

In the crystal structure of the title compound, almost planar [PtCl2{(C6H4)(NH2)2}] molecules are stacked into columns along the c axis, suggesting Pt⋯Pt interactions.
Keywords: crystal structure, columnar structure, infinite metal chain, platinum(II) complex, hydrogen bonding
Abstract
The PtII atom in the title compound, [PtCl2{(C6H4)(NH2)2}], lies on a twofold rotation axis and has a slightly distorted square-planar coordination environment defined by two N atoms of an 1,2-phenylenediamine ligand and two Cl− ions. In the crystal, the planar complex molecules are stacked parallel to the c axis, resulting in a columnar structure. In a column, an infinite almost straight Pt⋯Pt chain is formed, suggesting weak metal–metal interactions [Pt⋯Pt = 3.3475 (8) Å]. The crystal packing is stabilized by a three-dimensional N—H⋯Cl hydrogen-bonding network between the amino groups and the Cl ligands of adjacent molecules.
Chemical context
The title compound, dichlorido(1,2-phenylenediamine-κ2
N,N′)platinum(II) [PtCl2{(C6H4)(NH2)2}], (I), which was originally prepared by Connors et al. (1972 ▸), is a member of the family of derivatives of cis-diamminedichloridoplatinum(II), cis-[PtCl2(NH3)2] (cis-platin). Since the discovery of the antitumor activity of cis-platin (Rosenberg et al., 1965 ▸), numerous derivatives and analogues of cis-platin have been prepared and investigated. However, reports on the corresponding crystal structures are rather scarce, probably because of the difficulty in obtaining crystals suitable for X-ray analysis, in part owing to poor solubility. Although the antitumor activity (Connors et al., 1972 ▸; Meischen et al., 1976 ▸) and the chemical stabilities (Köckerbauer & Bednarski, 1996 ▸) of the title compound have been reported, its crystal structure has not been determined so far. In the course of our study of the deprotonation and redox properties of a platinum complex with 1,2-phenylenediamine as a ligand (Konno & Matsushita, 2006a
▸,b
▸), we have successfully obtained single crystals of the title compound and report here its crystal structure.
Structural commentary
The molecular structure of (I) is displayed in Fig. 1 ▸. The platinum compound (I) is isostructural with the palladium compound [PdCl2{(C6H4)(NH2)2}] reported previously (Konno & Matsushita, 2017 ▸). The PtII atom lies on a twofold rotation axis. Hence the asymmetric unit comprises half of a [PtCl2{(C6H4)(NH2)2}] molecule, the other half being completed by application of twofold rotation symmetry. The PtII atom is coordinated by two N atoms of an 1,2-phenylenediamine ligand and by two Cl− ions in a slightly distorted square-planar configuration (Table 1 ▸). The r.m.s. deviation of the least-squares plane formed by atoms Pt1, N1, C1, C2 and C3 is 0.0121 Å. The structural parameters of the coordination sphere around PtII in the crystal of (I) (Table 1 ▸) are consistent with those found in cis-[PtCl2(NH3)2] (Milburn & Truter, 1966 ▸), [PtCl2(en)] (en is ethylenediamine; Iball et al., 1975 ▸), cis-[PtCl2(L)2] (L is cyclohexylamine; Lock et al., 1980 ▸), [PtCl2(cis-dac)]·0.33-hydrate (dac is 1,2-diaminocyclohexane; Lock & Pilon, 1981 ▸), cis-[PtCl2(L′)(NH3)] (L′ is cyclobutylamine; Rochon & Melanson, 1986 ▸), [PtCl2(Me2en)] (Me2en is N,N-dimethylethylenediamine; Melanson et al., 1987 ▸), [PtCl2(tn)] (tn is 1,3-diaminopropane; Odoko & Okabe, 2006 ▸), [PtCl2(L′′)] (L′′ is 2-morpholinoethylamine; Shi et al., 2006 ▸), [PtCl2(Me4en)] (Me4en is N,N,N′,N′- tetramethylethylenediamine; Asiri et al., 2012 ▸). Bond lengths and angles of the 1,2-phenylenediamine moiety (Table 1 ▸) are not significantly different from those found in the bis(1,2-phenylenediamine)platinum(II) complex, [Pt(C6H8N2)2]Cl2·2H2O [N—C = 1.450 (2) Å, C—C = 1.365 (6)–1.389 (4) Å; Konno & Matsushita, 2006a ▸] or in isostructural dichlorido(1,2-phenylenediamine)palladium(II) [N—C = 1.458 (2) Å, C—C = 1.371 (3)–1.416 (8) Å; Konno & Matsushita, 2017 ▸].
Figure 1.

A view of the molecular structure of compound (I), showing the atomic numbering scheme. Displacement ellipsoids are drawn at the 50% probability level for non-H atoms. [Symmetry code: (i) −x + 1, y, −z +  .]
.]
Table 1. Selected geometric parameters (Å, °).
| Pt1—N1 | 2.040 (4) | C1—C2 | 1.372 (6) |
| Pt1—Cl1 | 2.3213 (13) | C1—C1ii | 1.386 (9) |
| Pt1—Pt1i | 3.3475 (8) | C2—C3 | 1.377 (11) |
| N1—C1 | 1.445 (6) | C3—C3ii | 1.38 (3) |
| N1—Pt1—N1ii | 83.6 (3) | Pt1i—Pt1—Pt1iii | 176.513 (11) |
| N1—Pt1—Cl1 | 91.39 (15) | C1—N1—Pt1 | 110.8 (3) |
| Cl1ii—Pt1—Cl1 | 93.69 (7) | C2—C1—C1ii | 119.8 (3) |
| N1—Pt1—Pt1i | 92.07 (14) | C2—C1—N1 | 122.7 (5) |
| Cl1—Pt1—Pt1i | 93.80 (4) | C1ii—C1—N1 | 117.4 (2) |
| N1—Pt1—Pt1iii | 85.32 (14) | C1—C2—C3 | 120.3 (8) |
| Cl1—Pt1—Pt1iii | 88.59 (4) | C2—C3—C3ii | 119.8 (6) |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
Supramolecular features
As shown in Fig. 2 ▸, the neutral planar molecules of (I) stack parallel to the c axis, resulting in a columnar structure. The planar [PtCl2{(C6H4)(NH2)2}] units are arranged in parallel and the 1,2-phenylenediamine moieties alternate with each other as a result of the c-glide operation. In the column, an infinite, almost straight [Pt⋯Pt⋯Pt = 176.513 (11)°] platinum chain is formed with a short interatomic distance [Pt⋯Pt = 3.3475 (8) Å], suggesting weak metal–metal interactions. The infinite palladium chain of the isostructural Pd complex is straighter [Pd⋯Pd⋯Pd = 179.232 (7)°] than the platinum chain. The Pt⋯Pt distance in (I) is slightly shorter than those of cis-[PtCl2(NH3)2] [3.372 (2) and 3.409 (2) Å; Milburn & Truter, 1966 ▸] or [PtCl2(en)] [3.381 Å; Iball et al., 1975 ▸], and is considerably shorter than that of [PtCl2(tn)] [3.646 Å; Odoko & Okabe, 2006 ▸], all of which have similar columnar structures.
Figure 2.

A view of the columnar structure of compound (I). Light-blue dashed lines represent hydrogen bonds between adjacent molecules in the column. Yellow dashed lines indicate the short contact between Pt atoms in the column. [Symmetry codes: (i) −x + 1, −y + 1, −z; (ii) −x + 1, −y + 1, −z + 1; (iii) x, y, z + 1.]
The intermolecular Pt⋯Pt distance of (I) suggests that the columnar structure is stabilized by weak metal–metal interactions. The columnar structure of (I) is further stabilized by intermolecular N—H⋯Cl hydrogen bonds between adjacent molecules in the column (Fig. 2 ▸ and Table 2 ▸). Intercolumnar hydrogen bonds also help to stabilize the crystal packing of the columns (Fig. 3 ▸, and Table 2 ▸).
Table 2. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯Cl1iv | 0.90 | 2.57 | 3.353 (4) | 146 |
| N1—H1B⋯Cl1v | 0.90 | 2.71 | 3.381 (4) | 133 |
| N1—H1B⋯Cl1vi | 0.90 | 2.73 | 3.320 (5) | 124 |
Symmetry codes: (iv) 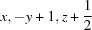 ; (v)
; (v)  ; (vi)
; (vi)  .
.
Figure 3.

The crystal packing of compound (I), viewed along the c axis. Light-blue dashed lines represent intercolumnar hydrogen bonds. Solid orange lines indicate the unit cell.
Synthesis and crystallization
Compound (I) was prepared using a method modified from that described by Connors et al. (1972 ▸) as follows. To an aqueous HCl solution (1.0 M, 15 ml) of K2[PtCl4] (0.241 mmol, 100 mg) was slowly added an aqueous HCl solution (1.0 M, 15 ml) of 1,2-phenylenediamine (0.241 mmol, 26 mg), and then the solution was sealed in a screw-cap vial and was kept at room temperature for one week in the dark. Pale-brown needle-like crystals suitable for X-ray analysis were obtained (yield 52%). Elemental analysis found: C 19.26, H 2.23, N 7.30%; calculated for C6H8Cl2N2Pt: C 19.26, H 2.16, N 7.49%. Elemental analysis was carried out by the Laboratory of Organic Elemental Analysis, Department of Chemistry, Graduate School of Science, The University of Tokyo.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. One reflection (010) was omitted in the final refinement because it was obstructed by the beam-stop. H atoms were placed in geometrically calculated positions and refined as riding, with C(aromatic)—H = 0.93 and N—H = 0.90 Å, and with U iso(H) = 1.2U eq(C,N). The maximum and minimum electron density peaks are located 0.80 and 0.74 Å, respectively, from atom Pt1.
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | [PtCl2(C6H8N2)] |
| M r | 374.13 |
| Crystal system, space group | Monoclinic, P2/c |
| Temperature (K) | 296 |
| a, b, c (Å) | 7.087 (2), 10.446 (3), 6.6920 (16) |
| β (°) | 116.61 (2) |
| V (Å3) | 442.9 (2) |
| Z | 2 |
| Radiation type | Mo Kα |
| μ (mm−1) | 16.38 |
| Crystal size (mm) | 0.26 × 0.13 × 0.07 |
| Data collection | |
| Diffractometer | Rigaku R-AXIS RAPID imaging-plate |
| Absorption correction | Multi-scan (ABSCOR; Higashi, 1995 ▸) |
| T min, T max | 0.116, 0.304 |
| No. of measured, independent and observed [F 2 > 2σ(F 2)] reflections | 10875, 1587, 1480 |
| R int | 0.030 |
| (sin θ/λ)max (Å−1) | 0.757 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.034, 0.099, 1.18 |
| No. of reflections | 1587 |
| No. of parameters | 52 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 4.62, −1.74 |
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989017008477/wm5396sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989017008477/wm5396Isup2.hkl
CCDC reference: 1055177
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| [PtCl2(C6H8N2)] | F(000) = 340 |
| Mr = 374.13 | Dx = 2.805 Mg m−3 |
| Monoclinic, P2/c | Mo Kα radiation, λ = 0.71075 Å |
| Hall symbol: -P 2yc | Cell parameters from 12924 reflections |
| a = 7.087 (2) Å | θ = 2.0–32.6° |
| b = 10.446 (3) Å | µ = 16.38 mm−1 |
| c = 6.6920 (16) Å | T = 296 K |
| β = 116.61 (2)° | Needle, pale brown |
| V = 442.9 (2) Å3 | 0.26 × 0.13 × 0.07 mm |
| Z = 2 |
Data collection
| Rigaku R-AXIS RAPID imaging-plate diffractometer | 1587 independent reflections |
| Radiation source: X-ray sealed tube | 1480 reflections with F2 > 2σ(F2) |
| Graphite monochromator | Rint = 0.030 |
| Detector resolution: 10.00 pixels mm-1 | θmax = 32.6°, θmin = 3.2° |
| ω scans | h = −10→10 |
| Absorption correction: multi-scan (ABSCOR; Higashi, 1995) | k = −15→15 |
| Tmin = 0.116, Tmax = 0.304 | l = −10→8 |
| 10875 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.034 | H-atom parameters constrained |
| wR(F2) = 0.099 | w = 1/[σ2(Fo2) + (0.0669P)2 + 0.3105P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.18 | (Δ/σ)max = 0.001 |
| 1587 reflections | Δρmax = 4.62 e Å−3 |
| 52 parameters | Δρmin = −1.74 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0039 (12) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. Least-squares planes (x,y,z in crystal coordinates) and deviations from them (* indicates atom used to define plane) - 2.7022 (0.0123) x - 0.0000 (0.0000) y + 6.6740 (0.0026) z = 0.3174 (0.0059) * 0.0000 (0.0000) Pt1 * -0.0185 (0.0028) Cl1 * 0.0206 (0.0042) N1 * 0.0050 (0.0039) C1 * -0.0017 (0.0044) C2 * 0.0031 (0.0116) C3 * 0.0185 (0.0028) Cl1_$6 * -0.0206 (0.0042) N1_$6 * -0.0050 (0.0039) C1_$6 * 0.0017 (0.0044) C2_$6 * -0.0031 (0.0116) C3_$6 Rms deviation of fitted atoms = 0.0121 |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Pt1 | 0.5000 | 0.504875 (16) | 0.2500 | 0.03029 (13) | |
| Cl1 | 0.23327 (19) | 0.65687 (13) | 0.1392 (2) | 0.0429 (3) | |
| N1 | 0.2863 (7) | 0.3592 (4) | 0.1666 (8) | 0.0407 (9) | |
| H1A | 0.2144 | 0.3653 | 0.2480 | 0.049* | |
| H1B | 0.1934 | 0.3657 | 0.0213 | 0.049* | |
| C1 | 0.3910 (7) | 0.2364 (4) | 0.2066 (6) | 0.0400 (8) | |
| C2 | 0.2835 (10) | 0.1225 (5) | 0.1621 (9) | 0.0563 (12) | |
| H2 | 0.1370 | 0.1224 | 0.1016 | 0.068* | |
| C3 | 0.391 (3) | 0.0081 (5) | 0.207 (2) | 0.068 (3) | |
| H3 | 0.3182 | −0.0690 | 0.1780 | 0.082* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Pt1 | 0.02635 (17) | 0.02835 (16) | 0.03208 (18) | 0.000 | 0.00944 (11) | 0.000 |
| Cl1 | 0.0335 (5) | 0.0383 (5) | 0.0513 (6) | 0.0058 (4) | 0.0140 (4) | 0.0012 (4) |
| N1 | 0.0347 (18) | 0.0371 (18) | 0.043 (2) | −0.0017 (14) | 0.0108 (16) | −0.0011 (15) |
| C1 | 0.050 (2) | 0.0328 (18) | 0.0365 (18) | −0.0028 (15) | 0.0185 (17) | −0.0012 (14) |
| C2 | 0.067 (3) | 0.046 (3) | 0.054 (3) | −0.018 (2) | 0.026 (2) | −0.005 (2) |
| C3 | 0.112 (10) | 0.037 (3) | 0.063 (6) | −0.017 (3) | 0.047 (6) | −0.006 (2) |
Geometric parameters (Å, º)
| Pt1—N1 | 2.040 (4) | N1—H1B | 0.9000 |
| Pt1—N1i | 2.040 (4) | C1—C2 | 1.372 (6) |
| Pt1—Cl1i | 2.3213 (13) | C1—C1i | 1.386 (9) |
| Pt1—Cl1 | 2.3213 (13) | C2—C3 | 1.377 (11) |
| Pt1—Pt1ii | 3.3475 (8) | C2—H2 | 0.9300 |
| Pt1—Pt1iii | 3.3475 (8) | C3—C3i | 1.38 (3) |
| N1—C1 | 1.445 (6) | C3—H3 | 0.9300 |
| N1—H1A | 0.9000 | ||
| N1—Pt1—N1i | 83.6 (3) | C1—N1—Pt1 | 110.8 (3) |
| N1—Pt1—Cl1i | 174.82 (12) | C1—N1—H1A | 109.5 |
| N1i—Pt1—Cl1i | 91.39 (15) | Pt1—N1—H1A | 109.5 |
| N1—Pt1—Cl1 | 91.39 (15) | C1—N1—H1B | 109.5 |
| N1i—Pt1—Cl1 | 174.82 (12) | Pt1—N1—H1B | 109.5 |
| Cl1i—Pt1—Cl1 | 93.69 (7) | H1A—N1—H1B | 108.1 |
| N1—Pt1—Pt1ii | 92.07 (14) | C2—C1—C1i | 119.8 (3) |
| N1i—Pt1—Pt1ii | 85.32 (14) | C2—C1—N1 | 122.7 (5) |
| Cl1i—Pt1—Pt1ii | 88.59 (4) | C1i—C1—N1 | 117.4 (2) |
| Cl1—Pt1—Pt1ii | 93.80 (4) | C1—C2—C3 | 120.3 (8) |
| N1—Pt1—Pt1iii | 85.32 (14) | C1—C2—H2 | 119.8 |
| N1i—Pt1—Pt1iii | 92.07 (14) | C3—C2—H2 | 119.8 |
| Cl1i—Pt1—Pt1iii | 93.80 (4) | C2—C3—C3i | 119.8 (6) |
| Cl1—Pt1—Pt1iii | 88.59 (4) | C2—C3—H3 | 120.1 |
| Pt1ii—Pt1—Pt1iii | 176.513 (11) | C3i—C3—H3 | 120.1 |
| Pt1ii—Pt1—N1—C1 | 84.8 (3) | Pt1iii—Pt1—N1—C1 | −92.9 (3) |
Symmetry codes: (i) −x+1, y, −z+1/2; (ii) −x+1, −y+1, −z; (iii) −x+1, −y+1, −z+1.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···Cl1iv | 0.90 | 2.57 | 3.353 (4) | 146 |
| N1—H1B···Cl1v | 0.90 | 2.71 | 3.381 (4) | 133 |
| N1—H1B···Cl1vi | 0.90 | 2.73 | 3.320 (5) | 124 |
Symmetry codes: (iv) x, −y+1, z+1/2; (v) x, −y+1, z−1/2; (vi) −x, −y+1, −z.
Funding Statement
This work was funded by Ministry of Education, Culture, Sports, Science and Technology, MEXT-Supported Program for the Strategic Research Foundation at Private Universities grant S1311027.
References
- Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994). J. Appl. Cryst. 27, 435.
- Asiri, A. M., Arshad, M. N., Ishaq, M., Alamry, K. A. & Bokhari, T. H. (2012). Acta Cryst. E68, m1562. [DOI] [PMC free article] [PubMed]
- Brandenburg, K. (2017). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Connors, T. A., Jones, M., Ross, W. C. J., Braddock, P. D., Khokhar, A. R. & Tobe, M. L. (1972). Chem. Biol. Interact. 5, 415–424. [DOI] [PubMed]
- Higashi, T. (1995). ABSCOR. Rigaku Corporation, Tokyo, Japan.
- Iball, J., MacDougall, M. & Scrimgeour, S. (1975). Acta Cryst. B31, 1672–1674.
- Köckerbauer, R. & Bednarski, P. J. (1996). J. Inorg. Biochem. 62, 281–298. [DOI] [PubMed]
- Konno, Y. & Matsushita, N. (2006a). Bull. Chem. Soc. Jpn, 79, 1046–1053.
- Konno, Y. & Matsushita, N. (2006b). Bull. Chem. Soc. Jpn, 79, 1237–1239.
- Konno, Y. & Matsushita, N. (2017). IUCrData, 2, x170144.
- Lock, C. J. L. & Pilon, P. (1981). Acta Cryst. B37, 45–49.
- Lock, C. J. L., Speranzinl, R. A. & Zvagulis, M. (1980). Acta Cryst. B36, 1789–1793.
- Meischen, S. J., Gale, G. R., Lake, L. M., Frangakis, C. J., Rosenblum, M. G., Walker, E. M., Atkins, L. M. & Smith, A. B. (1976). J. Natl Cancer Inst. 57, 841–845. [DOI] [PubMed]
- Melanson, R., de la Chevrotière, C. & Rochon, F. D. (1987). Acta Cryst. C43, 57–59.
- Milburn, G. H. W. & Truter, M. R. (1966). J. Chem. Soc. A, pp. 1609–1616.
- Odoko, M. & Okabe, N. (2006). Acta Cryst. C62, m136–m139. [DOI] [PubMed]
- Rigaku (1998). RAPID-AUTO. Rigaku Corporation, Tokyo, Japan.
- Rochon, F. D. & Melanson, R. (1986). Acta Cryst. C42, 1291–1294.
- Rosenberg, B., Van Camp, L. & Krigas, T. (1965). Nature, 205, 698–699. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shi, X.-F., Xie, M.-J. & Ng, S. W. (2006). Acta Cryst. E62, m2719–m2720.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989017008477/wm5396sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989017008477/wm5396Isup2.hkl
CCDC reference: 1055177
Additional supporting information: crystallographic information; 3D view; checkCIF report