

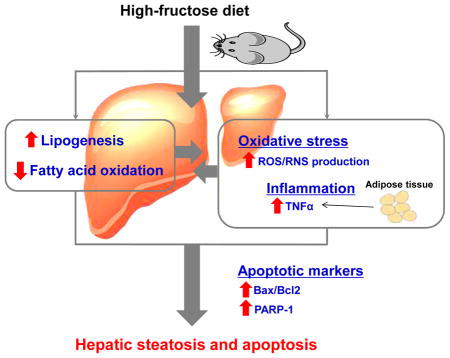
Graphical abstract
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) represents the most prominent form of chronic liver diseases, affecting numerous people at different ages (Wieckowska et al., 2007). Despite advances in this field, knowledge on the pathogenesis of NAFLD is still incomplete and the understanding of the mechanisms underlying the development of NAFLD is of extreme importance. According to the most widespread and prevailing model of “two-hit hypothesis”, the “first hit” involves lipid accumulation in the hepatocytes (Duvnjak et al., 2007). The first 'hit' is the accumulation of intrahepatic lipid (hepatic steatosis) owing to an imbalance of normal hepatic lipid metabolism of fat synthesis and oxidation, which results in either excessive lipid influx, decreased lipid clearance, or both (Duvnjak et al., 2007). The “first hit” increases the vulnerability of the liver to many additional factors that constitute the “second hit” and promote hepatic injury such as apoptosis, inflammation and fibrosis/cirrhosis. Increased oxidative stress, pro-inflammatory cytokines/chemokines, endotoxemia, insulin resistance and adipokines are considered “second hit” (Buzzetti et al., 2016).The resulting lobular inflammation leads to ballooning degeneration and perisinusoidal fibrosis, which are preceded by hepatic apoptosis and inflammation, with resulting scarring and progression to non-alcoholic steatohepatitis (NASH) (Syn et al., 2009). Understanding of the etiology and different dietary factors that induce the “two hits” is essential in the prevention and treatment of NAFLD.
There is strong evidence that the diet may affect the development of NAFLD (Le and Bortolotti, 2008). Dietary fructose is a major candidate for causing NAFLD. Fructose is a monosaccharide which is commonly used as a sweetener, e.g., in high fructose corn syrups. Industrially, it is frequently found in soft drinks and pre-packaged foods (Akar et al., 2012). Epidemiologic data suggest that there has been a significant rise in calories consumed from saturated fat and fructose rich foods (Bray et al., 2004). Fructose consumption accounts for approximately 10.2% of all calories in our average diet in the United States (Vos et al., 2008). Fructose intake is 2–3 fold higher in patients with NASH compared to BMI-matched controls and recently daily fructose ingestion has been associated with increased hepatic fibrosis (Ouyang et al., 2008). This has been paralleled by an increasing prevalence of obesity and its associated hepatic comorbidity, namely NAFLD (Cave et al., 2007). A correlation is observed between dietary fructose intake and the prevalence of metabolic syndrome and fatty liver (Bantle, 2009). Recent data suggest that increased fructose consumption elevates fat mass, de novo lipogenesis and inflammation while it promotes insulin resistance and post-prandial hypertriglyceridemia, particularly in overweight individuals (Cave et al., 2007). Further, studies have indicated that the development of NAFLD may be frequently associated with excessive dietary fructose consumption (Ouyang et al., 2008). Whether increased fructose consumption positively correlates with the development of NAFLD or promotes the transition from NAFLD to NASH and more advanced stages of liver damage remains unclear. The role of dietary saturated fat and fructose in triggering these mechanism(s) of fibrosis progression in NASH still need further investigations.
Even though growing evidence suggests that fructose contributes to the development and severity of NAFLD, the biological mechanism underlying fructose- caused NAFLD occurrence and progression to NASH is not clearly understood and is probably due to a number of other factors that are expressed in a context of genetic predisposition and sedentary life style. Increased oxidative stress is a major contributor in the pathogenesis of NAFLD (Browning and Horton, 2004). It is suggested that increased accumulation of liver triglycerides leads to increased oxidative stress in the hepatocytes of animals and humans (Browning and Horton, 2004). Data from animal models have shown increased oxidative stress due to increased fat influx into the liver (Hensley et al., 2000). In addition, pathological increases in cell death in the liver as well as in peripheral tissues have emerged as an important mechanism involved in the development and progression of NAFLD (Alkhouri et al., 2011). In fact, increased hepatocyte cell death mainly by apoptosis is frequently observed in patients with NAFLD (Alkhouri et al., 2011; Feldstein et al., 2003; Ribeiro et al., 2004). However, little has been reported about the fructose-induced oxidative stress and the development of hepatic apoptosis in NAFLD in the context of the “two-hit hypothesis.” Based on the current background, we aimed to evaluate the effects of dietary high fructose on hepatic lipid accumulation and increased oxidative stress with inflammation as the first and second hits, respectively, and then study the underlying mechanisms for the development of hepatocyte apoptosis and adipose inflammation in young female C57BL/6J mice.
2. Materials and methods
2.1. Animal model
Animal experiments were performed in accordance with the National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee. Age-matched (6 weeks) female C57BL/6 mice were obtained from Jackson Laboratories. After one week of acclimation, mice were randomly assigned to two groups (n≥6/group) fed either a regular chow as control diet (ContD, 7017 NIH-31 open formula mouse/rat sterilizable diet, http://www.envigo.com/resources/data-sheets/7017-datasheet-0915.pdf) or a high (H)-fructose (FR) diet (D) (HFRD, 35% fructose-derived calories, D15101101 specially formulated by Research Diets, New Brunswick, NJ, USA) for 3 weeks (Table 1). The NIH-31 rodent diet contains simple sugar content from natural ingredients at an estimated content of 2–5% by weight. There was also no additional mono- or disaccharide such as glucose, fructose and sucrose or otherwise added. In addition, generally rodent chow contains <0.5% fructose whereas HFRD contains 35% fructose. The mice were housed in groups of three per cage at 22°C with a 12 h light/dark cycle and given free access to diet and water. Body weight for each animal and their food intake were recorded weekly during the feeding period. After 3 weeks, the livers and adipose tissues were excised from the mice, which were fasted overnight (12 h) and immediately snap frozen, while individual trunk blood samples following decapitation of sedated mice were collected for serum preparation, as described (Choi et al., 2016a; Choi et al., 2016b). All samples were stored at −80°C until analysis.
Table 1.
Composition of standard chow control diet (ContD) and HFRD.
| ContD (3.0 kcal/g) | HFRD* (3.9 kcal/g) | ||
|---|---|---|---|
|
| |||
| % kcal from | % kcal from | ||
| Protein | 24 | 21 | |
| Carbohydrate | 62 | 68 | |
| Fat | 14 | 12 | |
|
| |||
| Ingredient | g | kcal | |
|
| |||
| Casein | 200 | 800 | |
| DL-Methionine | 3 | 12 | |
| Corn Starch | 318 | 1272 | |
| Fructose | 342 | 1368 | |
| Cellulose | 50 | 0 | |
| Corn Oil | 50 | 450 | |
| Mineral Mix | 35 | 0 | |
| Vitamin Mix | 1 | 4 | |
| Choline Bitartrate | 2 | 0 | |
| Total | 1001 | 3906 | |
HFRD: D15101101, a high-fructose rich diet specially formulated by Research Diets
2.2. Histopathology analysis
Liver tissue sections from the largest lobe were fixed in neutralized formalin (10%) before being stained with hematoxylin and eosin (H&E). Following staining, hepatic histological examination was performed with the histological scoring system for NAFLD blindly, as described (Abdelmegeed et al., 2017; Kleiner et al., 2005).
2.3. Measurements of the hepatic contents of triglyceride (TG) as well as serum levels of metabolic parameters
Liver tissues (50 mg wet weight) were homogenized in 5% Triton X-100 solution and heated in 80–100°C water bath for 2–5 min to solubilize the TG. The samples were then centrifuged at 10,000 × g for 10 min, and the resulting supernatants were used to determine the TG level by following the manufacturer’s protocol (BioVision Research products, Mountain View, CA, USA). The levels of serum TG, cholesterol and alanine aminotransferase (ALT) were measured for each mouse using the respective clinical IDEXX Vet Test Chemistry Analyzer system from IDEXX Laboratories (West brook, ME, USA). Serum adiponectin and tumor necrosis factor α (TNFα) concentrations were measured with the respective mouse adiponectin and TNFα ELISA kit by following the manufacturer’s protocols. (Life Technologies, Grand Island, NY, USA).
2.4. Western blotting
Hepatic cell lysates (50 μg proteins/lane) were separated by 12% SDS-PAGE and subjected to immunoblot analyses, as previously described (Abdelmegeed et al., 2012). Total liver lysates or nuclear proteins (50 μg/sample), prepared by differential centrifugation, were separated by 10 or 12 % SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. Upon completion of electrophoretic transfer of the proteins, membranes were blocked for 1 h in 4% bovine serum albumin in Tris-HCl buffered saline containing 0.01% Tween 20 (TBS-T). Membranes were then incubated with the specific primary antibodies at 4 °C overnight. The primary antibodies for hormone-sensitive lipase (HSL), fatty acid synthase (FAS), phospho-AMP-activated protein kinase (P-AMPK), AMPK, phospho-acetyl-CoA carboxylase (P-ACC), ACC, Bcl-2-associated X protein (Bax), B-cell lymphoma 2 (Bcl2), Histone H3 and β-actin were purchased from Cell Signaling Inc (Danvers, MA, USA). The specific antibody to poly-ADP-ribosyl polymerase-1 (PARP-1) or stearoyl-CoA desaturase-1 (SCD1) was obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Specific antibodies for cluster of differentiation 36 (CD36), fatty acid transport protein 2 (FATP2), adipose triglyceride lipase (ATGL), peroxisome proliferator-activated receptor α (PPARα), PPARγ, Retinoid X receptor α (RXRα), adiponectin receptor 2 (adiponectin R2), CYP2E1, inducible nitric oxide synthase (iNOS), or heme oxygenase 1 (HO-1) were from Abcam Inc. (Cambridge, MA, USA). After removal of the primary antibodies, the nitrocellulose membranes were washed with 1x PBS three times at 10-min intervals and then either incubated with the goat anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody (Santa Cruz Inc, Dallas, TX, USA) (1:5,000 dilutions in 5% milk powder in TBS-T) for image detection by enhanced chemiluminescence. Band densitometry was performed using NIH Image software.
2.5. Immunohistochemistry
To assess the macrophage infiltration in the adipose tissue, samples of the visceral adipose tissue were fixed in the 10% neutral buffered formalin, processed, embedded in paraffin, and sectioned. Briefly, deparaffinized adipose tissue sections were treated with 3% hydrogen peroxide followed by antigen retrieval. The sections were blocked with 2% non-fat skim milk solution, and incubated with the primary antibody against F4/80 (Abcam Inc., Cambridge, MA, USA). After incubation and subsequent washing steps, the attached primary antibody was then linked to the dextran polymer by following the manufacturer’s protocol (Envision kit, Dako, Carpinteria, CA, USA). The final reaction was performed by immersing the sections in a solution of 3,3’-diaminobenzidine (DAB). The sections were then counterstained with hematoxylin.
2.6. Real-time quantitative RT-PCR
Total RNA was isolated from frozen liver and fat tissues from each mouse using a TRIzol from Life Technologies (Grand Island, NY, USA), according to the manufacturer's recommendations. The concentration of RNA samples was measured by Nanodrop ND-1000 (Thermo Scientific, Wilmington, DE, USA). Real-time quantitative polymerase chain reaction (PCR) amplifications were carried out in 7900HT Sequence Detection System from Applied Biosystems (Foster City, CA, USA) and Eco Real-Time PCR system from Illumina (San Diego, CA) in 20 μL volume. The reaction was conducted using Power SYBR Green RNA-to-CT 1-Step Kit from Life Technologies (Grand Island, NY, USA) by following the manufacturer's suggestions. A total of 200 nM each of forward and reverse primers and 40 ng of template RNA were used. All amplification reactions were carried out in four biological replicates. To distinguish the specific amplicons from the nonspecific amplifications, a dissociation curve was generated and examined. The Ct values were calculated with Sequence Detection System 2.3, RQ Manager 1.2 (Applied Biosystems) and Eco software V4.0 (Illumina) with an automatic adjustment of base line and determination of Ct. The resulting Ct values were imported to Microsoft Excel worksheet for further analysis. The primer sequences were designed to span an intron region of the target gene of interest to avoid amplification of trace amounts of genomic DNA in the samples. Primers used for TNFα were: forward, 5’-GCTACGACGTGGGCTACA-3’ and reverse, 5’-CCCTCACACTCAGATCATCTTCT-3’; for IL6 were: forward, 5’-CCAGAGATACAAAGAAATGATGG-3’ and reverse, 5’-ACTCCAGAAGACCAGAGGAAAT-3’; and for cyclophilin were: forward, 5’-CAGACGCCACTGTCGCTTT-3’ and reverse, 5’-TGTCTTTGGAACTTTGTCTGCAA-3’.
2.7. Assays for measurements of hepatic glutathione level and anti-oxidant enzyme activities
Intrahepatic reduced glutathione (GSH) and oxidized glutathione disulfide (GSSG) (in 50 mg liver extracts) were determined by using commercially available kits (Cayman Chemical, Ann Arbor, Michigan, USA). The catalytic activities of total hepatic superoxide dismutase (SOD) and catalase (CAT) were also determined by using the kits from Cayman Chemical. The manufacturer's protocols were followed whenever commercial kits were used.
2.8. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
The ApopTag peroxidase in situ apoptosis detection kit from Millipore Corporation (Billerica, MA, USA) was used to identify apoptotic hepatocytes by labeling and detecting DNA strand breaks by the TUNEL method by following the manufacturer’s protocol. Numbers of TUNEL-positive hepatocytes were counted in 10 high-power (200x) microscope fields (HPF).
2.9. Data evaluation
The results are expressed as means ± SEM (n≥6/group). The significance of differences between groups was determined by the Student’s t test. Statistical analysis was conducted using Graphpad Prism software (GraphPad Software Inc.) and values with p < 0.05 were considered significant.
3. Results
3.1. Effects of HFRD on body weight and hepatic and serum TG levels
The histological analysis revealed accumulation of significantly greater levels lipid droplets in the livers of HFRD-fed mice after 3 weeks of feeding compared to the corresponding control. However, the increase of lipid droplets was moderate since the number and the size of the fat deposits indicated no massive hepatocyte ballooning (Fig. 1A). In addition, HFRD-fed mice had a significantly higher steatosis score than that of ContD-fed mice (Fig. 1B), whereas the inflammation score was not significantly increased in HFRD-fed mice, despite a tendency of an increased inflammation score, compared with ContD-fed mice (Fig. 1C). Consistent with histological assessment, HFRD-fed mice exhibited the significantly elevated levels of hepatic (Fig. 1D) and serum TG (Fig. 1E), serum cholesterol (Fig. 1E), and serum adiponectin (Fig. 1G) than the levels monitored in mice fed ContD. However, serum ALT levels in HFRD-fed mice were not significantly different from the control group (Fig. 1H). No significant changes in the body weight gain, liver weight, and food intake were observed between the two groups (Table 2).
Figure 1.

Effects of solid HFRD on hepatic steatosis and metabolic parameters in mice. (A) Representative liver histology with H&E staining, (B) steatosis score, (C) inflammation score, (D) concentration of hepatic TG, (E) serum TG, (F) serum cholesterol, (G) serum adiponectin and (H) serum ALT of experimental mice are shown. All results are presented as mean ± S.E.M. (n=6~7/group). Significant differences between the two groups are indicated by asterisks: *P < 0.05; **P < 0.01.
Table 2.
Body weight gain, liver weight and food intake from mice fed experimental diets.
| Group | ContD | HFRD |
|---|---|---|
| Final body weight (g) | 20.6 ± 0.3 | 19.9 ± 0.5 |
| Body weight gain (g/3 weeks) | 1.1 ± 0.2 | 1.0 ± 0.2 |
| Liver weight (g) | 0.85 ± 0.022 | 0.81 ± 0.021 |
| Liver index (g/100g body weight)* | 4.32 ± 0.076 | 4.21 ± 0.052 |
| Food intake (g/day) | 3.21 ± 0.15 | 3.55 ± 0.13 |
Each value represents the mean ± S.E.M. (n=6~7/group).
Liver index=liver weight/body weight.
3.2. Effects of HFRD on hepatic expressions of key proteins involved in lipid homeostasis
Hepatic lipid homeostasis can be maintained the balance between hepatic lipogenesis and lipid oxidation as well as uptake. Thus, we examined the hepatic expression of critical proteins involved in lipogenesis such as FAS and SCD1. Their levels were elevated in HFRD-fed mice compared with the control mice although the significance level was only observed for FAS (Fig. 2A).
Figure 2.

Effects of HFRD on hepatic levels of key proteins involved in lipid metabolism. (A) Representative images of the immunoblot analysis and the densitometric levels for SCD1 and FAS and (B) PPARα, PPARγ and RXRα in the livers of the indicated mouse groups are presented. Immunoblot results of the target proteins were normalized to β-actin (for SCD1 and FAS) or Histone H3 (for PPARα, PPARγ and RXRα), used as a loading control. All results are presented as mean ± S.E.M. (n=6~7/group). Significant differences between the two groups are indicated by asterisks: *P < 0.05; **P < 0.01.
We then evaluated the hepatic expressed levels of nuclear PPARα, PPARγ and RXRα proteins because the heterodimer PPARs-RXR transcriptional complex, where RXR is essential for the functions of PPARs, has been reported to play a critical role in lipid homeostasis and energy balance, including triglyceride metabolism including fatty acid transport and storage (Ziouzenkova et al., 2003). Hepatic PPARα protein levels were significantly up-regulated, while PPARγ change was not significant, in HFRD-fed mice compared to those of the control group (Fig. 2A and B). In contrast, HFRD exposure significantly decreased the hepatic levels of nuclear RXRα protein compared to control group (Fig. 2A and B). To further examine the effects of HFRD on critical genes or proteins involved in lipid oxidation leading to decreased fat accumulation, we evaluated the effects of HFRD on key elements involved in adiponectin signaling such as adiponectin receptor-2 (R2), AMPK and ACC. Hepatic expression of adiponectin R2 normalized to β-actin and phosphorylated AMPK normalized to AMPK were significantly decreased in HFRD-fed mice compared with those of the corresponding control mice (Fig. 3A and B, respectively). In addition, the ratio of phosphorylated (activated) ACC to ACC was significantly reduced in HFRD-fed mice compared with control mice (Fig. 3C).
Figure 3.

Effects of HFRD on hepatic levels of genes involved in adiponectin signaling. Representative images of the immunoblot analysis and densitometric levels for (A) Adiponectin R2, (B) P-AMPK and AMPK, (C) P-ACC and ACC are shown. Immunoblot results of the target protein was normalized to β-actin, used as a loading control. All results are presented as mean ± S.E.M. (n=6~7/group). Significant differences between the two groups are indicated by asterisks: *P < 0.05; **P < 0.01.
We next examined the effects of HFRD on key hepatic proteins involved in fatty acid uptake (CD36 and FATP2) and lipolysis (HSL and ATGL), respectively. The hepatic expression of both CD36 and FATP2 were significantly decreased in HFRD-fed mice compared with control mice (Fig. 4A). In contrast, mice fed HFRD exhibited a moderate but significant elevation in ATGL protein levels compared to control group, while HSL did not change significantly (Fig. 4B).
Figure 4.

Effects of dietary HFRD on hepatic levels of the key proteins involved in fatty acid uptake and lipolysis. Representative images of the immunoblot analysis and densitometric levels for (A) CD36 and FATP2 and (B) HSL and ATGL are provided. Immunoblot results of the target protein was normalized to β-actin, used as a loading control. All results are presented as mean ± S.E.M. (n=6~7/group). Significant differences between the two groups are indicated by asterisks: *P < 0.05; **P < 0.01.
3.3. Effects of HFRD on oxidative stress
To determine the effect of HFRD on oxidative stress, we evaluated the expressed levels of CYP2E1, an important source of oxidative stress, has been reported to increase in response to high fat diet (HFD) and to play a critical role in the development of HFD-mediated NAFLD (Abdelmegeed et al., 2012). However, western blot analysis showed that the hepatic levels of CYP2E1 protein were significantly decreased in HFRD-fed mice compared to those of control mice (Fig. 5A and B). We have also analyzed the hepatic levels of iNOS, which is known to promote nitroxidative stress in the cells and has been reported to increase in response to HFD or 30% fructose in drinking water in mouse models, leading to increased tissue injury (Song et al., 2013; Spruss et al., 2011). Indeed, HFRD exposure significantly increased the hepatic levels of iNOS compared with the control group (Fig. 5A and B). Also, the hepatic levels of the anti-oxidant HO-1 were significantly increased in the HFRD-fed mice compared with control mice, (Fig. 5A and B).
Figure 5.

Effects of HFRD on oxidative stress markers. (A) Representative images of the immunoblot analysis and the densitometric levels (B) for CYP2E1, iNOS and HO-1 in the livers of experimental mice are presented. Immunoblot results for the target protein was normalized to β-actin, used as a loading control. The levels of (C) GSH, (D) GSSG, and (E) GSSG/GSH as well as the activities of (F) SOD and (G) Catalase in HFRD-fed mice compared to control mice are shown. All results are presented as mean ± S.E.M. (n=6~7/group). Significant differences between the two groups are indicated by asterisks: *P < 0.05; **P < 0.01.
Oxidative stress may result from the imbalance between oxidative radical production and their removal by the antioxidant defense system including GSH and antioxidant enzymes such as SOD and CAT. Thus, we next evaluated the effect of HFRD on the hepatic levels of GSH, GSSG, and GSSG/GSH. Increased GSSG and GSSG/GSH levels were observed in HFRD-fed mice compared to the control group (Fig. 5C–E). However, no significant changes in the activities of liver SOD or catalase were observed in HFRD-fed mice compared to those of the control mice (Fig. 5F and G).
3.4. Effects of HFRD on adipose tissue inflammation
There is increasing evidence that visceral adipose tissue is a causative risk factor in promoting fatty liver. Adipose tissue inflammation in obesity is characterized by macrophage infiltration (Johnson et al., 2012). The infiltrated macrophages are responsible for the production of proinflammatory cytokines and the modulation of adipocyte-derived adipokines (Johnson et al., 2012). Therefore, we next determined the development of inflammation and macrophage infiltration in adipose tissues of HFRD-fed mice using immunohistochemistry to determine levels of macrophage marker F4/80 (Fig. 6A and B). In addition to the similar body weights, no significant changes in the fat mass were observed between the two groups (data not shown). However, the HFRD-fed mice showed significantly greater numbers of macrophages with increased inflammation in the adipose tissues compared to those of control (Fig. 6A and B). In confirmation of increased adipose tissue inflammation, the mRNA levels of inflammatory markers (TNFα and IL-6) were highly up-regulated in adipose tissues of HFRD-fed mice compared to control mice, although the significance level was only achieved for TNFα (Fig. 6C). Consistently, HFRD exposure increased the level of serum TNFα compared to control group, suggesting that adipose tissue might play at least a partial role in the increased levels of systemic TNFα (i.e. inflammation) (Fig. 6D).
Figure 6.

Effects of HFRD on adipose tissue inflammation. (A and B) Representative F4/80 immunohistochemistry showing macrophage infiltration and (C) visceral fat-pad weights. (D) Relative mRNA levels of inflammatory markers and (E) serum TNFα concentration are shown. All results are presented as mean ± S.E.M. (n=6~7/group). Significant differences between the two groups are indicated by asterisks: *P < 0.05; ** P < 0.01.
3.5. Effects of HFRD on hepatic apoptosis
To study the effects of HFRD-induced hepatic steatosis, oxidative stress, and inflammation on hepatocyte apoptosis, we used TUNEL assay to evaluate the number of apoptotic cells and performed immunoblot analyses to determine the levels of critical apoptotic marker proteins (Fig. 7). TUNEL analysis revealed that the number of apoptotic hepatocytes was significantly greater in HFRD-fed mice than control mice (Fig. 7A and B). In agreement with TUNEL analysis, significantly increased levels of Bax/Bcl2 and PARP-1, critical proteins in apoptosis, were observed in HFRD-fed mice than those of the control mice (Fig. 7C), confirming the increased hepatocyte apoptosis by HFRD.
Figure 7.

Effects of HFRD on hepatic apoptosis. (A) Representative images of TUNEL- positive apoptotic hepatocytes (marked with black arrows) in the livers of indicated groups are presented. (B) Number of TUNEL-positive hepatocyte in 10 high-powered fields (×200) was calculated. (C) Representative images and densitometric levels of the immunoblot analysis for Bax, Bcl2, and PARP-1 in the livers of the experimental groups. All results are presented as mean ± S.E.M. (n=6~7/group). Significant differences between the two groups are indicated by asterisks: *P < 0.05; **P < 0.01.
4. Discussion
One of the hallmarks of NAFLD is triglyceride accumulation in the cytoplasm of hepatocytes (Berlanga et al., 2014). Imbalances between the input, oxidation, de novo synthesis, lipolysis, and output of fatty acids could contribute to hepatic steatosis, as shown in animal models (Koteish and Diehl, 2001). Mice have been widely used as an experimental model to evaluate the development or progression of NAFLD and to study the underlying mechanism(s) of hepatic steatosis, apoptosis, and fibrosis because they share many similarities with human conditions (Gregg et al., 2012; Kim et al., 2015; Sheedfar et al., 2013). Females are more susceptible to the damaging effects of alcohol than their male counterparts (Strong et al., 2010) and many similarities between ethanol and fructose have been suggested (Byerley and Lee, 2010; Lustig, 2010). In addition, many previous studies involving NAFLD usually focus on using male mice, while ignoring females, and this disparity applies to the HFRD used in the current study compared to the recent report where male mice were used (Wang et al., 2015). In addition, it has been shown that female mice are more sensitive for nonalcoholic fatty liver disease than males when exposed to high fructose solution (30% in drinking water) for 16 weeks (Spruss et al., 2012). For these reasons, female mice were used in this study since it is essential to also evaluate female response to the fructose diet, and thus young female mice were used to determine the harmful effects of HFRD on the liver. It is important however to recognize that dissimilar outcomes might result from sex difference with hormonal variation.
De novo lipogenesis plays a substantial role in the pathogenesis of NAFLD (Reid et al., 2008). FAS and SCD1 catalyze the formation of monounsaturated fatty acids, which are the major fatty acid constituents of triglycerides (Reid et al., 2008). Indeed, we found that HFRD significantly increased the levels of hepatic FAS (Fig. 2) with decreased levels of P-AMPK and P-ACC (Fig. 3), suggesting an increased level of lipogenesis. It is interesting to note that we did not monitor a similar significant increase in either body or liver weight and adipose tissue indices, despite the significant accumulation of lipid droplets in the HFRD mice compared to the control group. These results suggest that the hepatic lipid droplets and adipose tissue fat accumulation were insufficient to significantly increase the liver or body weight since the overall fatty liver we monitored after 3 weeks of HFRD feeding was moderate. These results are different from the recent report [Wang et al., 2015] where significantly increased body weight and adiposity were observed within a shorter period in male mice fed the same diet. This difference might be due to variations of sex and age of the mice used in this study versus the Wang et al, 2015 report where 10~12 week old male mice were used. In support of this notion, it has been reported that mice weaned at age of 3 weeks and then fed a HFRD for additional 14 weeks failed to exhibit a significant change in body weight and increased adiposity at the end of the study (Tillman et al., 2014). The authors of this study suggested that one of the main reasons for the different results from other reports with a high fructose in solution or a solid diet might be due to the dissimilar ages of the mice being used at the starting dates. Tillman et al. also suggested that the metabolic rate might vary in mice at different ages, leading to variant effects and ultimately phenotypic changes. Thus, sex, age, metabolic rate, type of fructose diet (solution or solid HFRD) and environment should be taken into consideration when comparing the results of different studies and should be a focus of future research involving the fructose-containing diet.
PPARα is a nuclear hormone receptor that plays a central role in the transcriptional regulation of lipid and glucose metabolism (Mandard et al., 2004). In the liver, PPARα activation plays a role in the mitigation of hepatic steatosis and the progression of NAFLD (Abdelmegeed et al., 2011). In contrast, PPARγ is a master transcriptional regulator of adipogenesis and plays an important role in the process of lipid storage and insulin sensitivity (Okamura et al., 2010). The retinoid X receptors (RXRs) are unique among the nuclear receptors since they not only bind DNA as a homodimer but also are important as a heterodimeric partner for a number of other nuclear receptors to bind DNA (Mangelsdorf and Evans, 1995) . In the adult liver, RXRα is the most abundant of the three RXRs (Mangelsdorf et al., 1992), PPARα forms a heterodimer with RXRα, which binds to specific DNA sequences known as peroxisome proliferator response elements (PPREs). This transcriptional complex promotes the expression of the genes that mediate fatty acid transport to mitochondria and oxidation (Mandard et al., 2004). Intriguingly, although we observed a significant increase of PPARα with a modest increase in PPARγ levels in HFRD-fed mice, the decrease RXRα levels were more profound (Fig. 2), suggesting an alteration of the functions of both PPARα and PPARγ, potentially contributing to fat accumulation. The novel finding of the decline of RXRα levels in HFRD-fed mice might provide an explanation of fat accumulation (Fig. 1), despite the significantly increased levels of PPARα which may reflect a compensatory mechanism.
Adiponectin, a major product of adipocytes, is a prototypic anti-inflammatory and anti-diabetic adipokine, which can exert its fatty oxidation effects via activating P-AMPK and PPARα. Adiponectin has two specific receptors: adiponectin receptor type 1 and 2 (AdipoR1 and 2). AdipoR1 is widely expressed in many tissues, whereas AdipoR2 is mainly produced in the liver (Tilg, 2010). It has been shown that AdipoR2 expression was inversely related to increased hepatic injury and the development of fibrosis stage (Kaser et al., 2005). Our results revealed that HFRD caused a noticeable decrease in the levels of hepatic AdipoR2 (Fig. 3) while there was an increased levels of serum adiponectin in this group compared to the control group (Fig. 1). These results suggest potential development of adiponectin resistance in mice fed HFRD through significant downregulation of AdipoR2. To test the molecules downstream to the AdipoR2, we monitored the changes in phosphorylation of the lipogenic enzyme ACC on Ser79, a down-stream substrate of P-AMPK, both of which are down-stream metabolic targets of adiponectin. Phosphorylation of ACC on Ser79 blocks the enzymatic activity of ACC, leading to suppressed conversion of acetyl-CoA to malonyl-CoA, an important intermediate of fatty acid synthesis (Yamauchi et al., 2002). On the other hand, ACC can be over-expressed in response to fructose load (Nagai et al., 2002) and observed (Fig. 3C). In support of our suggestion of adiponectin resistance development, HFRD-fed mice showed significantly decreased levels of the active P-AMPK (Fig. 3B), which could consequently result in decreased levels of inactive P-ACC (Fig. 3C) with elevated fat synthesis along with increased FAS (Fig. 2A), ultimately contributing to lipid accumulation (Fig. 1).
As mentioned earlier, fatty acid uptake into the liver contributes to the steady balance of hepatic triglycerides in the liver (Bradbury, 2006). FATPs and CD36 are key regulators of the transmembrane process of fat uptake. Of these FATP isoforms, adenovirus-mediated knockdown of FATP2 significantly decreases the rates of fatty acid uptake into mouse hepatocytes (Falcon et al., 2010). Elevated hepatic expression levels of CD36 have been observed in NAFLD and appear to mediate enhanced uptake of non-esterified fatty acids (Miquilena-Colina et al., 2011). Both ATGL and HSL can contribute to the overall hepatic TG hydrolase activities and likely play a direct role in liver lipolysis under normal physiological conditions (Reid et al., 2008). Interestingly, both CD36 and FATP2 (Fig. 4A) were significantly decreased while ATGL was slightly but significantly up-regulated (Fig. 4B) by HFRD. These unexpected results shown in Fig. 4 can be explained by a cellular defense mechanism via decreased fatty acid uptake and increased lipolysis to compensate, at least partially, against the increased free fatty acid load that was developed due to the combined action of increased de novo lipid synthesis and decreased fatty acid oxidation, as discussed earlier. However, the development of increased hepatic TG levels (“1st hit”) would prime the liver to more deleterious effects following additional exposure to oxidative stress and inflammation of the “2nd hits” (Day and James, 1998). The development of hepatic steatosis in response to fructose in this model is in agreement with many other studies using fructose regardless of 30% solution in drinking water (Spruss et al., 2011) or a solid diet as HFRD (Spruss et al., 2011; Wang et al., 2015).
Oxidative stress, which can also increase liver fat accumulation via inhibiting the secretion of very low density lipoprotein (VLDL) by hepatocytes, can play a contributing role in the progression from simple steatosis to NASH (Gambino et al., 2011). Oxidative stress is a cellular state in which the production of harmful molecules such as reactive oxygen species (ROS) and reactive nitrogen species (RNS) can decrease cell viability and thus promote cell death (Hardwick et al., 2010). ROS can promote necro-inflammation and activate several intracellular signaling pathways that can lead to hepatocyte apoptosis (Gambino et al., 2011). CYP2E1 has been proposed to play a role in AFLD and NAFLD via increased ROS levels (Lieber, 2004). However, CYP2E1 levels were actually decreased in response to HFRD in our model, although its levels were shown to be increased by experimental models of NAFLD and patients (Aljomah et al., 2015). Inducible nitric oxide synthase (iNOS) (Saha and Pahan, 2006), which is known to produce greater amounts of nitric oxide (NO) than other NOS isoforms (Akbar et al., 2016), has also been reported to play a critical role in the development and/or progression of alcoholic fatty liver disease (AFLD) and NAFLD (Abdelmegeed and Song, 2014), since iNOS-null mice are resistant to AFLD (McKim et al., 2003) and NAFLD (Nozaki et al., 2015). In addition, iNOS was reported to play an important role in the onset of hepatic steatosis and inflammation in response to chronic exposure to 30% fructose solution in drinking water (Spruss et al., 2011). Indeed, we monitored a significantly higher levels of iNOS in HFRD-fed mice, suggesting iNOS as an important source of ROS/RNS in this model.
Hepatocytes possess a variety of non-enzymatic and enzymatic anti-oxidant systems to protect against oxidative liver injury (Stocker et al., 1987). The anti-oxidant defense includes GSH, which is one of the major cellular antioxidants, and GSH depletion has been reported in AFLD (Han et al., 2016) and NAFLD (Lieber et al., 2004). Anti-oxidant defense also includes SOD, which converts highly reactive superoxide radical to H2O2 and CAT, which converts H2O2 to water and molecular oxygen (Abdelmegeed et al., 2009). HO-1, which converts heme into biliverdin, which, is then transformed into bilirubin, is also a potent anti-oxidant protein (Stocker et al., 1987). In the current study, we monitored a significant increase in the levels of GSSG/GSH ratio mainly due to the significant increase of GSSG levels in HFRD compared to those of control. This is in agreement with previous reports indicated that GSSG levels may indeed be much higher than GSH levels in NAFLD (Pastore et al., 2003). The levels of HO-1, which is known to be induced under increased oxidative stress, were elevated in HFRD, further confirming the increased oxidative stress levels, since the increased levels might be considered as a protective compensatory mechanism by the cells. However, we did not monitor any significant changes in the levels of SOD or catalase activities. Collectively, this data suggest that the increased oxidative stress levels as evidenced by increased GSSG/GSH is mainly due to increased production of ROS/RNS and that iNOS is one of the sources of this ROS/RNS, although other pro-oxidant enzymes such as NADPH-oxidase and xanthine oxidase could have been activated or upregulated. Alternatively, CYP2E1, despite little induction in HFRD-fed mice, could have provided ROS, needed for the production of toxic peroxynitrite especially in the presence of elevated NO by iNOS, through the permissive role of CYP2E1, as recently reviewed (Abdelmegeed et al., 2015).
Abnormal adipose tissue metabolism has been identified as a critical mechanistic link between HFD-induced obesity and NAFLD (Choi et al., 2016a). For instance, in patients with severe obesity, the mRNA up-regulation of IL6 and TNFα in the adipose tissues is more prominent than that the livers (Moschen et al., 2010). Higher serum levels of TNFα have been found in patients with NASH compared with healthy subjects and these differences were independent of higher insulin resistance (Hui et al., 2004). Furthermore, it has been shown, using mice fed HFD with different proportions that there is a strong link between inflammatory and morphological changes in adipose tissue and progression of steatosis to NASH. Consequently, adipose tissue inflammation or dysfunction may signal or initiate the development and/or progression of hepatic steatosis toward NASH (Promrat et al., 2010). In agreement, we monitored an increased levels of macrophage markers F4/80 as well as the significant increase of TNFα and marginal increase of IL-6 levels in the adipose tissue of HFRD in comparison to that of the control.
Pathological increases in cell death in the liver as well as in peripheral tissues have emerged as an important mechanism involved in the development and progression of NAFLD. An increase in hepatocyte cell death by apoptosis is typically present in patients with NAFLD and in experimental models of steatohepatitis (Alkhouri et al., 2011; Feldstein et al., 2003; Feldstein and Gores, 2005; Ribeiro et al., 2004; Rust and Gores, 2000; Sellmann et al., 2015). In the mitochondrial-regulating apoptosis system, Bcl-2 and its family members together constitute a very complex interaction network regulating apoptosis (Begriche et al., 2006). Bcl-2 inhibits apoptosis while Bax promotes apoptosis, and the ratio of Bax/Bcl-2 is directly related to the occurrence of apoptosis. (Akcali et al., 2004). For instance, when the Bax protein is dominant, apoptosis occurs, whereas when the Bcl-2 protein is dominant, the cell survives (Ramalho et al., 2006). This is in accordance in this study where TUNEL assay revealed increased levels of apoptotic hepatocytes in HFRD and that ratio of Bax/Bcl-2 was higher in this group compared to the control (Fig. 7). We also evaluated PARP-1, as a confirmatory marker of increased hepatocytes apoptosis (Pacher and Szabo, 2008), in HFRD-fed mice. Upon DNA damage (for example induced by ROS/RNS or ionizing radiation), PARP1 activity increases and the ADP-ribose part of NAD+ is transferred to proteins, giving rise to formation of poly(ADP-ribose) polymers (Jagtap and Szabo, 2005). This post-translational modification alters the function of many enzymes and structural proteins and may initiate caspase-independent cell death (Jagtap and Szabo, 2005). Indeed, PARP-1 levels were increased inn HFRD-fed mice in this model (Fig. 7C), further confirming the occurrence of hepatocyte apoptosis. Studies from our laboratory, as well as others, have demonstrated that saturated fatty acids (SFAs), as well as free cholesterol, are key mediators of lipotoxicity by triggering specific signaling pathways resulting in apoptotic cell death (Li et al., 2009), which seems to be the case in this model.
5. Conclusion
Our results revealed that HFRD exposure increased hepatic steatosis via disturbance of lipid homeostasis with a significantly increased lipogenesis and decreased lipid oxidation, which could not be overcome by decreased fatty acid uptake. Increased lipid accumulation in the liver constitutes the “1st hit”. Hepatic steatosis primed the liver to the deleterious effects of increased hepatic oxidative stress a “2nd hit” and systemic inflammation, as evidenced by increased serum TNFα levels, as another “2nd hit”, which might be originated from adipose tissue, leading to increased hepatocytes apoptosis (Fig. 8). To our knowledge, our mechanistic studies of the alteration of lipid homeostasis, partly via the decreased levels of RXRα, and the development of hepatocyte apoptosis as shown by various parameters in response to increased oxidative stress and inflammation, originated partly from the adipose tissue of HFRD-fed mice, are novel and can serve as a springboard for further investigations.
Figure 8.

Proposed mechanisms for the induction of hepatic apoptosis and steatosis by high-fructose diet (HFRD).
Supplementary Material
Highlights.
HFRD-fed mice exhibited higher levels of hepatic lipogenesis as evidenced by increased FAS and SCD1.
Hepatic fatty acid oxidation was inhibited as evidenced by decreased adiponection-R2, P-AMPK, P-ACC, and RXR-α.
HFRD increased the hepatic levels of iNOS and GSSG/GSH, suggesting the development of oxidative stress.
HFRD-fed mice elevated the levels of macrophages and pro-inflammatory cytokine TNF-α in the adipose tissue.
HFRD-fed mice exhibited increased hepatocyte apoptosis, as shown by the elevated levels of Bax/Bcl2 ratio and PARP-1 protein.
Acknowledgments
This work was supported by the Intramural Research Program of National Institute on Alcohol Abuse and Alcoholism and a grant to Youngshim Choi from the KRIBB Research Initiative Program (Korean Biomedical Scientist Fellowship Program), Korea Research Institute of Bioscience and Biotechnology, Republic of Korea. We are thankful to Dr. Klaus Gawrisch for supporting this study.
Abbreviations
- ACC
acetyl-CoA carboxylase
- Adiponectin R2
adiponectin receptor 2
- AFLD
alcoholic fatty liver disease
- ALT
alanine aminotransferase
- AMPK
AMP-activated protein kinase
- ATGL
adipose triglyceride lipase
- Bax
Bcl-2-associated X protein
- Bcl2
B-cell lymphoma 2
- CAT
catalase
- CD36
cluster of differentiation 36
- ContD
regular chow control diet
- FAS
fatty acid synthase
- FATP2
fatty acid transport protein 2
- GSH
reduced glutathione
- GSSG
oxidized glutathione disulfide
- HFD
high fat diet
- HFRD
high (H)-fructose (FR) diet (D)
- HO-1
heme oxygenase-1
- HSL
hormone-sensitive lipase
- iNOS
inducible nitric oxide synthase
- NAFLD
Non-alcoholic fatty liver disease
- P-ACC
phospho-acetyl-CoA carboxylase
- P-AMPK
phospho-AMP-activated protein kinase
- PARP-1
poly-ADP-ribosyl polymerase-1
- PPARα
peroxisome proliferator-activated receptor α
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RXRα
Retinoid X receptor α
- SCD1
stearoyl-CoA desaturase-1
- SOD
superoxide dismutase
- TG
triglyceride
- TNFα
tumor necrosis factor α
- VLDL
very low density lipoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelmegeed MA, Banerjee A, Yoo SH, Jang S, Gonzalez FJ, Song BJ. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J Hepatol. 2012;57:860–866. doi: 10.1016/j.jhep.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmegeed MA, Choi Y, Godlewski G, Ha SK, Banerjee A, Jang S, Song BJ. Cytochrome P450-2E1 promotes fast food-mediated hepatic fibrosis. Sci Rep. 2017;7:39764. doi: 10.1038/srep39764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmegeed MA, Ha SK, Choi Y, Akbar M, Song BJ. Role of CYP2E1 in mitochondrial dysfunction and hepatic tissue injury in alcoholic and non-alcoholic diseases. Curr Mol Pharmacol. 2015 doi: 10.2174/1874467208666150817111114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmegeed MA, Moon KH, Hardwick JP, Gonzalez FJ, Song BJ. Role of peroxisome proliferator-activated receptor-alpha in fasting-mediated oxidative stress. Free Radic Biol Med. 2009;47:767–778. doi: 10.1016/j.freeradbiomed.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmegeed MA, Song BJ. Functional roles of protein nitration in acute and chronic liver diseases. Oxid Med Cell Longev. 2014;2014:149627. doi: 10.1155/2014/149627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr. 2011;141:603–610. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akar F, Uludag O, Aydin A, Aytekin YA, Elbeg S, Tuzcu M, Sahin K. High-fructose corn syrup causes vascular dysfunction associated with metabolic disturbance in rats: protective effect of resveratrol. Food Chem Toxicol. 2012;50:2135–2141. doi: 10.1016/j.fct.2012.03.061. [DOI] [PubMed] [Google Scholar]
- Akbar M, Essa MM, Daradkeh G, Abdelmegeed MA, Choi Y, Mahmood L, Song BJ. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016;1637:34–55. doi: 10.1016/j.brainres.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akcali KC, Dalgic A, Ucar A, Haj KB, Guvenc D. Expression of bcl-2 gene family during resection induced liver regeneration: comparison between hepatectomized and sham groups. World J Gastroenterol. 2004;10:279–283. doi: 10.3748/wjg.v10.i2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljomah G, Baker SS, Liu W, Kozielski R, Oluwole J, Lupu B, Baker RD, Zhu L. Induction of CYP2E1 in non-alcoholic fatty liver diseases. Exp Mol Pathol. 2015;99:677–681. doi: 10.1016/j.yexmp.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhouri N, Carter-Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol Hepatol. 2011;5:201–212. doi: 10.1586/egh.11.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantle JP. Dietary fructose and metabolic syndrome and diabetes. J Nutr. 2009;139:1263S–1268S. doi: 10.3945/jn.108.098020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Berlanga A, Guiu-Jurado E, Porras JA, Auguet T. Molecular pathways in non-alcoholic fatty liver disease. Clin Exp Gastroenterol. 2014;7:221–239. doi: 10.2147/CEG.S62831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury MW. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: possible role in steatosis. Am J Physiol Gastrointest Liver Physiol. 2006;290:G194–198. doi: 10.1152/ajpgi.00413.2005. [DOI] [PubMed] [Google Scholar]
- Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. 2004;79:537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD) Metabolism. 2016;65:1038–1048. doi: 10.1016/j.metabol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Byerley LO, Lee WN. Are ethanol and fructose similar? J Am Diet Assoc. 2010;110:1300–1301. doi: 10.1016/j.jada.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, McClain C. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184–195. doi: 10.1016/j.jnutbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Choi Y, Abdelmegeed MA, Akbar M, Song BJ. Dietary walnut reduces hepatic triglyceride content in high-fat-fed mice via modulation of hepatic fatty acid metabolism and adipose tissue inflammation. J Nutr Biochem. 2016a;30:116–125. doi: 10.1016/j.jnutbio.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Abdelmegeed MA, Song BJ. Preventive effects of dietary walnuts on high-fat-induced hepatic fat accumulation, oxidative stress and apoptosis in mice. J Nutr Biochem. 2016b;38:70–80. doi: 10.1016/j.jnutbio.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- Duvnjak M, Lerotic I, Barsic N, Tomasic V, Virovic Jukic L, Velagic V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J Gastroenterol. 2007;13:4539–4550. doi: 10.3748/wjg.v13.i34.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon A, Doege H, Fluitt A, Tsang B, Watson N, Kay MA, Stahl A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am J Physiol Endocrinol Metab. 2010;299:E384–393. doi: 10.1152/ajpendo.00226.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front Biosci. 2005;10:3093–3099. doi: 10.2741/1765. [DOI] [PubMed] [Google Scholar]
- Gambino R, Musso G, Cassader M. Redox balance in the pathogenesis of nonalcoholic fatty liver disease: mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2011;15:1325–1365. doi: 10.1089/ars.2009.3058. [DOI] [PubMed] [Google Scholar]
- Gregg SQ, Gutierrez V, Robinson AR, Woodell T, Nakao A, Ross MA, Michalopoulos GK, Rigatti L, Rothermel CE, Kamileri I, Garinis GA, Stolz DB, Niedernhofer LJ. A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology. 2012;55:609–621. doi: 10.1002/hep.24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KH, Hashimoto N, Fukushima M. Relationships among alcoholic liver disease, antioxidants, and antioxidant enzymes. World J Gastroenterol. 2016;22:37–49. doi: 10.3748/wjg.v22.i1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick RN, Fisher CD, Canet MJ, Lake AD, Cherrington NJ. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2010;38:2293–2301. doi: 10.1124/dmd.110.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, Kotake Y, Sang H, Pye QN, Wallis GL, Kolker LM, Tabatabaie T, Stewart CA, Konishi Y, Nakae D, Floyd RA. Dietary choline restriction causes complex I dysfunction and increased H(2)O(2) generation in liver mitochondria. Carcinogenesis. 2000;21:983–989. doi: 10.1093/carcin/21.5.983. [DOI] [PubMed] [Google Scholar]
- Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. 2012;249:218–238. doi: 10.1111/j.1600-065X.2012.01151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser S, Moschen A, Cayon A, Kaser A, Crespo J, Pons-Romero F, Ebenbichler CF, Patsch JR, Tilg H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut. 2005;54:117–121. doi: 10.1136/gut.2003.037010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IH, Kisseleva T, Brenner DA. Aging and liver disease. Curr Opin Gastroenterol. 2015;31:184–191. doi: 10.1097/MOG.0000000000000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ Nonalcoholic Steatohepatitis Clinical Research N. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21:89–104. doi: 10.1055/s-2001-12932. [DOI] [PubMed] [Google Scholar]
- Le KA, Bortolotti M. Role of dietary carbohydrates and macronutrients in the pathogenesis of nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care. 2008;11:477–482. doi: 10.1097/MCO.0b013e328302f3ec. [DOI] [PubMed] [Google Scholar]
- Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem. 2009;284:5637–5644. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber CS. CYP2E1: from ASH to NASH. Hepatol Res. 2004;28:1–11. doi: 10.1016/j.hepres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–509. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- Lustig RH. Fructose: metabolic, hedonic, and societal parallels with ethanol. J Am Diet Assoc. 2010;110:1307–1321. doi: 10.1016/j.jada.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61:393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, Evans RM. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992;6:329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- McKim SE, Gabele E, Isayama F, Lambert JC, Tucker LM, Wheeler MD, Connor HD, Mason RP, Doll MA, Hein DW, Arteel GE. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125:1834–1844. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- Miquilena-Colina ME, Lima-Cabello E, Sanchez-Campos S, Garcia-Mediavilla MV, Fernandez-Bermejo M, Lozano-Rodriguez T, Vargas-Castrillon J, Buque X, Ochoa B, Aspichueta P, Gonzalez-Gallego J, Garcia-Monzon C. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut. 2011;60:1394–1402. doi: 10.1136/gut.2010.222844. [DOI] [PubMed] [Google Scholar]
- Moschen AR, Molnar C, Geiger S, Graziadei I, Ebenbichler CF, Weiss H, Kaser S, Kaser A, Tilg H. Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut. 2010;59:1259–1264. doi: 10.1136/gut.2010.214577. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Nishio Y, Nakamura T, Maegawa H, Kikkawa R, Kashiwagi A. Amelioration of high fructose-induced metabolic derangements by activation of PPARalpha. Am J Physiol Endocrinol Metab. 2002;282:E1180–1190. doi: 10.1152/ajpendo.00471.2001. [DOI] [PubMed] [Google Scholar]
- Nozaki Y, Fujita K, Wada K, Yoneda M, Kessoku T, Shinohara Y, Imajo K, Ogawa Y, Nakamuta M, Saito S, Masaki N, Nagashima Y, Terauchi Y, Nakajima A. Deficiency of iNOS-derived NO accelerates lipid accumulation-independent liver fibrosis in non-alcoholic steatohepatitis mouse model. BMC Gastroenterol. 2015;15:42. doi: 10.1186/s12876-015-0269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura M, Inagaki T, Tanaka T, Sakai J. Role of histone methylation and demethylation in adipogenesis and obesity. Organogenesis. 2010;6:24–32. doi: 10.4161/org.6.1.11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, Johnson RJ, Abdelmalek MF. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. 2008;48:993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore A, Federici G, Bertini E, Piemonte F. Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta. 2003;333:19–39. doi: 10.1016/s0009-8981(03)00200-6. [DOI] [PubMed] [Google Scholar]
- Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, Fava JL, Wing RR. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–129. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho RM, Cortez-Pinto H, Castro RE, Sola S, Costa A, Moura MC, Camilo ME, Rodrigues CM. Apoptosis and Bcl-2 expression in the livers of patients with steatohepatitis. Eur J Gastroenterol Hepatol. 2006;18:21–29. doi: 10.1097/00042737-200601000-00005. [DOI] [PubMed] [Google Scholar]
- Reid BN, Ables GP, Otlivanchik OA, Schoiswohl G, Zechner R, Blaner WS, Goldberg IJ, Schwabe RF, Chua SC, Jr, Huang LS. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem. 2008;283:13087–13099. doi: 10.1074/jbc.M800533200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro PS, Cortez-Pinto H, Sola S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME, Rodrigues CM. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- Rust C, Gores GJ. Apoptosis and liver disease. Am J Med. 2000;108:567–574. doi: 10.1016/s0002-9343(00)00370-3. [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem Int. 2006;49:154–163. doi: 10.1016/j.neuint.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellmann C, Priebs J, Landmann M, Degen C, Engstler AJ, Jin CJ, Garttner S, Spruss A, Huber O, Bergheim I. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J Nutr Biochem. 2015;26:1183–1192. doi: 10.1016/j.jnutbio.2015.05.011. [DOI] [PubMed] [Google Scholar]
- Sheedfar F, Di Biase S, Koonen D, Vinciguerra M. Liver diseases and aging: friends or foes? Aging Cell. 2013;12:950–954. doi: 10.1111/acel.12128. [DOI] [PubMed] [Google Scholar]
- Song BJ, Abdelmegeed MA, Henderson LE, Yoo SH, Wan J, Purohit V, Hardwick JP, Moon KH. Increased nitroxidative stress promotes mitochondrial dysfunction in alcoholic and nonalcoholic fatty liver disease. Oxid Med Cell Longev. 2013;2013:781050. doi: 10.1155/2013/781050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruss A, Henkel J, Kanuri G, Blank D, Puschel GP, Bischoff SC, Bergheim I. Female mice are more susceptible to nonalcoholic fatty liver disease: sex-specific regulation of the hepatic AMP-activated protein kinase-plasminogen activator inhibitor 1 cascade, but not the hepatic endotoxin response. Mol Med. 2012;18:1346–1355. doi: 10.2119/molmed.2012.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruss A, Kanuri G, Uebel K, Bischoff SC, Bergheim I. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxid Redox Signal. 2011;14:2121–2135. doi: 10.1089/ars.2010.3263. [DOI] [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Strong MN, Yoneyama N, Fretwell AM, Snelling C, Tanchuck MA, Finn DA. "Binge" drinking experience in adolescent mice shows sex differences and elevated ethanol intake in adulthood. Horm Behav. 2010;58:82–90. doi: 10.1016/j.yhbeh.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syn WK, Choi SS, Diehl AM. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin Liver Dis. 2009;13:565–580. doi: 10.1016/j.cld.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H. Adipocytokines in nonalcoholic fatty liver disease: key players regulating steatosis, inflammation and fibrosis. Curr Pharm Des. 2010;16:1893–1895. doi: 10.2174/138161210791208929. [DOI] [PubMed] [Google Scholar]
- Tillman EJ, Morgan DA, Rahmouni K, Swoap SJ. Three months of high-fructose feeding fails to induce excessive weight gain or leptin resistance in mice. PLoS One. 2014;9:e107206. doi: 10.1371/journal.pone.0107206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos MB, Kimmons JE, Gillespie C, Welsh J, Blanck HM. Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J Med. 2008;10:160. [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun RQ, Zeng XY, Zhou X, Li S, Jo E, Molero JC, Ye JM. Restoration of autophagy alleviates hepatic ER stress and impaired insulin signalling transduction in high fructose-fed male mice. Endocrinology. 2015;156:169–181. doi: 10.1210/en.2014-1454. [DOI] [PubMed] [Google Scholar]
- Wieckowska A, McCullough AJ, Feldstein AE. Noninvasive diagnosis and monitoring of nonalcoholic steatohepatitis: present and future. Hepatology. 2007;46:582–589. doi: 10.1002/hep.21768. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Ziouzenkova O, Perrey S, Asatryan L, Hwang J, MacNaul KL, Moller DE, Rader DJ, Sevanian A, Zechner R, Hoefler G, Plutzky J. Lipolysis of triglyceride-rich lipoproteins generates PPAR ligands: evidence for an antiinflammatory role for lipoprotein lipase. Proc Natl Acad Sci U S A. 2003;100:2730–2735. doi: 10.1073/pnas.0538015100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.