.1111/liv.12331. [DOI] [PubMed] [Google Scholar]
- 80.Bikman BT, et al. Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J Biol Chem. 2012;287:17426–17437. doi: 10.1074/jbc.M112.359950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holland WL, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:1858–1870. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang Q, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 83.Kim JK, et al. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108:437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Holland WL, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15:585–594. doi: 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 86.Yang G, et al. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2009;297:E211–224. doi: 10.1152/ajpendo.91014.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Salinas M, et al. Inhibition of PKB/Akt1 by C2-ceramide involves activation of ceramide-activated protein phosphatase in PC12 cells. Mol Cell Neurosci. 2000;15:156–169. doi: 10.1006/mcne.1999.0813. [DOI] [PubMed] [Google Scholar]
- 88.Schubert KM, et al. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J Biol Chem. 2000;275:13330–13335. doi: 10.1074/jbc.275.18.13330. [DOI] [PubMed] [Google Scholar]
- 89.Zhou H, et al. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem. 1998;273:16568–16575. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- 90.Stratford S, et al. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem. 2004;279:36608–36615. doi: 10.1074/jbc.M406499200. [DOI] [PubMed] [Google Scholar]
- 91.Galbo T, et al. PP2A inhibition results in hepatic insulin resistance despite Akt2 activation. Aging. 2013;5:770–781. doi: 10.18632/aging.100611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Caro JF, et al. Studies on the mechanism of insulin resistance in the liver from humans with noninsulin-dependent diabetes. Insulin action and binding in isolated hepatocytes, insulin receptor structure, and kinase activity. J Clin Invest. 1986;78:249–258. doi: 10.1172/JCI112558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Watarai T, et al. Alteration of Insulin-Receptor Kinase Activity by High-Fat Feeding. Diabetes. 1988;37:1397–1404. doi: 10.2337/diab.37.10.1397. [DOI] [PubMed] [Google Scholar]
- 94.Henao-Mejia J, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vandanmagsar B, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Holland WL, et al. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab. 2017;6:267–275. doi: 10.1016/j.molmet.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fedak KM, et al. Applying the Bradford Hill criteria in the 21 st century: how data integration has changed causal inference in molecular epidemiology. Emerg Themes Epidemiol. 2015;12 doi: 10.1186/s12982-015-0037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peverill W, et al. Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation. Int J Mol Sci. 2014;15:8591–8638. doi: 10.3390/ijms15058591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Farese RV, Jr, et al. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 2012;15:570–573. doi: 10.1016/j.cmet.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aburasayn H, et al. Targeting ceramide metabolism in obesity. Am J Physiol Endocrinol Metab. 2016;311:E423–435. doi: 10.1152/ajpendo.00133.2016. [DOI] [PubMed] [Google Scholar]
- 101.Ader M, Bergman RN. Peripheral effects of insulin dominate suppression of fasting hepatic glucose production. Am J Physiol. 1990;258:E1020–1032. doi: 10.1152/ajpendo.1990.258.6.E1020. [DOI] [PubMed] [Google Scholar]
- 102.Cherrington AD, et al. The direct and indirect effects of insulin on hepatic glucose production in vivo. Diabetologia. 1998;41:987–996. doi: 10.1007/s001250051021. [DOI] [PubMed] [Google Scholar]
- 103.Edgerton DS, et al. Insulin’s direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight. 2017;2:e91863. doi: 10.1172/jci.insight.91863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:1131–1141. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- 105.Centers For Disease Control . 2014 Diabetes Report Card. 2014. [Google Scholar]
- 106.Zhang D, et al. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hoy AJ, et al. Adipose triglyceride lipase-null mice are resistant to high-fat diet-induced insulin resistance despite reduced energy expenditure and ectopic lipid accumulation. Endocrinology. 2011;152:48–58. doi: 10.1210/en.2010-0661. [DOI] [PubMed] [Google Scholar]
- 108.Turpin SM, et al. Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia. 2011;54:146–156. doi: 10.1007/s00125-010-1895-5. [DOI] [PubMed] [Google Scholar]
- 109.Kumashiro N, et al. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatol Baltim Md. 2013;57:1763–1772. doi: 10.1002/hep.26170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Soufi N, et al. Inhibiting monoacylglycerol acyltransferase 1 ameliorates hepatic metabolic abnormalities but not inflammation and injury in mice. J Biol Chem. 2014;289:30177–30188. doi: 10.1074/jbc.M114.595850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Samuel VT, et al. Targeting Foxo1 in Mice Using Antisense Oligonucleotide Improves Hepatic and Peripheral Insulin Action. Diabetes. 2006;55:2042–2050. doi: 10.2337/db05-0705. [DOI] [PubMed] [Google Scholar]
- 112.Choi CS, et al. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Natl Acad Sci U S A. 2007;104:16480–16485. doi: 10.1073/pnas.0706794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Matsuzaka T, et al. Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat Med. 2007;13:1193–1202. doi: 10.1038/nm1662. [DOI] [PubMed] [Google Scholar]
- 114.Jaworski K, et al. AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat Med. 2009;15:159–168. doi: 10.1038/nm.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jornayvaz FR, et al. A high-fat, ketogenic diet causes hepatic insulin resistance in mice, despite increasing energy expenditure and preventing weight gain. Am J Physiol Endocrinol Metab. 2010;299:E808–815. doi: 10.1152/ajpendo.00361.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jornayvaz FR, et al. Thyroid hormone receptor-α gene knockout mice are protected from diet-induced hepatic insulin resistance. Endocrinology. 2012;153:583–591. doi: 10.1210/en.2011-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jurczak MJ, et al. Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J Biol Chem. 2012;287:2558–2567. doi: 10.1074/jbc.M111.316760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brown WH, et al. Fatty acid amide hydrolase ablation promotes ectopic lipid storage and insulin resistance due to centrally mediated hypothyroidism. Proc Natl Acad Sci U S A. 2012;109:14966–14971. doi: 10.1073/pnas.1212887109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun Z, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. 2012;18:934–942. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu L, et al. Estrogen signaling prevents diet-induced hepatic insulin resistance in male mice with obesity. Am J Physiol Endocrinol Metab. 2014;306:E1188–1197. doi: 10.1152/ajpendo.00579.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chan SMH, et al. Fenofibrate insulates diacylglycerol in lipid droplet/ER and preserves insulin signaling transduction in the liver of high fat fed mice. Biochim Biophys Acta. 2015;1852:1511–1519. doi: 10.1016/j.bbadis.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 122.Jordy AB, et al. Analysis of the liver lipidome reveals insights into the protective effect of exercise on high-fat diet-induced hepatosteatosis in mice. Am J Physiol Endocrinol Metab. 2015;308:E778–791. doi: 10.1152/ajpendo.00547.2014. [DOI] [PubMed] [Google Scholar]
- 123.Aroor AR, et al. Dipeptidyl peptidase-4 inhibition ameliorates Western diet-induced hepatic steatosis and insulin resistance through hepatic lipid remodeling and modulation of hepatic mitochondrial function. Diabetes. 2015;64:1988–2001. doi: 10.2337/db14-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shang J, et al. Duodenal-jejunal bypass surgery induces hepatic lipidomic alterations associated with ameliorated hepatic steatosis in mice. Obes Silver Spring Md. 2016;24:1938–1945. doi: 10.1002/oby.21583. [DOI] [PubMed] [Google Scholar]
- 125.Wilson CG, et al. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology. 2016;157:570–585. doi: 10.1210/en.2015-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Popov VB, et al. Second-generation antisense oligonucleotides against β-catenin protect mice against diet-induced hepatic steatosis and hepatic and peripheral insulin resistance. FASEB J Off Publ Fed Am Soc Exp Biol. 2016;30:1207–1217. doi: 10.1096/fj.15-271999. [DOI] [PMC free article] [PubMed] [Google Scholar]