

Abstract
S-Adenosyl methionine (SAM)-dependent C-methyltransferases are responsible for the C2-methylation of 3-ketoacyl-acyl carrier protein (ACP) intermediates to give the corresponding 2-methy-3-ketoacyl-ACP products during bacterial polyketide biosynthesis mediated by trans-AT polyketide synthases that lack integrated acyl transferase (AT) domains. A coupled ketoreductase (KR) assay was used to assign the stereochemistry of the C-methyltransferase-catalyzed reaction. Samples of chemoenzymatically-generated 3-ketopentanoyl-ACP (9) were incubated with SAM and BonMT2 from module 2 of the bongkrekic acid polyketide synthase. The resulting 2-methyl-3-ketopentanoyl-ACP (10) was incubated separately with five (2R)- or (2S)-methyl specific KR domains. Analysis of the derived 2-methyl-3-hydroxypentanoate methyl esters (8) by chiral GC-MS established that the BonMT2-catalyzed methylation generated exclusively (2R)-2-methyl-3-ketopentanoyl-ACP ((2R)-10). Identical results were also obtained with three additional C-methyltransferases – BaeMT9, DifMT1, and MupMT1 – from the bacillaene, difficidin, and mupirocin trans-AT polyketide synthases
Graphical abstract
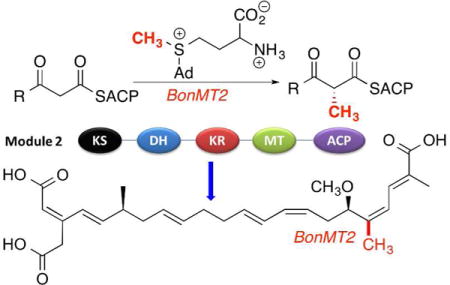
Modular polyketide synthases (PKSs) are polyfunctional megaproteins that are responsible for the bacterial synthesis of an enormous range of physiologically active natural products, including many important antibiotics, antitumor agents, and immunosuppressants. Within a typical PKS, each module is responsible for a separate round of polyketide chain elongation and modification. PKS modules fall into two broad functional classes. In cis-AT polyketide synthases, typified by the extensively studied 6-deoxyerythronolide B synthase of erythromycin biosynthesis,1 each module harbors a dedicated acyl transferase (AT) domain that is responsible for loading a specific chain elongation substrate, most often malonyl-CoA or methylmalonyl-CoA, onto the pantetheinyl arm of a paired acyl carrier protein (ACP) domain within the same module. The resultant malonyl- or methylmalonyl-ACP then serves as the substrate for chain elongation by decarboxylative condensation catalyzed by the paired ketosynthase (KS) domain so as to generate the respective 3-ketoacyl-ACP or (2R)-2-methyl-3-ketoacyl-ACP intermediate. Different degrees of further functional group modification catalyzed by variable combinations of ketoreductase (KR), dehydratase (DH), and enoyl-reductase (ER) domains may then occur before transfer to the KS domain of the proximal downstream module for a further round of chain elongation and functional group modification. By contrast, trans-AT polyketide synthases lack integrated AT domains, relying instead on one or more discrete AT proteins to supply the requisite building blocks, most often malonyl-CoA, to each PKS module.2 As an alternative to methylmalonyl-CoA, certain modules harbor an integrated, S-adenosylmethionine (SAM)-dependent C-methyltransferase (C-MT) that converts the initially generated 3-ketoacyl-ACP intermediate to the corresponding 2-methyl-3-ketoacyl-ACP. Of 48 trans-AT PKSs for which the resultant natural product has been assigned, 47 (98%) harbor 1–10 C-methyltransferases (Tables S1 and S2), as represented by bongkrekic acid (1),3 bacillaene (2),4 difficidin (3),4a and mupirocin (4)5 (Figure 1). By contrast, embedded C-methyltransferases are considerably less common in cis-AT PKS systems, with only 9 out of 168 (5%) harboring 1 or 2 C-methyltransferases and only gephyronic acid synthase containing as many as 6 (Tables S1 and S2).
Figure 1.
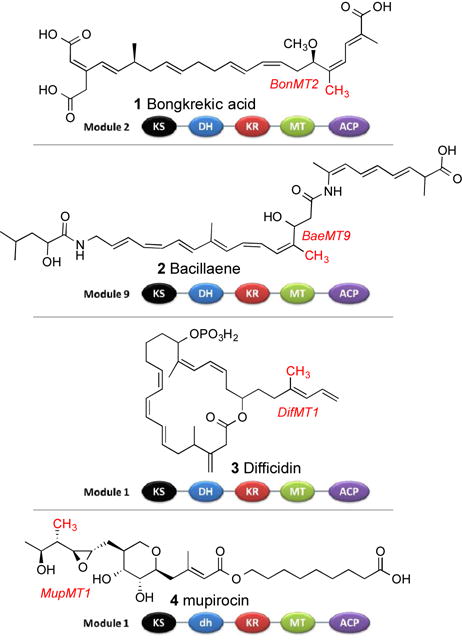
Representative polyketides produced by trans-AT polyketide synthases and domain organization of selected modules harboring C-methyltransferase domains, with the resultant methyl groups highlighted in red.
The stereochemistry of the various C-methylation reactions cannot be inferred from the structure and stereochemistry of the ultimately formed polyketide products, since the configuration of each of the initially generated 2-methyl-3-ketoacyl-ACP intermediates is obscured by a variety of intervening PKS-catalyzed transformations, including possible epimerization of the unreduced 2-methyl-3-ketoacyl-ACP intermediate, catalyzed by a redox-inactive KR0 or DH domain,6 by KR-catalyzed reduction, with or without epimerization,7 or DH-catalyzed dehydration. Although a number of excised PKS C-methyltranferases have been expressed as recombinant proteins and shown to catalyze the expected SAM-dependent methylation of 3-ketoacylthioesters, the stereochemistry of the resulting 2-methyl-3-ketoacyl thioester products has not been determined.8,9
Recently, we described a versatile coupled enzyme assay utilizing a combination of naturally occurring and engineered, epimerase-inactive KR domains that are strictly specific for either (2R)- or (2S)-2-methyl-3-ketoacyl thioesters.6b Using this assay, we established unambiguously that the decarboxylative condensation catalyzed by the NanKS1 domain of nanchangmycin synthase generates exclusively a (2R)-2-methyl-3-ketoacyl-ACP product. In fact, the coupled KR-assay is entirely general and can be used to assign the stereochemistry of any chemoenzymatically generated 2-methyl-3-ketoacyl-ACP. We report below the determination of the stereospecificity of C-methyltransferases from four different trans-AT PKS systems illustrated in Figure 1, each of which was shown to catalyze the exclusive formation of the (2R)-2-methyl-3-ketoacyl-ACP product.
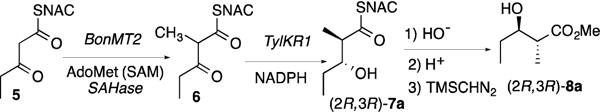
BonMT2, the prospective C-methyltransferase from module 2 of the bongkrekic acid (1) PKS (Figure 1), was expressed in Escherichia coli as an N-terminal His6-tagged protein from a codon-optimized synthetic gene using consensus domain boundaries (Figures S1 and S2, Table S3). Incubation of BonMT2 with the N-acetylcysteamine thioester 3-ketopentanoyl-SNAC (5) and SAM, in the presence of S-adenosyl homocysteine (SAH) nucleosidase to relieve potent product inhibition by SAH,8,10 gave 2-methyl-3-ketopentanoyl-SNAC (6), as established by HPLC-MS (Scheme 1, Figure S3). The product was further characterized by reduction with TylKR1 from module 1 of the tylactone synthase6,7b,7e in the presence of NADPH to give (2R,3R)-2-methyl-3-hydroxypentanoyl-SNAC (7a), whose structure and stereochemistry were established by base-catalyzed thioester hydrolysis and treatment with trimethylsilyldiazomethane (TMS-CHN2). Chiral GC-MS analysis of the derived methyl esters confirmed predominant formation of the characteristic reduction product, methyl (2R,3R)-2-methyl-3-hydroxypentanoate (8a) (Figure S4).
Scheme 1.
Further biochemical characterization of BonMT2 required 3-ketopentanoyl-BonACP2 (9), free of potentially interfering 3-ketopentanoyl-SNAC (5) or 3-ketopentanoyl-CoA.11 To this end, malonyl-BonACP2, prepared from recombinant apo-BonACP2 and malonyl-CoA using the phosphopantetheinyl transferase Sfp,12 was incubated with recombinant BonKS2 which had been preincubated with propionyl-SNAC (Scheme 2a). The formation of 9 was established by LC-ESI-MS analysis of an aliquot after desalting and concentration by ultrafiltration. After addition of BonMT2, SAM, and SAH nucleosidase, the incubation was continued for an additional 4 h. Following removal of BonKS2 and BonMT2 by ultrafiltration, the mixture was analyzed by LC-ESI-MS, confirming formation of the predicted 2-methyl-3-ketopentanoyl-BonACP2 (10), MD 15556, corresponding to an increase of 14 amu over the substrate 3-ketopentanoyl-BonACP2 (9), MD 15542 (Table S6, Figure S8).
Scheme 2.
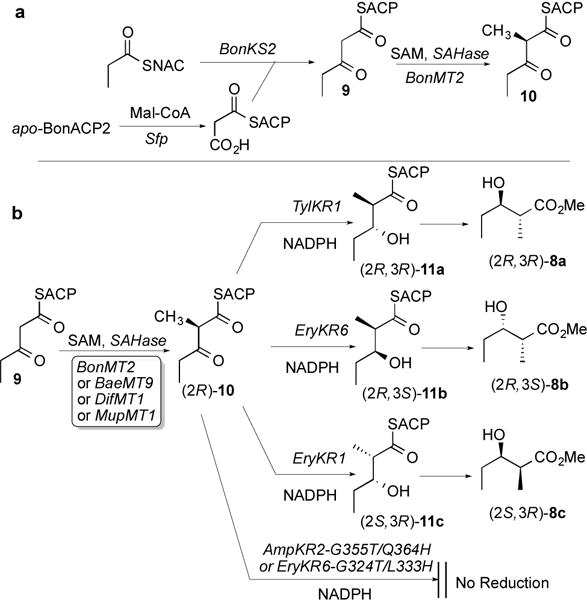
Having established that BonMT2 can catalyze the predicted SAM-dependent, C2-methylation of 3-ketopentanoyl-BonACP2 (9), we then determined the stereochemistry of the reaction using the coupled ketoreductase assay. To this end, BonMT2, in the presence of SAH nucleosidase, was used to convert a mixture of SAM and 3-ketopentanoyl-BonACP2 (9), chemoenzymatically generated in situ as described above, to the corresponding 2-methyl-3-ketopentanoyl-BonACP2 (10) (Scheme 2b). After 15-min incubation at room temperature, 800-μL aliquots of the reaction mixture were added to individual solutions containing excess NADPH and each of the following five ketoreductases: TylKR1, EryKR6, EryKR1, EryKR6-G324T/L333H, and AmpKR2-G355T/Q364H.6b,13 Aliquots of each KR incubation mixture were withdrawn after 15, 30, and 60 min, then quenched and hydrolyzed with aq. NaOH. After acidification the resulting 2-methyl-3-hydroxypentanoic acids were converted using TMS-CHN2 to the corresponding methyl esters 8 which were analyzed by chiral GC-MS (Figures S9 and S10). Reduction with the individual epimerase-inactive, (2R)-2-methyl-3-ketoacyl-ACP-specific ketoreductases TylKR1 and EryKR6 resulted in time-dependent, diastereospecific formation of exclusively methyl (2R,3R)-2-methyl-3-hydroxypentanoate (8a) and methyl (2R,3S)-2-methyl-3-hydroxypentanoate (8b), respectively, consistent with the established substrate and product specificity of each KR domain (Table S7).7a,7c In like manner, incubation with epimerase-active EryKR1 gave the corresponding methyl (2S,3R)-2-methyl-3-hydroxypentanoate (8c).7c,14 By contrast, neither of the epimerase-inactive, (2S)-2-methyl-3-ketoacyl-ACP-specific, mutant ketoreductases, EryKR6-G324T/L333H or AmpKR2-G355T/Q364H, produced any detectable methyl 2-methyl-3-hydroxypentanoate (8), even after 60 min incubation. These combined results therefore conclusively demonstrate that the BonMT2-catalyzed C2-methylation of 3-ketopentanoyl-BonACP2 (9) is completely stereospecific for formation of (2R)-2-methyl-3-ketopentanoyl-BonACP2 ((2R)-10).
We also carried out the analogous sets of coupled KR assays with each of the previously characterized C-methyltransferases from trans-AT PKSs: BaeMT9, from module 9 of the bacillaene PKS, DifMT1 from module 1 of the difficidin PKS, and MupMT1 from module 1 of the mupirocin PKS (Scheme 2b, Figure S10, Table S7).8 In each series of incubations, only the (2R)-2-methyl-3-ketoacyl-ACP-specific KR domains gave rise to time-dependent formation of the corresponding reduced diketide acid methyl esters (8a, 8b, and 8c from TylKR1, EryKR6, and EryKR1, respectively), while neither of the two (2S)-2-methyl-3-ketoacyl-ACP-specific KR domains produced any reduced 2-methyl-3-hydroxy diketide products. It can therefore confidently be concluded that BaeMT9, DifMT1, and MupMT1 are each completely stereospecific for conversion of their 3-ketoacyl-ACP substrate to the corresponding (2R)-2-methyl-3-ketoacyl-ACP product.15
In bacterial polyketide biosynthesis, nature has evolved two complementary strategies for the generation of 2-methyl-3-ketoacyl-ACP intermediates. For canonical cis-AT PKSs, (2S)-methylmalonyl-CoA serves as the substrate for each integrated AT domain, resulting in the formation of a (2R)-2-methyl-3-ketoacyl-ACP intermediate by decarboxylative condensation of methylmalonyl-ACP with the electrophilic acyl-ACP primer or partially elaborated polyketide chain.6b,7a,7c,16 By contrast, trans-AT PKSs rely on one or more discrete, primarily malonyl-CoA-specific AT domains, leading to the generation of a 3-ketoacyl-ACP intermediate as the product of the KS-catalyzed chain elongation reaction. Those trans-AT modules that harbor an integrated SAM-dependent C-methyltransferase can then generate the corresponding 2-methyl-3-ketoacyl-ACP. Using the coupled ketoreductase assay, we have now demonstrated that all four of the individual C-methyltransferases analyzed – BonMT2, BaeMT9, DifMT1, MupMT1 – catalyze stereospecific methylation to yield exclusively the (2R)-2-methyl-3-ketoacyl-ACP product. Furthermore, multiple sequence alignment of a representative set of 38 PKS C-methyltransferase domains reveals a high level of sequence conservation and, most importantly, no significant sequence segregation according to whether the eventually formed polyketide displays a trisubstituted Z-double bond (6 C-MTs, including BonMT2 and BaeMT9), a trisubstituted E-double bond (12 C-MTs, including DifMT1), or a 2-methyl-3-hydroxyacyl-ACP-derived architecture (20 C-MTs, including MupMT1) (Figure S14), suggesting that the formation of the (2R)-2-methyl-3-ketoacyl-ACP intermediate is likely to be general across all PKS systems.
Unlike cis-AT PKSs, the KR domains in trans-AT PKS modules harboring C-methyltransferases must not reduce the initially generated 3-ketoacyl-ACP before the requisite formation of the 2-methyl-3-ketoacyl-ACP. We surmise that this discrimination results from the relative values of kcat/Km for processing of the 3-ketoacyl-ACP intermediate by the C-MT and KR domains.17
In mupirocin, the substructure derived from Mup module 1 has the (12S,13S)-12-methyl-13-hydroxy configuration (Figure 1), suggesting that MupKR1 converts the (2R)-2-methyl-3-ketobutyryl-ACP intermediate generated by MupMT1 to the corresponding (2S,3S)-2-methyl-3-hydroxybutyryl-ACP product (epimerizing A2-type KR domain). Interestingly, in sequence alignments MupKR1 lacks the characteristic A2-type sequence markers7d,7e and appears to cluster instead with EryKR6 and other non-epimerizing A1-type domains that generate (2R,3S)-2-methyl-3-hydroxybutyryl-ACP products (Figure S15).
In bongkrekic acid (1), the configuration of the intermediate 2-methyl-3-hydroxy-4-methylhex-3-enoyl-ACP generated by BonKR2 is obscured in the final product by the BonDH2-catalyzed dehydration. To address this issue, we have recently expressed BonKR2 and established that it is a non-epimerizing A1-type KR that stereospecifically converts (2R)-2-methylpentanoyl-ACP to the corresponding (2R,3S)-2-methyl-3-hydroxypentanoyl-ACP.18 Notably, syn dehydration by BonDH2 would be expected to generate the (Z)-3-methyl-2-enoyl-ACP intermediate from which bongkrekic acid (1) is directly derived. This and related transformations are currently under investigation.
Supplementary Material
Acknowledgments
This work was supported by grants from the U. S. National Institutes of Health, GM022172 to D.E.C. and GM087934 to C.K. We thank Prof. Adrian Keatinge-Clay and Drew T. Wagner of the University of Texas, Austin for a generous gift of the expression plasmids for BaeMT9, DifMT1, and MupMT1.
Footnotes
Supporting Information.
Experimental methods, including C-MT design and expression, coupled KR assays, kinetic assays, and GC-MS and ESI-MS analysis. This material is available free of charge at http://pubs.acs.org.
Notes
No competing financial interests have been declared.
References
- 1.a) Cane DE. J Biol Chem. 2010;285:27517–27523. doi: 10.1074/jbc.R110.144618. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hopwood DA. Methods in Enzymology Complex enzymes in microbial natural product biosynthesis, part B: polyketides, aminocoumarins and carbohydrates. Academic Press; United States: 2009. p. 459. [DOI] [PubMed] [Google Scholar]
- 2.a) Cheng YQ, Tang GL, Shen B. Proc Natl Acad Sci U S A. 2003;100:3149–3154. doi: 10.1073/pnas.0537286100. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Helfrich EJ, Piel J. Nat Prod Rep. 2016;33:231–316. doi: 10.1039/c5np00125k. [DOI] [PubMed] [Google Scholar]
- 3.Moebius N, Ross C, Scherlach K, Rohm B, Roth M, Hertweck C. Chem Biol. 2012;19:1164–1174. doi: 10.1016/j.chembiol.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 4.a) Chen XH, Vater J, Piel J, Franke P, Scholz R, Schneider K, Koumoutsi A, Hitzeroth G, Grammel N, Strittmatter AW, Gottschalk G, Sussmuth RD, Borriss R. J Bacteriol. 2006;188:4024–4036. doi: 10.1128/JB.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moldenhauer J, Chen XH, Borriss R, Piel J. Angew Chem Int Ed Engl. 2007;46:8195–8197. doi: 10.1002/anie.200703386. [DOI] [PubMed] [Google Scholar]; c) Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. Proc Natl Acad Sci U S A. 2007;104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Sayed AK, Hothersall J, Cooper SM, Stephens E, Simpson TJ, Thomas CM. Chem Biol. 2003;10:419–430. doi: 10.1016/s1074-5521(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 6.a) Garg A, Xie X, Keatinge-Clay A, Khosla C, Cane DE. J Am Chem Soc. 2014;136:10190–10193. doi: 10.1021/ja5056998. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xie X, Garg A, Khosla C, Cane DE. J Am Chem Soc. 2017;139:3283–3292. doi: 10.1021/jacs.7b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Castonguay R, He W, Chen AY, Khosla C, Cane DE. J Am Chem Soc. 2007;129:13758–13769. doi: 10.1021/ja0753290. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Castonguay R, Valenzano CR, Chen AY, Keatinge-Clay A, Khosla C, Cane DE. J Am Chem Soc. 2008;130:11598–11599. doi: 10.1021/ja804453p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Valenzano CR, Lawson RJ, Chen AY, Khosla C, Cane DE. J Am Chem Soc. 2009;131:18501–18511. doi: 10.1021/ja908296m. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) You YO, Khosla C, Cane DE. J Am Chem Soc. 2013;135:7406–7409. doi: 10.1021/ja4014776. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Keatinge-Clay AT. Chem Biol. 2007;14:898–908. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Stevens DC, Wagner DT, Manion HR, Alexander BK, Keatinge-Clay AT. J Antibiot. 2016;69:567–570. doi: 10.1038/ja.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skiba MA, Sikkema AP, Fiers WD, Gerwick WH, Sherman DH, Aldrich CC, Smith JL. ACS Chem Biol. 2016;11:3319–3327. doi: 10.1021/acschembio.6b00759. These authors stated that the acidity of the H-2 proton of the 2-methyl-3-ketoacyl-ACP generated by CurJ precludes chemoenzymatic assignment of the C2-methyl stereochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hendricks CL, Ross JR, Pichersky E, Noel JP, Zhou ZS. Anal Biochem. 2004;326:100–105. doi: 10.1016/j.ab.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 11.The use of the ACP-conjugated substrate is essential since the rate of epimerization of the 2-methyl-3-ketoacyl-ACP has been shown to be <0.003 min−1, compared to a kepim for the considerably more labile -SNAC derivative of ~0.15 min−1 (refs 7a and 7c).
- 12.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 13.Zheng J, Piasecki SK, Keatinge-Clay AT. ACS Chem Biol. 2013;8:1964–1971. doi: 10.1021/cb400161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garg A, Khosla C, Cane DE. J Am Chem Soc. 2013;135:16324–16327. doi: 10.1021/ja408944s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Although competing reduction of non-methylated 3-ketopentanoyl-ACP would not have affected the validity of the assignment of the stereochemistry of the C2-methylation using the coupled KR assay, the corresponding 3-hydroxypentanoyl-ACP was notably absent from the assay products, as analyzed by chiral GC-MS of the derived methyl esters. Control kinetic assays and stereochemical analysis showed that while each of the KR domains used in the assay was capable of reducing the unmethylated 3-ketopentanoyl-SNAC substrate, the relative kcat/Km for reduction of (±)-2-methyl-3-ketopentanoyl-SNAC by TylKR1, EryKR6, or EryKR1 was typically 3–5 times greater than that for reduction of 3-ketopentanoyl-SNAC, with the relative kcat/Km values for the (2S)-methyl-specific EryKR6-G324T/L333H and AmpKR2-G355T/Q364H being 55 and 2, respectively (Tables S4, S5, and S8, Figures S5–S7, S11–S13). Both TylKR1 and AmpKR2-G355T/Q364H reduced 3-ketopentanoyl-SNAC exclusively to (3R)-3-hydroxypentanoyl-SNAC, while reduction by either EryKR6 or EryKR6-G324T/L333H gave essentially racemic 3-hydroxypentanoyl-SNAC. A further control showed that the 3-ketopentanoyl-ACP intermediate was generated continuously over the 1-h time course of the ketoreductase assay.
- 16.Marsden AF, Caffrey P, Aparicio JF, Loughran MS, Staunton J, Leadlay PF. Science. 1994;263:378–380. doi: 10.1126/science.8278811. [DOI] [PubMed] [Google Scholar]
- 17.Cacho RA, Thuss J, Xu W, Sanichar R, Gao Z, Nguyen A, Vederas JC, Tang Y. J Am Chem Soc. 2015;137:15688–15691. doi: 10.1021/jacs.5b11814. The kinetics and relative substrate specificity of the competition between C-MT and KR domains have been analyzed in detail for the iterative fungal PKS lovastatin synthase: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie X. unpublished observation [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.