

Abstract
Background
Enhancers are non-coding DNA sequences, which when they are bound by specific proteins increase the level of gene transcription. Enhancers activate unique gene expression patterns within cells of different types or under different conditions. Enhancers are key contributors to gene regulation, and causative variants that affect quantitative traits in humans and mice have been located in enhancer regions. However, in the bovine genome, enhancers as well as other regulatory elements are not yet well defined. In this paper, we sought to improve the annotation of bovine enhancer regions by using publicly available mammalian enhancer information. To test if the identified putative bovine enhancer regions are enriched with functional variants that affect milk production traits, we performed genome-wide association studies using imputed whole-genome sequence data followed by meta-analysis and enrichment analysis.
Results
We produced a library of candidate bovine enhancer regions by using publicly available bovine ChIP-Seq enhancer data in combination with enhancer data that were identified based on sequence homology with human and mouse enhancer databases. We found that imputed whole-genome sequence variants associated with milk production traits in 16,581 dairy cattle were enriched with enhancer regions that were marked by bovine-liver H3K4me3 and H3K27ac histone modifications from both permutation tests and gene set enrichment analysis. Enhancer regions that were identified based on sequence homology with human and mouse enhancer regions were not as strongly enriched with trait-associated sequence variants as the bovine ChIP-Seq candidate enhancer regions. The bovine ChIP-Seq enriched enhancer regions were located near genes and quantitative trait loci that are associated with pregnancy, growth, disease resistance, meat quality and quantity, and milk quality and quantity traits in dairy and beef cattle.
Conclusions
Our results suggest that sequence variants within enhancer regions that are located in bovine non-coding genomic regions contribute to the variation in complex traits. The level of enrichment was higher in bovine-specific enhancer regions that were identified by detecting histone modifications H3K4me3 and H3K27ac in bovine liver tissues than in enhancer regions identified by sequence homology with human and mouse data. These results highlight the need to use bovine-specific experimental data for the identification of enhancer regions.
Electronic supplementary material
The online version of this article (doi:10.1186/s12711-017-0331-4) contains supplementary material, which is available to authorized users.
Background
Genomic selection is a powerful tool that has rapidly accelerated genetic gains in the dairy industry [1]. Genomic estimated breeding values (GEBV) for ranking selection candidates are calculated as the sum of the individual effects of genome-wide single nucleotide polymorphisms (SNPs). Genomic prediction for a given trait of interest would be most accurate if all causative variants that affect a trait were known and used in the prediction. For most complex traits, such as milk production in dairy cattle, very few causal variants are known [2] and therefore it is unlikely that the full set of causative variants are contained within the SNP panels used for routine evaluation. The task of identifying causative variants for complex traits is challenging since it is likely that a very large number of causative mutations with small effects contribute to the total genomic variation of the trait [3].
Recent research has indicated that much of the variation that affects complex traits lies in the non-coding genome [2], particularly transcriptional regulatory elements. Enhancers, which are also called locus control regions (LCR) or upstream activating sequences (UAS) [4], are non-coding DNA sequences, which when they are bound by specific proteins, enhance the transcriptional level of a related gene or set of genes [4]. To date, the identification of genomic regulatory elements including enhancers has followed two main approaches. Firstly, evolutionarily conserved non-coding sequences between mammalian species or higher vertebrates [5, 6] have been used to identify the more conserved developmental enhancers [7, 8]. Secondly, a more recent approach that uses chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-Seq) can detect candidate enhancers on a genome-wide scale. This approach uses antibodies to snapshot transcriptional proteins that are bound to DNA sequences in vivo, and has revealed a much larger number of candidate enhancers [4, 9] than the previous approach, the majority of which were detected only in a specific physiological context [10]. Examples of biological signals that allow the identification of enhancers are mono-methylation of lysine 4 on histone H3 (H3K4me1) [11–13], p300-CBP coactivator protein family [14–16], tri-methylation of lysine 4 on histone H3 (H3K4me3) and acetylated lysine 27 on histone H3 (H3K27ac) [17–19]. The histone mark H3K4me3 displays a bimodal distribution that flanks the transcription start sites (TSS) of active or to be expressed genes in eukaryotes [20]. It is a prevalent histone mark for promoters [21, 22] and is also found in the coding regions of genes [21, 22], and occasionally it marks active enhancers [13, 20, 23, 24]. The histone mark H3K27ac distinguishes active enhancers from poised enhancers at a tissue-specific level and in a developmental-specific manner [12, 25]. It also marks active promoters [12] and displays broader profiles than H3K4me3, which is in line with its association with open chromatin [12, 13]. The number of histone marks and co-occupation of other cellular elements collaboratively define the transcriptional state of a genomic region [20].
The aim of this study was to identify bovine enhancer regions in silico based on sequence homology with functional annotation data in other species in addition to bovine ChIP-Seq data. We evaluated the influence of mutations in enhancer regions on complex production traits by performing a multi-breed genome-wide association study (GWAS) with imputed whole-genome sequence data in 16,581 cattle followed by meta-analysis and enrichment analysis.
Methods
Mammalian enhancer sets
We used four public mammalian enhancer datasets to search for bovine putative enhancers, i.e. VISTA [26], FANTOM5 [27], dbSUPER [28] and the Villar et al. [29] study. The VISTA enhancer browser [26] selects evolutionarily ultra-conserved sequences between vertebrates and validates enhancer activities in transgenic mouse reporter assays [6]. The functional annotation of the mammalian genome 5 project (FANTOM5) [27] provides a repository of active enhancers from various human and mouse tissues. FANTOM5 enhancers are defined by bidirectional transcription signals at the 5′ end of the transcription start site (TSS) using single-molecule HeliScope cap analysis of gene expression (CAGE) [30]. The database dbSUPER collects data on super-enhancers from various human or mouse tissues across multiple studies [28]. A super-enhancer (also known as a stretch enhancer) is a group of active enhancers that are densely clustered in a 10 to 30-kb region and are highly associated with cell identity genes and disease-associated genomic variations [31]. We combined these sets of homologous enhancers with predicted bovine enhancers from Villar et al. [29] who used ChIP-Seq to detect, in the bovine genome, binding sites to H3K4me3 and H3K27ac [17–19] in the liver tissue of four bulls. Finally, the library of enhancers that we used to identify bovine putative enhancers includes 4481 VISTA (2235 human and 2246 mouse), 109,882 FANTOM5 (65,423 human and 44,459 mouse), 1745 dbSUPER (607 human and 1138 mouse), 13,797 Villar H3K4me3 (13,797 bull) and 45,784 Villar H3K27ac (45,784 bull) sequences, which were downloaded from their respective host sites on 10 August 2015, 6 October 2015, 14 September 2015, 4 September 2015 and 4 September 2015.
Genotypes
Illumina BovineHD BeadChip 800 K SNPs (real or imputed) were available for 3311 Holstein bulls, 8478 Holstein cows, 875 Jersey bulls and 3917 Jersey cows. Among these individuals, 145 Holstein bulls and 47 Jersey bulls were from the 1000 Bull Genomes Project [32], and most of the cows were from the 10,000 Holstein Cow Genomes Project and Jer-nomics Project [33]. Quality control and imputation were performed as described in [34], with an additional filter to retain only the SNPs that overlapped with sequence variants discovered in the 1000 Bull Genomes Project (run 4). The genotypes of all animals were then imputed to whole-genome sequence (WGS) using Fimpute [35] with a reference population of 1147 individuals with whole-genome sequences from the 1000 Bull Genomes Project (run 4). After imputation, 28,899,038 WGS variants were available. All genomic loci were mapped to the bovine genome assembly UMD3.1 (bostau6) [36].
Phenotypes
Phenotypes for the genotyped animals were available for milk production traits including fat yield (FY), milk yield (MY) and protein yield (PY) from the national dairy database operated by DataGene (Melbourne, Australia). The phenotypes used in the analyses were trait deviations (TD) for cows and daughter trait deviations (DTD) for bulls. TD were calculated based on cows’ lactation records (three lactations on average) and corrected for known fixed effects as per DataGene routine evaluations from the April 2013 official breeding value run. DTD were generated from nationwide progeny test data collected on many bulls’ daughters, and were corrected for known fixed effects such as herd, year and season. The animals used in our study were the same as or overlapped with those in previous publications [34, 37, 38].
Other data
The reference genomes used in this analysis, GRCh38.p4 (hg38), GRCm38.p4 (mm10) and UMD3.1.1 (bostau8), along with their annotation files, were downloaded from the National Centre for Biotechnology Information (NCBI) Reference Sequence Database (RefSeq) [39] on 20 August 2015. Genomic coordinate conversion files (chain file format) were downloaded from the UCSC (University of California, Santa Cruz) database [40] on 29 February 2016. Annotations for the sequence variants were collated using NGS-SNP [41]. The bovine quantitative trait loci (QTL) annotation file was downloaded from the Animal QTL database (Animal Qtldb) [42] on 17 May 2016.
Mapping bovine candidate enhancers
The human and mouse enhancer regions from VISTA, FANTOM5 and dbSUPER were mapped to the bovine reference genome assembly UMD3.1.1 via command line applications Nucleotide Basic Local Alignment Search Tool (BLASTn) [43] (default settings except for the e-value were ) and UCSC Batch Coordinate Conversion (liftOver) [40] (default settings), respectively. The BLASTn approach measures local sequence similarity to identify which query segments can be matched to different parts of the target genome [43]. The liftOver approach measures global sequence similarity where the query sequence is optimised to the best matching location in the target genome, although the best matching location may be stretched out on a much longer region than the query sequence [40]. The BLASTn software returned specific genomic coordinates for mapped query segments, whereas the liftOver command application returned a mapped file for all the genomic coordinates that were found in the target genome, and an unmapped file for all the query sequences that were partially or fully unmapped. We considered all the returned queries with full or partial hits as mapped input sequences in BLASTn, and all the queries that were not marked as fully unmapped were considered as mapped input sequences in liftOver. LiftOver outputs were combined with BLASTn results. All regions from the combined set that overlapped over more than one bp were merged into a longer and non-overlapping genomic interval. Bovine enhancer data from ChIP-Seq H3K4me3 and H3K27ac signals [29] were directly merged into the non-overlapping set, respectively.
Genome-wide association study
A multi-breed genome-wide association study (GWAS) was performed to detect imputed WGS variants that were associated with FY, MY and PY. Following the approach described by [37], Holstein and Jersey data were combined, but the analyses were separated by gender, because phenotype measurements in bulls and cows have different degrees of uncertainty [34]. The efficient mixed-model association expedited (EMMAX) analysis software package [44] was used to fit the 28,899,038 WGS variants one by one in the linear mixed model:
where is a vector of phenotypes (DTD for bulls and TD for cows); is the design matrix that allocates phenotypes to fixed effects accounting for overall mean and breeds; is a vector of fixed effect solutions; is a vector of animal genotypes; is a vector of genotype effects; is a matrix that allocates phenotypic records to animals and is a vector of polygenic breeding values fitted as a random effect and assumed to follow a normal distribution , where is the genetic variance of the trait, and is the genomic relationship matrix calculated from the 800 K genotypes as in [45]; and is a vector of residual errors distributed , where is the error variance. The polygenic breeding values were included in the model to avoid false positive SNP effects due to population structure and sub-structure [44].
Meta-analysis
The multi-breed GWAS results from bull and cows were combined using an inverse-variance weighting meta-analysis within a fixed effect model as described by [46]. We did not perform a joint analysis since inclusion of different accuracies for the phenotypes of bulls and cows was not possible in EMMAX. For the inverse-variance weighted meta-analysis, the following were calculated:
-
The standard error of SNP effects is calculated as follows:
where indicates the SNP at position , indicates the gender cohort, is the standard error of the SNP effect, is the SNP effect output from EMMAX, is the quantile function of the standard normal distribution and is the GWAS P value output from EMMAX.1 -
The inverse-variance weight for each SNP is then calculated as:
where and are as defined above, is the inverse-variance weight, and is the standard error of the SNP effect calculated from Eq. (1).2 -
The inverse-variance weighted effect for each SNP is then calculated as:
where and are as defined above, is the weighted effect, is the SNP effect output from EMMAX.3 -
The SNP effect from the meta-analysis that combines gender cohorts is calculated as:
-
The variance of the SNP effect from the meta-analysis that combines gender cohorts is calculated as:
where is as above, is the variance of the SNP effect from the meta-analysis, is the number of cohorts being combined in the meta-analysis (here, is equal to 2 because the bull and cow cohorts were combined), and is the weight calculated from Eq. (2).5 -
The P value from the meta-analysis that combines gender cohorts is calculated as:
Variants with no effect or with a P value of 1 were removed from the downstream analysis. Of the 28,899,038 imputed WGS variants input for FY cohort, after the meta-analysis, 23,462,606 variants remained. Of the 28,899,038 imputed WGS variants input for MY cohort, after the meta-analysis 23,462,606 variants remained. Of the 28,899,038 imputed WGS variants input for PY cohort, after the meta-analysis 23,470,573 variants remained. Significant variants from the meta-analysis were selected using the same threshold as in the GWAS .
Enrichment analysis
The bovine candidate enhancers were categorised into five enhancer sets based on their input databases: VISTA, FANTOM5, dbSUPER, Villar H3K4me3 or Villar H3K27ac. Two enrichment analyses, i.e. permutation test and gene set enrichment analysis (GSEA), were performed to examine if any of the bovine candidate enhancer sets were enriched with variants associated with FY, MY or PY. The permutation test compared the number of significant SNPs in an enhancer set with the null distribution sampled from the rest of the genome. However, the need for a predefined threshold for statistical significance in the permutation tests may result in not detecting relevant biological differences that are modest relative to the noise that is inherent to the data [47]. This insensitivity of the permutation test was partly overcome by GSEA, which considered the distribution of all effects and tested if SNPs in an enhancer set were responsible for the enrichment signal, without applying any significant threshold [47].
The permutation test was run for 10,000 random repeats to test if the number of significant SNPs in an enhancer set was significantly larger than that obtained by random chance. The numbers of SNPs and of significant SNPs in an enhancer set, and the number of SNPs in a random draw are denoted as , and , respectively. In each permutation, a significant SNP was determined by a global P value cut-off of . The fold change of the enrichment was defined as the ratio of to the mean of all in random samples. The ranking position of within the distribution of all over all random samples, denoted as , was determined, and a P value to test the significance of the ranking was computed. For the largest among all , the P value was set to <0.0001 and otherwise it was . Our permutation tests resulted in 15 independent analyses (3 phenotypes 5 enhancer databases).
The GSEA statistics was the cumulative sum of the effects of SNPs in putative enhancers computed from the sorted list of all SNP effects. Here, the effect was assessed by –log10 (P value). At each point in the GSEA algorithm, the test statistic was computed as follows:
where is the position of the effect of an enhancer SNP in the sorted list of all SNP effects, and are respectively the cumulative probability of observing all enhancer SNPs and all non-enhancer SNPs up to position , thus denotes the level of enrichment of enhancer SNPs up to position . The position at which reaches the maximum deviation from 0, , defines the strength of the enrichment signal in the enhancer set. All enhancer SNPs that are identified before reaches are assigned to the candidate core enhancer set.
The significance of each GSEA was determined in a similar way as that for the permutation test described above. We randomly shuffled the SNPs within the sorted list while retaining the sorted positions of −log10 (P value) and recalculated the value. The shuffle was repeated 10,000 times and 10,000 values were obtained. A GSEA result was considered significant if the value was larger than all values. Our GSEA resulted in 15 independent analyses (3 phenotypes 5 enhancer databases), but the sets of core enhancer SNPs were only those from the significant GSEA cohorts.
Results
Mapping bovine candidate enhancers
Two aligners, BLASTn and liftOver, were used to map human and mouse enhancers on the bovine genome (Table 1). All sets of bovine putative enhancer regions covered bovine chromosome 1 to 30. The bovine reference genome assembly bostau6 and bostau8 do not include chromosome Y. The mapping rate was defined as the ratio between the number of query sequences found in the bovine genome and the number of query sequences input for search. Cross-species mapping rates were equal to, in decreasing order, 96% for VISTA, 92% for dbSUPER and 87% for FANTOM5. The number of overlaps between BLASTn and liftOver results was small for FANTOM5 (<10%), moderate for dbSUPER (16%) and high for VISTA (71%). Over 93 and over 95% of the dbSUPER hits were within 10 and 30 kb to each other, respectively. As expected, homologous enhancer sequences were on average shorter than their respective query sequences (Table 1).
Table 1.
Mapping of bovine candidate enhancers
| dba | Queryb | Metf | m%g | Hitsh | eSNPl | ||||
|---|---|---|---|---|---|---|---|---|---|
| ISeqc | u (bp)d | σ (bp)e | OSeqi | µ (bp)j | σ (bp)k | ||||
| VISTA | 4481 | 1959 | 1395 | COM | 4285 (96%) | 9945 | 896 | 710 | 82,865 |
| LO | 3808 (85%) | 964 | 1399 | 883 | |||||
| BN | 3627 (81%) | 9945 | 896 | 710 | |||||
| FANTOM5 | 109,882 | 277 | 158 | COM | 95,123 (87%) | 30,371 | 231 | 115 | 50,447 |
| LO | 94,302 (86%) | 6061 | 245 | 503 | |||||
| BN | 10,054 (9%) | 30,389 | 231 | 115 | |||||
| dbSUPER | 1745 | 45,750 | 56,541 | COM | 1605 (92%) | 50,938 | 739 | 763 | 282,285 |
| LO | 1549 (88%) | 32 | 1916 | 2447 | |||||
| BN | 1113 (64%) | 50,938 | 739 | 763 | |||||
| H3K4me3 | 13,797 | 2393 | 879 | NA | 13,797 (100%) | 13,660 | 2394 | 879 | 302,659 |
| H3K27ac | 45,784 | 2304 | 1910 | NA | 45,784 (100%) | 42,963 | 2305 | 1910 | 965,716 |
aDatabase from which input query sequences were obtained
bQuery sequence downloaded from respective host sites (may include overlapping regions)
cNumber of input enhancer query sequences
dMean length of input enhancer query sequences (measured in bp)
eStandard deviation length of input enhancer query sequences (measured in bp)
fMethod that returned values in column m%; COM: a set of non-overlapping regions from the combined results of liftOver and BLASTn; LO: liftOver; BN: BLASTn
gNumber of mapped query sequences (ratio of mapped query sequences); NA not applicable
hHit is a non-overlapping genomic interval in the bovine genome that matches with at least one query sequence from the respective input database
iNumber of non-overlapping candidate bovine enhancer genomic intervals; all hits, i.e. output from the respective software were merged into non-overlapping genomic intervals; some OSeq values were larger than corresponding ISeq values because one query sequence was found at multiple locations in the bovine genome
jMean length of bovine putative enhancer sequences (measured in bp)
kStandard deviation length of bovine putative enhancer sequences (measured in bp)
lNumber of imputed whole-genome sequence variants in OSeq genomic intervals
A pair-wise comparison was performed to examine the degree of overlap between the sets of bovine putative enhancer regions (Fig. 1). Villar H3K27ac and dbSUPER were the two major enhancer sets, because the Villar H3K27ac set covered 82% of the Villar H3K4me3 bovine genomic intervals, and the dbSUPER set covered 71% of the VISTA and 52% of the FANTOM5 bovine genomic intervals (Table 2; Fig. 1). However, the Villar H3K27ac and dbSUPER sets differed substantially (less than 5% overlaps; Table 2; Fig. 1).
Fig. 1.
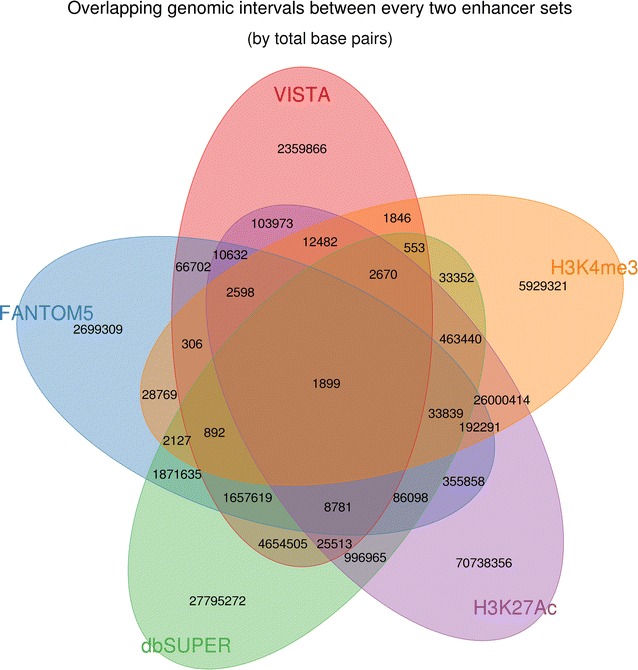
Degree of overlap between bovine enhancer sets (measured by total base pairs)
Table 2.
Degree of overlap between the sets of bovine enhancers analysed
| VISTAa (%) | FANTOM5b (%) | dbSUPERc (%) | H3K27acd (%) | H3K4me3e (%) | |
|---|---|---|---|---|---|
| VISTA | 100 | 19.63 | 71.29 | 1.89 | 0.26 |
| FANTOM5 | 24.92 | 100 | 52.18 | 9.86 | 3.74 |
| dbSUPER | 16.88 | 9.73 | 100 | 4.30 | 1.43 |
| H3K27ac | 0.17 | 0.70 | 1.63 | 100 | 26.97 |
| H3K4me3 | 0.07 | 0.80 | 1.65 | 81.66 | 100 |
Each value in the table represents the ratio, expressed as a percentage, of the total overlapping base pairs between the two enhancer sets listed in a row and column, relative to the total number of base pairs in the enhancer set listed in the corresponding row
aVISTA is a database for evolutionarily ultra-conserved sequences between vertebrates
bFANTOM5 is a database for active enhancers from various human and mouse tissue
cdDbSUPER is a database for super-enhancers from various human or mouse tissues across multiple studies
dH3K27ac represents the dataset from the Villar et al. [29] study, which used ChIP-Seq profiling to detect the regions of the bovine genome that contained the histone modification signal H3K27ac from four bulls’ liver tissues
eH3K4me3 represents the dataset from the Villar et al. [29] study which used ChIP-Seq profiling to detect the regions of the bovine genome that contained the histone modification signal H3K4me3 from four bull’s liver tissues
Given that enhancers are highly tissue-specific, we compared only liver-specific enhancers from homologous enhancer sets and Villar ChIP-Seq enhancer set. Only eight VISTA enhancers were liver-specific, which generated 236 bovine putative liver enhancers. We could not determine from which tissue FANTOM5 sequences originated. No liver data was available in the dbSUPER database. The bovine putative VISTA-liver enhancers overlapped very little with the bovine-liver H3K27ac (27%) and did not overlap at all with the bovine-liver H3K4me3 enhancers (Fig. 2).
Fig. 2.
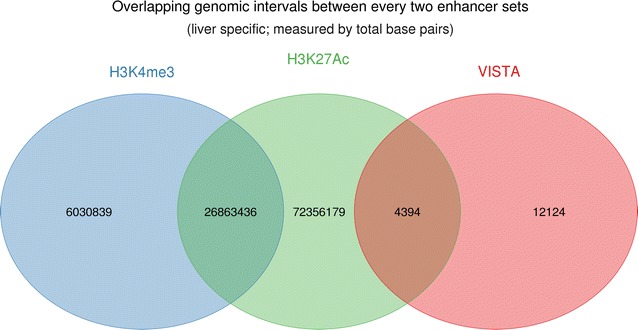
Degree of overlap between enhancer sets from liver (measured by total base pairs)
Genome-wide association study
The number of significant variants for each trait is in Table 3. Bulls and cows demonstrated similar GWAS profiles for the respective phenotype cohorts (Fig. 3).
Table 3.
Number of imputed whole-genome sequence variants and of significant variants
| Phenotype | GWAS | Meta-analysis | |||
|---|---|---|---|---|---|
| Gender | Variants tested | Significant variants | Filtered variants tested | Significant variants | |
| FY | Bulls | 28,899,038 | 3720 (0.013%) | 23,462,193 | 6967 (0.030%) |
| Cows | 3474 (0.012%) | ||||
| MY | Bulls | 4408 (0.015%) | 23,455,977 | 10,071 (0.043%) | |
| Cows | 6801 (0.024%) | ||||
| PY | Bulls | 1786 (0.006%) | 23,470,099 | 4804 (0.020%) | |
| Cows | 2981 (0.010%) | ||||
Fig. 3.
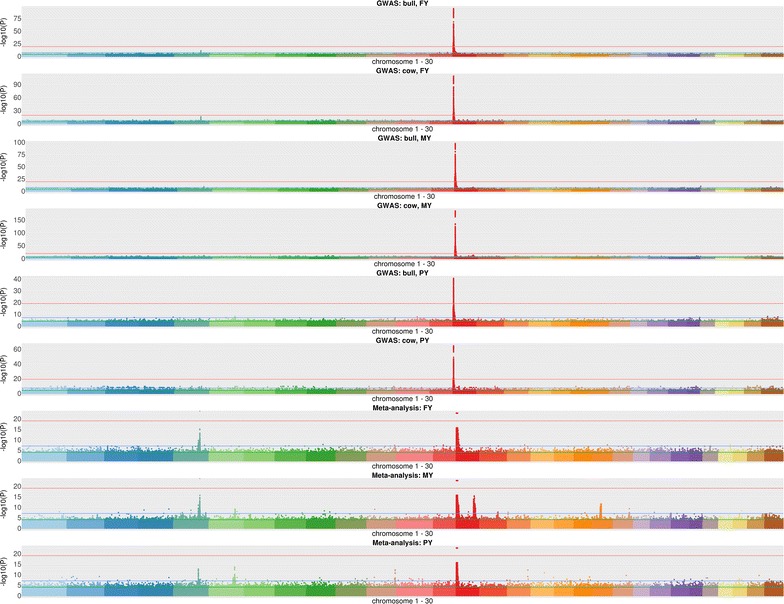
Manhattan plots: all GWAS and meta-analysis cohorts
Meta-analysis
The meta-analysis recovered 92% of the significant variants from GWAS, and revealed additional variants that were not significant in the separate bull and cow GWAS (Table 3) and [see Additional file 1: Figure S1]. Significant variants were clustered on chromosomes 5, 14 and 27 for FY, chromosomes 5, 6, 14, 15, 20 for MY and chromosomes 5, 6, 11, 12, 14 and 16 for PY (Fig. 3). The Villar H3K27ac enhancer set had the largest number of significant variants (FY: 419, MY: 538, and PY: 289 variants) that spread across chromosomes 5, 6, 14, 15, 16, 20 and 27, followed by the Villar H3K4me3 set (FY: 260, MY: 273, and PY: 191 variants) that spread across chromosomes 5 and 14, dbSUPER (FY: 19, MY: 43, and PY: 19 variants) that spread across chromosomes 5, 6, 14, 15 and 20, VISTA (FY: 4, and MY: 12 variants) that spread across chromosomes 5 and 14, and FANTOM5 (FY: 3, MY: 9, and PY: 1 variants) that spread across chromosomes 14 and 15. Villar H3K27ac and dbSUPER were the two major enhancer sets that captured significant variants, with the Villar H3K27ac set covering 78% of the Villar H3K4me3 significant variants, and dbSUPER covering 50% of the VISTA and 38% of the FANTOM5 significant variants (Table 4; Fig. 4). However, the significant variants in the Villar H3K27ac and dbSUPER enhancer sets differed significantly, with less than 2% of the dbSUPER and less than 0.1% of the H3K27ac significant variants being identical (Table 4; Fig. 4).
Table 4.
Degree of overlap between significant variants in the sets of bovine enhancers analysed
| VISTA (%) | FANTOM5 (%) | dbSUPER (%) | H3K4me3 (%) | H3K27Ac (%) | |
|---|---|---|---|---|---|
| VISTA | 100 | 31.25 | 50 | 0 | 0 |
| FANTOM5 | 38.46a | 100 | 38.46 | 0 | 0 |
| dbSUPER | 9.88 | 6.17 | 100 | 0 | 1.23 |
| H3K4me3 | 0 | 0 | 0 | 100 | 77.90 |
| H3K27Ac | 0 | 0 | 0.08 | 46.04 | 100 |
aEach value in the table represents the ratio, expressed as a percentage, of the total overlapping variants between the two enhancer sets listed in a row and column, relative to the total number of significant variants in the enhancer set listed in the corresponding row
Fig. 4.
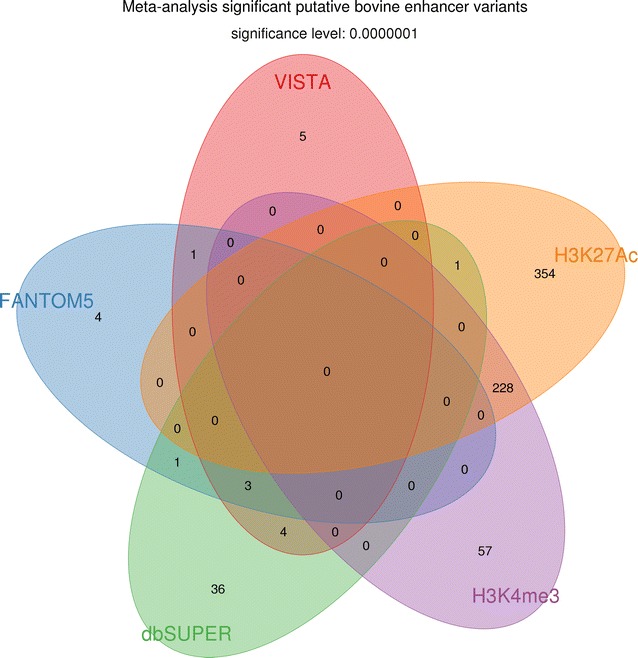
Degree of overlap between significant imputed WGS SNPs in bovine enhancer sets
Enrichment analysis
A permutation test with 10,000 repeats was performed to examine the enhancer sets for their global sequence wide significance. Only the Villar H3K4me3 and H3K27ac enhancers demonstrated genome-wide significance across all phenotypes , whereas the homology-based enhancers did not show such a high level of enrichment in significant variants associated with milk production traits (Table 5; Fig. 5). Since dbSUPER comprised clusters of enhancers, we expanded the length of dbSUPER putative bovine enhancers, such that any sequences that were within less than 30 kb to each other were merged into a single longer enhancer sequence. The permutation test was then applied to the expanded dbSUPER enhancer sequences but the enrichment signal remained low.
Table 5.
Enrichment of significant enhancer SNPs for milk production traits in the permutation tests
| Phenotype | Database | All SNPs/a subset of SNPs | Fold changea | Rankb |
|---|---|---|---|---|
| FY | VISTA | All | 0.16235 | 0.0001 |
| MY | 0.336793 | 0.0001 | ||
| PY | 0 | 0.0001 | ||
| FY | FANTOM5 | 0.200888 | 0.0001 | |
| MY | 0.414773 | 0.0018 | ||
| PY | 0.096781 | 0.0001 | ||
| FY | dbSUPER | 0.226716 | 0.0001 | |
| MY | 0.354855 | 0.0001 | ||
| PY | 0.329258 | 0.0001 | ||
| FY | Villar (H3K4me3) | 2.892337 | <0.0001 | |
| MY | 2.100798 | <0.0001 | ||
| PY | 3.081699 | <0.0001 | ||
| FY | Villar (H3K27ac) | 1.459357 | <0.0001 | |
| MY | 1.29579 | <0.0001 | ||
| PY | 1.458739 | <0.0001 | ||
| FY | Villar:H3K4me3 | H3K4me3-specific only | 3.358042 | <0.0001 |
| MY | 2.440737 | <0.0001 | ||
| PY | 4.414017 | <0.0001 | ||
| FY | Villar:H3K27ac | H3K27ac-sepcific only | 0.967677 | 0.3063 |
| MY | 1.023431 | 0.647 | ||
| PY | 0.953471 | 0.2794 | ||
| FY | Villar:H3K4me3 and H3K27ac | Overlaps: H3K4me3 and H3K27ac | 2.795722 | <0.0001 |
| MY | 2.025759 | <0.0001 | ||
| PY | 2.801491 | <0.0001 |
aFold change is the ratio between the actual number of significant SNPs in an enhancer set and the mean number of all significant SNPs in the 10,000 random samples
bRanking position of the actual number of significant SNPs in an enhancer set within the distribution of all the numbers of significant SNPs for the 10,000 random samples; if the actual number of significant SNPs was the largest among all the numbers of the 10,000 random significant SNPs, the rank was set to <0.0001; otherwise it was denoted as the ranking position of the actual number of significant SNPs among the number of random significant SNPs
Fig. 5.
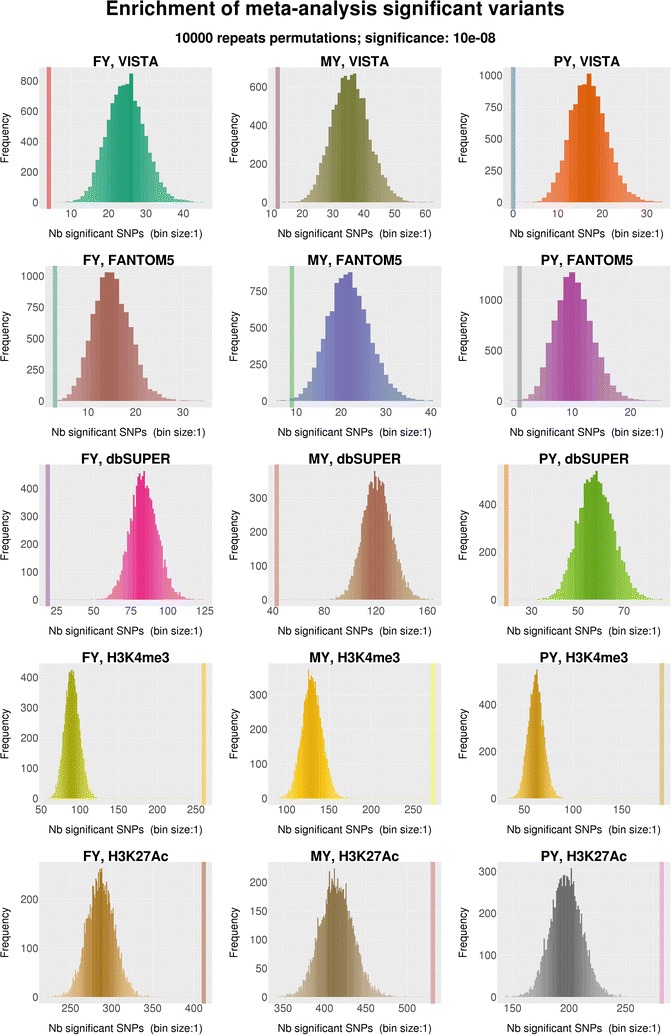
Enrichment of significant SNPs in all enhancer sets. The vertical line indicates the number of significant variants in the original analysis. The histograms represent the number of significant variants in random samplings. If an analysis was significant, the vertical line would be on the right to the histogram and clearly separated from the histogram
Only the bovine-specific Villar H3K4me3 and H3K27ac enhancer sets demonstrated high levels of enrichment whereas homology-based enhancer sets all showed low levels of enrichment in GSEA. Around 29% of the SNPs in the Villar H3K4me3 enhancer set and 35% of the SNPs in the H3K27ac enhancer set accounted for the enrichment signals in milk production traits [see Additional file 4: Table S1]. These GSEA core enhancer SNPs were located across all the chromosomes regardless of the phenotype cohorts or histone modification signals. The number of core H3K4me3 SNPs were, in decreasing order, within intronic, upstream, intergenic, 5′-UTR, downstream, 3′-UTR, splicing, non-coding exonic and stop regions (Figs. 6, 7), whereas the core H3K27ac SNPs followed a slightly different order, i.e. within intergenic, intronic, upstream, downstream, 5′-UTR, 3′-UTR, splicing, non-coding exonic and stop regions (Figs. 6, 7).
Fig. 6.
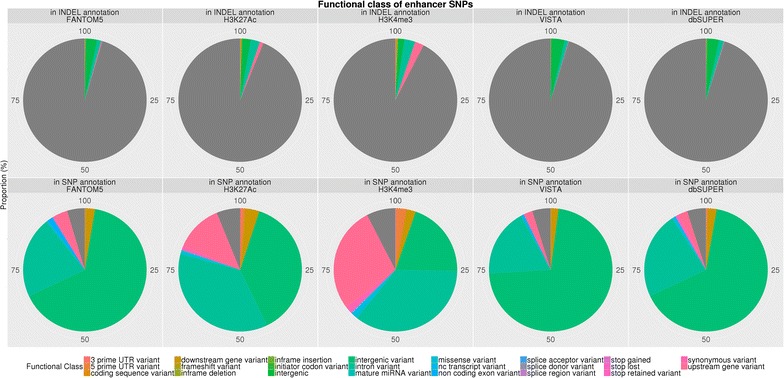
Functional class of enhancer SNPs. Each pie chart demonstrates the proportion of the total number of SNPs in the putative enhancer regions in the functional classes. The grey area represents variants without functional class annotation
Fig. 7.
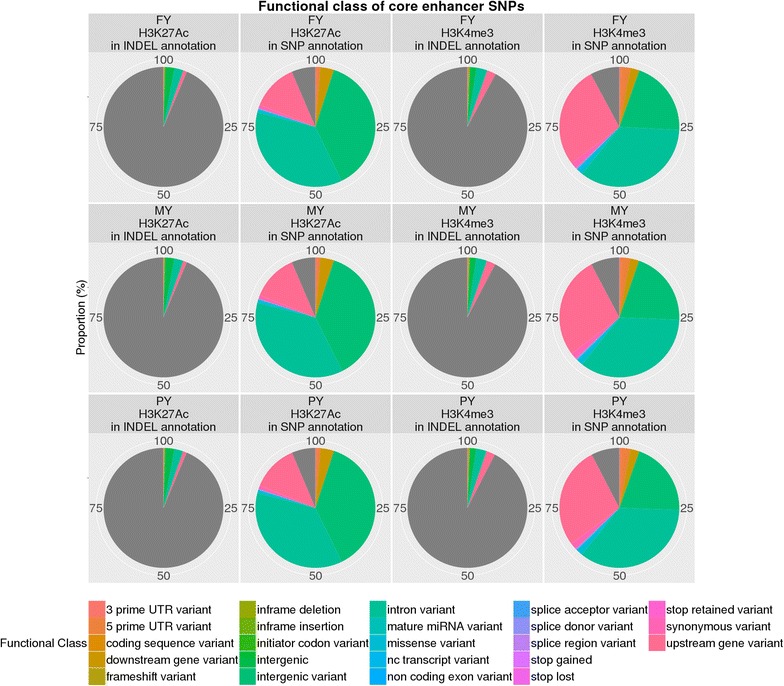
Functional class of core enhancer SNPs. Each pie chart demonstrates the proportion of the total number of SNPs in the Villar identified regions in the functional classes. The grey area represents variants without functional class annotation
To demonstrate the power of GSEA over the permutation test, we examined the relationship between P value threshold and SNP location. We found that the SNPs located close to a gene tended to be more significant than their counterparts in intergenic regions. Most H3K27ac-specific SNPs were intergenic and H3K4me3-specific SNPs were located in the vicinity of transcription start sites (TSS) [see Additional file 2: Figure S2 and Additional file 3: Figure S3]. As a result, the H3K4me3-specific SNPs tended to show a higher level of enrichment in the permutation test [see Additional file 4: Table S1]. However, the GSEA analysis revealed that more than 82% of the core SNPs responsible for the GSEA signal in the H3K4me3 set were also in the H3K27ac set, but more than 74% of the core SNPs in the H3K27ac set were not in the H3K4me3 set [see Additional file 5: Table S2]. This means that the H3K27ac-specific SNPs contributed some additional enrichment signal although their P values did not pass the threshold.
Discussion
The first goal of this study was to identify and improve the annotation of enhancer regions in the bovine genome. To create a library of bovine enhancers, we used publicly available human and mouse enhancer databases from VISTA, FANTOM5 and dbSUPER, along with the bovine enhancer data that were detected by ChIP-Seq from the Villar D et al. (2015) study. VISTA contains ultra-conserved developmental enhancer sequences with more than 96% of these being mapped to the bovine genome (Table 1). DbSUPER included more than 92% sequences that were mapped to the bovine genome (Table 1) probably because it contains long genomic sequences from clusters of enhancers that are closely located, which increases their chances of being mapped. The FANTOM5 data comprises very short sequences that were mapped very sparsely to the bovine genome when searched by sequence similarity in BLASTn (9.15%; Table 1) but were well recovered by liftOver which uses information from whole-genome comparisons to tolerate more frequent changes between query and target sequences (88.77%; Table 1). We exploited homologous mammalian enhancer data to identify bovine enhancers and the results are in agreement with previous findings [25, 29, 48–50] that showed that enhancer sequences, particularly the short and function-specific enhancers, are poorly conserved across species.
The second goal of this study was to validate our candidate bovine enhancer sites. We used a multi-breed GWAS followed by meta-analysis and enrichment analysis approach to examine if significant variants associated with milk production traits from meta-analyses are enriched in bovine putative enhancer sets. The genome-wide significant variants that were detected by this procedure are located in genes that affect milk production traits in cattle, in novel candidate genes, and in our candidate bovine enhancer sets. Both the permutation test and GSEA showed that only the Villar H3K4me3 and Villar H3K27ac predicted enhancer regions were significantly enriched with SNPs that are associated with the complex traits analysed here. The Villar H3K4me3 and H3K27ac enhancer sets were respectively 2.0 to 3.0-fold and 1.3 to 1.5-fold more enriched with variants that affect milk production traits than the rest of the genome (Table 5). Furthermore, the results of the permutation test and GSEA showed that the enriched H3K4me3 SNPs had significant effects within narrow genomic intervals close to genes. In addition, we observed that, in general, the H3K27ac enhancer regions encompassed the H3K4me3 enhancer regions but that most of the signals in the H3K27ac enhancer regions were located far from genes, and had small but significant effects. This finding is in line with existing literature that reports that the H3K4me3 enhancer regions display sharper peaks around TSS [51], the H3K27ac enhancer regions cover broader domains that are roughly equally distributed between intergenic and intronic regions [12], and that the proportion of SNPs at TSS reaching a significance level of −log10 (P value) higher than 10 is 50 to 100 times greater than that of SNPs in intergenic regions [52].
Our analysis did not show enrichment with enhancer regions for any production trait in any homology-based enhancer sets from VISTA, FANTOM5 and dbSUPER. There are two possible reasons for this finding. First, none of the VISTA, FANTOM5 and dbSUPER enhancer sets were sampled from a tissue that is directly linked to milk production (an example of tissue that is directly linked to milk production is the lactating mammary gland tissue). Therefore, the homology-based enhancers that are relevant to milk production may not be present in our downloaded databases and therefore cannot be considered in this study. Second, although VISTA, FANTOM5 and dbSUPER may contain sets from tissues that are involved in the physiological processes that are fundamental for the regulation of milk production, the procedure to map these sequences to the bovine genome is based on the identification of conserved sequences with human and mouse sequences, and as a result, the bovine-specific mutations within the homology-based enhancers cannot be captured [53]. Our results support the hypothesis of a rapid evolution of the enhancer sequences since the bovine-specific liver enhancer regions differed substantially from all homology-based liver enhancer regions (Fig. 2), which suggests that bovine-specific enhancers are more likely to be enriched with causative mutations that affect complex traits, in this case milk production. Our results, combined with the above reasons, highlight the complexity of the genomic regulatory machinery and the importance of analysing enhancers specific to the species under investigation [4]. The success of this study based on regulatory landscape data from one tissue type (liver) using two epigenetic marks (H3K4me3 and H3K27ac) indicates that our results might be even more convincing if we had data from more tissue types.
On chromosome 14, the observed enrichment signal in enhancer regions could be due to SNPs in linkage disequilibrium (LD) with the well-known mutation in the DGAT1 gene [54]. To account for LD confounding around the DGAT1 mutation, we re-ran our GWAS on chromosome 14 by correcting for the effect of the DGAT1 gene by including the causative mutation in the model as a fixed effect. The correlations of the SNP effects (P values) between before and after the correction were 85% (59%), which showed that there were other significant SNPs on chromosome 14 apart from the DGAT1 mutation. After correction, no significant SNPs remained in the VISTA and FANTOM5 enhancer sets for all milk production traits, but 34 to 67% significant SNPs remained in the Villar H3K4me3, Villar H3K27ac and dbSUPER enhancer sets [see Additional file 6: Table S3]. The SNPs that remained significant after the correction in the putative enhancer sets were located in regions up to 10 Mb around the DGAT1 gene. In addition, while the Villar H3K4me3 and dbSUPER enhancer sets had no corrected significant variants within the DGAT1 gene, the Villar H3K27ac enhancer set include one such significant variant (Chr14: 1797137 in FY and MY cohorts) in the first intron of the DGAT1 gene.
Several candidate regulatory variants that affect the expression of MGST1 have been reported to be responsible for the QTL effect on chromosome 5 for milk production traits [38, 55]. We found that they were within or close to the Villar H3K4me3 and H3K27ac enhancer regions, which provided evidence that the causal mutation is in fact a regulatory variant [see Additional file 7: Table S4 and Additional file 8: Figure S4].
Several studies have reported that the variant Chr6:88741762 is significantly associated with milk production traits [38]. This variant was significant in both our MY and PY cohorts, within the H3K27ac set, and is located 2470 bp upstream of the GC gene. An RNA-Seq analysis [56] showed that GC was most highly expressed in the liver and over-expressed in the mammary gland, and that there was a strong allele-specific expression in liver compared to 17 other bovine tissues [see Additional file 8: Figure S4].
Conclusions
This study used mammalian enhancer prediction data and bovine trait association to provide a functional variance analysis of candidate bovine enhancer regions. Overall, our findings agree with previous research that enhancer sequences are species-specific and rarely conserved across species. We conclude that bovine-specific histone data such as H3K4me3 and H3K27ac are essential for the successful functional annotation of bovine enhancer regions. Although the amount of bovine enhancer information is limited, we have successfully identified many genomic regions as potential enhancers and demonstrated that variation in these regions is associated with variation in animal production traits. Future studies will benefit from the combination of information from topological domain association, expression quantitative trait loci and bovine ChIP-Seq data, such as that generated from the Functional Annotation of Animal Genomes (FAANG) consortium [57], to accelerate the identification of mutations that affect complex traits.
Additional files
{kind=link}
Additional file 1: Figure S1. Comparison of the similarity between GWAS significant SNPs and meta-analysis significant SNPs (P ≤ 10−8). A meta-analysis significant variant was counted twice, one for the bull and one for the cow to account for the identity of a GWAS significant variant.
{kind=link}
Additional file 2: Figure S2. Comparison of the significant SNPs (P ≤ 10−8) between the Villar2015 H3K27ac and H3K4me3 enhancer sets.
{kind=link}
Additional file 3: Figure S3. Comparison of the functional class of significant SNPs (P ≤ 10−8) between the bovine liver H3K4me3 and H3K27ac enhancer sets.
Additional file 4: Table S1. Properties of GSEA core SNPs in the Villar (H3K4me3 and H3K27ac) enhancer sets. The proportion of significant GSEA core SNPs in the H3K27ac set doubled when the P value threshold decreased from 10−8 to 10−5 whereas the proportion of GSEA core SNPs in the H3K4me3 set did not increase as much.
Additional file 5: Table S2. Proportion of GSEA core SNPs in the H3K4me3 set that are also in the H3K27ac set, and proportion of the GSEA core SNPs in the H3K27ac set that are also in the H3K4me3 set.
Additional file 6: Table S3. Number of significant enhancer SNPs (P ≤ 10−8) that remained on chromosome 14 after correcting for the effect of the well-known DGAT1 mutation.
Additional file 7: Table S4. SNPs highlighted in previous studies [38, 55] that were also our GSEA core SNPs in the Villar (H3K4me3 and H3K7ac) sets.
{kind=link}
Additional file 8: Figure S4. Manhattan plot showing the enhancer signals around the MGST1 and GC genes.
Authors’ contributions
MW performed all the analysis and drafted the manuscript. TPH and BJH conceived the study and provided technical consultation. IMM provided insights into candidate causative mutations for MGST1 and GC using Bayesian analysis. BGC and JEP assisted with the design of the study and coordination. All authors read and approved the final manuscript.
Acknowledgements
We are tremendously grateful to Agriculture Victoria, Australia for funding this project. We gratefully thank DataGene for the provision of cattle genotypes and phenotypes. We are also immensely grateful to Dr Paul Stothard and his team at the University of Alberta for collating annotation information of sequence variants used in this study. We acknowledge other partners in the 1000 Bull Genomes Project for access to the dairy cattle genomes.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Programs, scripts and information for setting up the analysis can be obtained from the authors upon request.
Consent for publication
The authors agree for the publication of this manuscript to Genetic Selection Evolution.
Ethics approval and consent to participate
No animal experiments were performed specifically for this work. For the data that were obtained from existing sources, references for these experiments are provided.
Funding
The authors thank the DairyBio Initiative, which is funded by the Victorian Government (Melbourne, Australia), for the generous funding for this project.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12711-017-0331-4) contains supplementary material, which is available to authorized users.
Contributor Information
Min Wang, Email: 18589817@students.latrobe.edu.au.
Timothy P. Hancock, Email: timothy.hancock@ecodev.vic.gov.au
Iona M. MacLeod, Email: iona.macleod@ecodev.vic.gov.au
Jennie E. Pryce, Email: jennie.pryce@ecodev.vic.gov.au
Benjamin G. Cocks, Email: ben.cocks@ecodev.vic.gov.au
Benjamin J. Hayes, Email: ben.hayes@ecodev.vic.gov.au
References
- 1.Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME. Invited review: genomic selection in dairy cattle: progress and challenges. J Dairy Sci. 2009;92:433–443. doi: 10.3168/jds.2008-1646. [DOI] [PubMed] [Google Scholar]
- 2.Raven LA, Cocks BG, Kemper KE, Chamberlain AJ, Vander Jagt CJ, Goddard ME, et al. Targeted imputation of sequence variants and gene expression profiling identifies twelve candidate genes associated with lactation volume, composition and calving interval in dairy cattle. Mamm Genome. 2016;27:81–97. doi: 10.1007/s00335-015-9613-8. [DOI] [PubMed] [Google Scholar]
- 3.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G. Enhancers: five essential questions. Nat Rev Genet. 2013;14:288–295. doi: 10.1038/nrg3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 6.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 7.Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, et al. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet. 2008;40:158–160. doi: 10.1038/ng.2007.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13:469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, et al. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet. 2012;44:148–156. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]
- 12.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Nat Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26:909–922. doi: 10.1016/j.ccell.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Kimura H. Histone modifications for human epigenome analysis. J Hum Genet. 2013;58:439–445. doi: 10.1038/jhg.2013.66. [DOI] [PubMed] [Google Scholar]
- 22.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NCT, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 23.Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, et al. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011;30:4198–4210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 2013;155:1521–1531. doi: 10.1016/j.cell.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan A, Zhang X. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016;44:D164–D171. doi: 10.1093/nar/gkv1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, et al. Enhancer evolution across 20 mammalian species. Cell. 2015;160:554–566. doi: 10.1016/j.cell.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kodzius R, Kojima M, Nishiyori H, Nakamura M, Fukuda S, Tagami M, et al. CAGE: cap analysis of gene expression. Nat Methods. 2006;3:211–222. doi: 10.1038/nmeth0306-211. [DOI] [PubMed] [Google Scholar]
- 31.Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 32.Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brondum RF, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet. 2014;46:858–865. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 33.Pryce J, Hayes B. A review of how dairy farmers can use and profit from genomic technologies. Anim Prod Sci. 2012;52:180–184. doi: 10.1071/AN11172. [DOI] [Google Scholar]
- 34.Kemper KE, Reich CM, Bowman PJ, vander Jagt CJ, Chamberlain AJ, Mason BA, et al. Improved precision of QTL mapping using a nonlinear Bayesian method in a multi-breed population leads to greater accuracy of across-breed genomic predictions. Genet Sel Evol. 2015;47:29. doi: 10.1186/s12711-014-0074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15:478. doi: 10.1186/1471-2164-15-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10:R42. doi: 10.1186/gb-2009-10-4-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raven LA, Cocks BG, Hayes BJ. Multibreed genome wide association can improve precision of mapping causative variants underlying milk production in dairy cattle. BMC Genomics. 2014;15:62. doi: 10.1186/1471-2164-15-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacLeod IM, Bowman PJ, Vander Jagt CJ, Haile-Mariam M, Kemper KE, Chamberlain AJ, et al. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genomics. 2016;17:144. doi: 10.1186/s12864-016-2443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform. 2013;14:144–161. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grant JR, Arantes AS, Liao X, Stothard P. In-depth annotation of SNPs arising from resequencing projects using NGS-SNP. Bioinformatics. 2011;27:2300–2301. doi: 10.1093/bioinformatics/btr372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu ZL, Park CA, Wu XL, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41:D871–D879. doi: 10.1093/nar/gks1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borenstein M, Hedges LV, Higgins JPT, Rothstein HR. A basic introduction to fixed-effect and random-effects models for meta-analysis. Res Synth Methods. 2010;1:97–111. doi: 10.1002/jrsm.12. [DOI] [PubMed] [Google Scholar]
- 47.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Nat Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet. 2010;42:806–810. doi: 10.1038/ng.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, et al. Large-scale discovery of enhancers from human heart tissue. Nat Genet. 2012;44:89–93. doi: 10.1038/ng.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benayoun BA, Pollina EA, Uçar D, Mahmoudi S, Karra K, Wong ED, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158:673–688. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9:e1003449. doi: 10.1371/journal.pgen.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prabhakar S, Poulin F, Shoukry M, Afzal V, Rubin EM, Couronne O, et al. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006;16:855–863. doi: 10.1101/gr.4717506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grisart B, Farnir F, Karim L, Cambisano N, Kim JJ, Kvasz A, et al. Genetic and functional confirmation of the causality of the DGAT1 K232A quantitative trait nucleotide in affecting milk yield and composition. Proc Nat Acad Sci USA. 2004;101:2398–2403. doi: 10.1073/pnas.0308518100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Littlejohn MD, Tiplady K, Fink TA, Lehnert K, Lopdell T, Johnson T, et al. Sequence-based association analysis reveals an MGST1 eQTL with pleiotropic effects on bovine milk composition. Sci Rep. 2016;6:25376. doi: 10.1038/srep25376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chamberlain AJ, Vander Jagt CJ, Hayes BJ, Khansefid M, Marett LC, Millen CA, et al. Extensive variation between tissues in allele specific expression in an outbred mammal. BMC Genomics. 2015;16:993. doi: 10.1186/s12864-015-2174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andersson L, Archibald AL, Bottema CD, Brauning R, Burgess SC, Burt DW, et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015;16:57. doi: 10.1186/s13059-015-0622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Comparison of the similarity between GWAS significant SNPs and meta-analysis significant SNPs (P ≤ 10−8). A meta-analysis significant variant was counted twice, one for the bull and one for the cow to account for the identity of a GWAS significant variant.
Additional file 2: Figure S2. Comparison of the significant SNPs (P ≤ 10−8) between the Villar2015 H3K27ac and H3K4me3 enhancer sets.
Additional file 3: Figure S3. Comparison of the functional class of significant SNPs (P ≤ 10−8) between the bovine liver H3K4me3 and H3K27ac enhancer sets.
Additional file 4: Table S1. Properties of GSEA core SNPs in the Villar (H3K4me3 and H3K27ac) enhancer sets. The proportion of significant GSEA core SNPs in the H3K27ac set doubled when the P value threshold decreased from 10−8 to 10−5 whereas the proportion of GSEA core SNPs in the H3K4me3 set did not increase as much.
Additional file 5: Table S2. Proportion of GSEA core SNPs in the H3K4me3 set that are also in the H3K27ac set, and proportion of the GSEA core SNPs in the H3K27ac set that are also in the H3K4me3 set.
Additional file 6: Table S3. Number of significant enhancer SNPs (P ≤ 10−8) that remained on chromosome 14 after correcting for the effect of the well-known DGAT1 mutation.
Additional file 7: Table S4. SNPs highlighted in previous studies [38, 55] that were also our GSEA core SNPs in the Villar (H3K4me3 and H3K7ac) sets.
Additional file 8: Figure S4. Manhattan plot showing the enhancer signals around the MGST1 and GC genes.
Data Availability Statement
Programs, scripts and information for setting up the analysis can be obtained from the authors upon request.