

SUMMARY
Constitutive activation of Gαq signaling by mutations in GNAQ or GNA11 occurs in over 80% of uveal melanomas (UMs) and activates MAPK. Protein kinase C (PKC) has been implicated as a link, but the mechanistic details remained unclear. We identified PKC δ and ε as required and sufficient to activate MAPK in GNAQ mutant melanomas. MAPK activation depends on Ras and is caused by RasGRP3, which is significantly and selectively overexpressed in response to GNAQ/11 mutation in UM. RasGRP3 activation occurs via PKC δ- and ε-dependent phosphorylation and PKC-independent, DAG-mediated membrane recruitment, possibly explaining the limited effect of PKC inhibitors to durably suppress MAPK in UM. The findings nominate RasGRP3 as a therapeutic target for cancers driven by oncogenic GNAQ/11.
In Brief
Chen et al. find that Ras is required for GNAQ-mediated MAPK activation and identify PKC δ, ε and RasGRP3 as components of a signaling module necessary and sufficient to activate the Ras/MAPK pathway in GNAQ mutant uveal melanoma (UM). RasGRP3 is selectively overexpressed in response to GNAQ/11 mutations in UM.
INTRODUCTION
Uveal melanoma (UM) is the most common intraocular malignancy in adults and cannot be effectively treated once metastatic (Singh et al., 2005). UM is a genetically and biologically distinct type of melanoma with a significantly lower 10-year survival rate than cutaneous melanoma. It originates from melanocytes of the choroidal plexus, ciliary body, or iris of the eye and does not harbor mutations in BRAF, NRAS, and KIT that prevail in cutaneous melanomas arising from intraepithelial melanocytes of the skin or mucosa. Instead, UM harbors mutually exclusive mutations in GNAQ, GNA11, PLCB4, or cysteinyl leukotriene receptor 2 (CYSLTR2) (Johansson et al., 2016; Moore et al., 2016; Van Raamsdonk et al., 2009; Van Raamsdonk et al., 2010). GNAQ and GNA11 encode closely related, large GTPases of the Gαq family, which are α subunits of heterotrimeric G proteins that operate downstream of G protein-coupled receptors (GPCRs). Relevant Gαq-coupled receptors in melanocytes include endothelin and WNT receptors, which play a critical role in melanocyte differentiation and survival and have been associated with invasion and metastasis in melanoma, and the CYSLTR2 (Dissanayake et al., 2007; Moore et al., 2016; Sheldahl et al., 1999; Shin et al., 1999). Approximately 95% of GNAQ and GNA11 mutations in melanoma affect codons 209 (Q209) of the G proteins with only 5% affecting codon 183 (R183) in the Ras-like domain. The respective mutations result in complete or partial loss of GTPase activity, thereby locking GNAQ/11 into its active protein, GTP-bound conformation, resulting in a dominant acting oncogene that transforms melanocytes (Van Raamsdonk et al., 2009; Van Raamsdonk et al., 2010). Similar to the mutations in RAS oncoproteins, the defects in GNAQ or GNA11 GTPases are difficult to target directly, making it important to delineate the oncogenic effector pathways downstream to identify opportunities for targeted therapy. Recently, mutations in the CYSLTR2, a Gq-coupled GPCR, and the downstream effector of Gαq PLCB4, encoding a phospholipase C β(PLC β) isoform, have been reported in the small percentage of UMs without GNAQ or GNA11 mutations (Johansson et al., 2016; Moore et al., 2016). In the TCGA, 78 out of 80 human UMs have mutations in either GNAQ, GNA11, PLCB4, or CYSLTR2, indicating that UM is defined by activating mutations in the GNAQ/11 pathway.
MAP-kinase pathway activation has been shown as one contributing factor to GNAQ-mediated oncogenesis (Van Raamsdonk et al., 2009). However, how exactly mutant GNAQ/11 relays signals to the MAPK pathway in UM remains to be clarified. Recent studies have demonstrated MAP-kinase pathway activation is at least in part mediated by protein kinase C (PKC) (Chen et al., 2014; Wu et al., 2012). PLC β, a direct downstream effector of mutant GNAQ/11, hydrolyzes the membrane phospholipid phosphatidylinositol 4,5-bisphosphate to release two potent second messengers: inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). DAG provides a docking site within the inner leaflet of the plasma membrane for enzymes, including specific PKC isoforms (Hubbard and Hepler, 2006) and IP3 releases calcium from the smooth endoplasmic reticulum. Calcium also activates some PKC isoforms. The PKC family consists of at least ten serine/threonine kinases, which are subdivided into classic, novel, and atypical isoforms (Griner and Kazanietz, 2007). The classical PKC isoforms (α, βI, βII, γ) can be activated by both DAG and calcium, while the novel PKCs (β, ε, θ, η) are only DAG dependent. By contrast, the atypical PKCs (ζ, ι) are not responsive to DAG or calcium. The specific PKC isoforms that mediate the activating effect on the MAPK pathway in the context of GNAQ or GNA11 mutations remain unclear. Recent studies have also demonstrated that mutant GNAQ/11 promote UM tumorigenesis by activating YAP independent of PLC β (Feng et al., 2014; Yu et al., 2014). Furthermore, prior studies have shown that PKC inhibition alone in UM cell lines is not sufficient to completely suppress MAPK signaling (Chen et al., 2014), suggesting that PKC-independent effectors may be involved that mediate MAPK signaling in GNAQ/11 mutant cells. Alternative effectors that may connect PLCβ-generated IP3 and DAG to MAPK signaling in UM and have recently been implicated in various types of cancer are members of the RasGRP family of RasGEFs (Ras guanine nucleotide exchange factors) (Ksionda et al., 2013). Studies in BRAFV600E melanomas suggest that therapeutically meaningful responses in patients can only be expected if marked suppression of the MAPK pathway is achieved (Bollag et al., 2010; Flaherty et al., 2012). To achieve this goal in UM, a more refined understanding of the connection between MAPK signaling and GNAQ/11 mutant in uveal melanoma is required to develop more effective targeting strategies.
Here, the goal of the current study is to investigate which PKC isoforms activate MAPK signaling and how PKC signaling relays to the MAPK pathway in GNAQ/11 mutant melanoma, thus identifying specific therapeutic targets for cancers driven by oncogenic GNAQ/11.
RESULTS
PKC δ and PKC ε Are the Critical PKC Isoforms that Mediate MAP-Kinase Signaling in UM Cells with GNAQ/11 Mutation
Oncogenic GNAQ activates MAPK in part via PKC (Chen et al., 2014). However, which of the numerous isoforms are involved in this signaling is not entirely resolved. The significant homology among the different isoforms α, βI, βII, γ, δ, ε, θ, η, ζ, and ι poses challenges for their specific identification. To overcome this limitation we tested the specificity of commercially available PKC antibodies by transfecting HA-tagged full-length PKC isoforms or their catalytic subunits (Figures S1A and S1B) into 293FT cells and identified a panel of specific antibodies that was used to screen a panel of melanoma cells with or without GNAQ/11 mutations. As shown in Figure 1A, five PKC isoforms (PKC α, δ, ε, ζ, and ι) were consistently expressed throughout all six UM cells with GNAQ/11 mutations tested (MEL202, 92-1, OMM1.3, and MEL270 with GNAQ mutations, and OMM-GN11 and UPMD-1 with GNA11 mutations). These isoforms were also expressed at similar levels in UM cell lines (MEL285, MEL290, and MUM2C) that are commonly used as GNAQ/11 wild-type UM cell lines in the research community. We expanded the panel of GNAQ/11 wild-type lines with other cell lines of melanocytic lineage, stemming from cutaneous melanomas (C8161, MM415, MM485, and SK-MEL-28). These cutaneous melanoma cell lines had similar PKC isoform expression patterns to the UM cell lines, concordant with prior reports (Denning, 2012; Oka and Kikkawa, 2005). By contrast, PKC βI, PKC θ, PKC η, and PKC γ expression was not detected in any of the 13 melanoma cells, mostly consistent with previous studies (Denning, 2012; Gilhooly et al., 2001; Oka and Kikkawa, 2005; Selzer et al., 2002), although PKC η expression has been described in some melanoma cells and tissues (Oka and Kikkawa, 2005). While PKC β II showed differential expression between melanoma cell lines with and without GNAQ/11 mutations, it was not expressed in all GNAQ/11 mutant cell lines, ruling it out as a universal effector downstream of mutant GNAQ/11. While PKC ζ and ι, where found to be expressed in GNAQ/11 mutant cell lines, are classified as atypical PKCs as they are not activated by either calcium or DAG, making them unlikely candidate effectors of mutant Gαq. By contrast, PKC α, δ, and ε, which were consistently expressed in all UM cell lines with GNAQ or GNA11 mutations, are candidates to mediate MAPK signaling in GNAQ mutant melanomas.
Figure 1. PKC δ and ε Are Important Downstream Effectors of Oncogenic GNAQ in Mediating MAPK Signaling.

(A) Expression of PKC isoforms in melanoma cell lines with and without GNAQ or GNA11 mutations. Previously validated antibodies against various PKC isoforms (Figures S1A and S1B) were tested on a broad panel of melanoma cell lines. Suitable positive controls for isoforms found not to be expressed are shown on the rightmost lane. PC, positive control.
(B) Knockdown of GNAQ and PKC isoforms δ and ε reduced levels of pMEK and pERK in the GNAQ-mutated uveal melanoma cell line 92-1, but not in the GNAQ wild-type cell line C8161.
(C) Cells were transfected with respective siRNAs either singly or in combination and then lysed. −, no siRNA; NT, non-targeting siRNA.
(D) PKC δ and ε synergized with GNAQQ209L in activating MAPK in 293FT cells. 293FT cells were transfected for 24 hr. PKC isoforms expression was detected using HA tags and GNAQQ209L using a Glu-Glu tag (EE).
(E) Effect of knockdown of PKC α, δ, ε, and GNAQ or GNA11, on the proliferation of GNAQ mutant melanoma cells (92-1 and OMM1.3) and GNA11 mutant cell line (UPMD-2) and GNAQ/11 wild-type melanoma cells (C8161 and SK-MEL-28 from cutaneous origin and MEL285 from uveal origin). Cells were transfected with various combination of siRNAs as indicated. After transfection (48 hr), cells were counted and seeded at equal density. Cells were counted at days 1, 3, and 5 after plating. −, no siRNA control; NT, non-targeting siRNA control. Error bars represent the SEM. See also Figures S1 and S2.
To determine which of these PKC isoforms mediate MAPK signaling in GNAQ/11 mutant cells, we performed small interfering RNAs (siRNA)-mediated knockdown of PKC isoforms (PKC α, δ, ε as well as ζ used as control) alone or in combination in three GNAQ mutant cell lines (92-1, OMM1.3, and MEL202) and examined MAPK signaling at the level of pMEK and pERK. As shown in Figures 1B and S1C, knockdown of either PKC δ or ε alone resulted in partial inhibition of pMEK and pERK, whereas knockdown of PKC α and PKC ζ had no significant effect. Combined knockdown of PKC δ and ε inhibited pMEK and pERK levels similar to knockdown of GNAQ itself. Similar results were observed in the GNA11 mutant cell line UPMD-2 (Figure S1C). In contrast to the suppressive effect on MAP-kinase pathway activation in GNAQ mutant cell lines, knockdown of GNAQ, PKC δ, and PKC ε, singly or in combination had no effect in three melanoma cell lines without GNAQ mutations (C8161 from cutaneous origin, and MUM2C and MEL285 from uveal origin) (Figures 1C and S1D). By contrast, knockdown of PKC βII in 92-1 with GNAQ mutation (Figure 1B) and OMM-GN11 and UPMD-2 cell lines with GNA11 mutation (Figure S1E) had no effect on pMEK and pERK.
To establish an initial connection between specific PKC isoforms and mutant GNAQ, we co-transfected PKC isoforms (α, δ, ε, ζ, ι) together with GNAQQ209L into the 293FT model cell line. As shown in Figure 1D, only PKC δ and ε strongly synergized with GNAQQ209L in activating the MAP-kinase pathway. We confirmed that the synergistic effect was dependent on the kinase activity of PKC δ and ε, by using kinase-dead mutants of PKC δ and ε (Figure S2A). Furthermore, only GNAQQ209L, but not wild-type GNAQ, synergized with PKC δ and ε, indicating that the synergy depended on active GNAQ (Figure S2B).
PKC is recruited by DAG to the plasma membrane which contributes to its activation. In normal melanocytes, both PKC α and δ were localized in the cytosolic fraction, while PKC ε were localized in both cytosolic and membrane fractions (Figure S2C). After stimulation with the stable DAG analog TPA, the PKC isoforms α, δ, and ε, but not ζ, translocated to the membrane (Figure S2D). In UM cell lines with GNAQ/11 mutations, PKC α and ζ were localized in the cytosolic fraction, while a considerable portion of the PKC δ and ε signal was localized at the membrane (Figure S2E). These data further support the notion that the main active isoforms in the context of GNAQ/11 mutations are PKC δ and ε.
As previously shown, cell proliferation of GNAQ mutant UM cells depends on GNAQ and PKC as knockdown of GNAQ or PKC inhibition suppress cell proliferation (Chen et al., 2014; Khalili et al., 2012; Wu et al., 2012). To determine whether this inhibition is mediated by PKC δ and PKC ε, we investigated their contribution by knocking them down both singly and in combination. Combined knockdown of PKC δ and ε, but not α, reduced cell proliferation similar to knockdown of GNAQ in GNAQ mutant cell lines (92-1 and OMM1.3), while the knockdown had no effect in cell lines without GNAQ mutations (Figure 1E). Similar results were obtained in the GNA11 mutant cell line (UPMD-2) (Figure 1E). We confirmed that expression levels of the targeted genes remained suppressed during the course of these experiments (Figures S2F and S2G). Taken together, these experiments indicate that PKC δ and ε are essential for MAPK signaling and proliferation of UM cell lines with GNAQ mutations, but not in related cell lines without GNAQ mutations.
Ras Is Required for Oncogenic GNAQ to Activate the MAP-Kinase Pathway
To determine at which level mutant GNAQ activates the MAP-kinase pathway, we investigated whether the activation depended on Ras. We found that 293FT cells transfected with GNAQQ209L had increased levels of Ras-GTP compared with controls, which could be further increased by co-transfection of PKC δ and ε compared with co-transfection of GFP (Figure 2A), while PKC α or ζ together with GNAQQ209L also increased Ras-GTP level, albeit to a lesser extent (Figure S2H). TPA also increased Ras-GTP levels. All three Ras isoforms (Hras, Kras, and Nras) were activated by oncogenic GNAQ (Figure 2B). Co-depletion of Kras and Nras was the minimum requirement for pMEK/pERK downregulation in 92-1 and Omm1.3 cells, while only simultaneous knockdown of all three Ras isoforms decreased pMEK and pERK levels, comparable with knockdown of GNAQ and inhibited cell proliferation in Mel202 cells (Figures 2C and S2I). These results indicate that activation of MAPK signaling in GNAQ mutant cells requires the presence of Ras, with little or no specific preference of any specific Ras family member.
Figure 2. Oncogenic GNAQ Activates Ras.

(A) GNAQQ209L transfected alone or combined with PKC δ or ε into 293FT cells increased Ras-GTP levels. 293FT cells were transfected with indicated cDNA plasmids for 24 hr. Ras-GTP pull-down was performed and detected by western blot with a pan-Ras antibody. TPA stimulation was used a positive control. Expression levels of GNAQQ209L were monitored with Glu-Glu (EE) tag. PKC δ and ε were detected via HA tags.
(B) Oncogenic GNAQ activates all three Ras isoforms. 293FT cells were transfected with indicated cDNA plasmids for 24 hr. Ras-GTP pull-down was performed and detected by western blot with Ras isoform-specific antibodies.
(C) Depletion of Ras isoforms reduced MAPK signaling as demonstrated by decreased pMEK and pERK in GNAQ mutant melanoma cell lines. Three human uveal melanoma cell lines with GNAQ mutation (92-1, OMM1.3, and MEL202) were transfected with mock (−), non-targeting siRNAs (NT), siRNAs against GNAQ, Hras (H), Kras (K), Nras (N), or indicated combinations for 72 hr and lysed. See also Figure S2.
The RasGEF, RasGRP3, Is Implicated as a Signaling Module in UM
To determine how mutant GNAQ activates Ras/MAPK signaling, we compared gene expression profiles between five GNAQ or GNA11 mutant melanoma cells (GNAQmt: 92-1, MEL202, OMM1.3; GNA11mt: UPMD-1, OMM-GN11) and five GNAQ/11 wild-type melanoma cells with either NRAS (SK-MEL-2, MM415, and MM485, all from cutaneous origins) or BRAF mutations (SK-MEL-5 from cutaneous origin and MUM2C from uveal origin). As shown in Figure 3A, RasGRP3, a RasGEF, ranked at the top of 487 differentially expressed genes between (cut off: p < 0.05; fold change >2 or < 2). In addition, RasGRP3 spiked our particular interest because Ras-MAPK activation via RasGRP3 is regulated by the second messenger DAG and by phosphorylation by PKC (Ksionda et al., 2013). qRT-PCR experiments confirmed that RasGRP3 mRNA is markedly upregulated (>100-fold) in melanoma cell lines with GNAQ or GNA11 mutations, compared with normal melanocytes or melanoma cell lines with other mutations. Other RasGRP family members, as well as the RasGEF SOS1, did not show any increased expression levels in melanoma cell lines with GNAQ or GNA11 mutations or showed differential expression differences in these cells (Figure S3A). A comparison of RasGRP3 mRNA levels across TCGA datasets (Figure 3C) revealed that RasGRP3 expression in UM tissues is consistently increased and considerably higher than in other cancer types, including cancers reported to have elevated RasGRP3 expression, such as breast cancer (Nagy et al., 2014), prostate cancer (Yang et al., 2010), lymphoma (Teixeira et al., 2003), and cutaneous melanoma (Yang et al., 2011). Interestingly, the four samples in the 478 samples of cutaneous melanoma samples that harbor GNAQ or GNA11 hotspot mutations, also had elevated RasGRP3 mRNA with levels comparable with those present in UM (Figure 3D). These data suggest that higher expression of RasGRP3 is specifically associated with the presence of oncogenic GNAQ/11.
Figure 3. RasGRP3 Is Upregulated in Uveal Melanoma Cells with GNAQ/11 Mutations.

(A) Scatterplot of top 487 genes (left panel) and heatmap of top 30 genes (right panel) of differentially expressed genes between 5 GNAQ/11 mutant (mt) and 5 GNAQ/11 wild-type (WT). The black arrow indicates RasGRP3.
(B) qRT-PCR shows markedly elevated RasGRP3 mRNA levels in 6 GNAQ mutant cells (92-1, MEL202, OMM1.3, MEL270, UPMM2, and UPMM3) and 3 GNA11 mutant cells (OMM-GN11, UPMD-1, and UPMD-2), compared with three types of human melanocytes (IHM, immortalized human melanocytes; NHM, normal human melanocytes) and seven human melanoma cell lines without GNAQ/11 mutations. Three (MUM2C, MEL285, and MEL290) melanoma cell lines without GNAQ/11 mutations are from uveal origin.
(C) RasGRP3 mRNA expression across different human cancers from TCGA RNA sequencing data. RasGRP3 expression is highest in uveal melanoma.
(D) Cutaneous melanomas with GNAQQ209P or GNA11Q209L mutations in the TCGA have increased RasGRP3 mRNA expression, comparable with uveal melanomas.
(E) Western blot shows increased RasGRP3 protein levels in human melanoma cells with GNAQ/11 mutations compared with cell lines with other mutations (MEL285, MUM2C, and MEL290 are of uveal and the other GNAQ/11 WT cell lines are of cutaneous origin). RasGRP1, RasGRP2, and RasGRP4 were not detectable in melanoma cells with GNAQ/11 mutations. 293FT lysates transfected with RasGRPs cDNA were used as positive control. PC, positive control.
(F) RasGRP3 immunohistochemistry in human uveal melanoma cell lines. RasGRP3 immunohistochemistry of pellets of formalin-fixed paraffin-embedded GNAQ mutant uveal melanoma cell lines (92-1 and MEL270) and GNAQ/11 wild-type lines (CRMM1 and CRMM2). Scale bars, 100 μm.
(G) RasGRP3 immunohistochemistry in four uveal melanoma tissues with GNAQ mutation and four with GNA11 mutations. Scale bars, 100 μm. See also Figure S3.
Western blotting with validated antibodies (Figure S3B) demonstrated that the increased RasGRP3 mRNA levels lead to elevated protein levels (Figures 3E and S3C). Similarly, the lymphoblastoid cell line Ramos B (Teixeira et al., 2003), and the cutaneous melanoma cell lines SK-MEL-5 and UACC257 (Yang et al., 2011), have been reported to express elevated RasGRP3 levels. We found that both RasGRP3 and p-RasGRP3 (T133) protein levels in melanoma cell lines with GNAQ mutations were significantly higher than in cell lines without GNAQ mutations, including SK-MEL-5 and UACC257 (Figure S3D). We also examined RasGRP3 protein expression in formalin-fixed, paraffin-embedded UM tissues by immunohistochemistry. As shown in Figure S3E, two conjunctival melanoma cell lines that had no GNAQ/11 mutations (CRMM1 and CRMM2) had no detectable expression of RasGRP3 protein by western blot and immunohistochemistry (Figure 3F), while UM cell lines with GNAQ mutation (92-1, MEL270) or GNA11 mutation (OMM1) showed strong RasGRP3 immunoreactivity (Figures 3F and S3F). RasGRP3 protein expression was also detected by immunohistochemistry in UM tissues with GNAQ and GNA11 mutations (Figure 3G).
Elevated RasGRP3 Expression and RasGRP3 Phosphorylation Is a Consequence of Mutant GNAQ Signaling and Is Mediated by PKC δ and PKC ε
We next investigated the connections between GNAQ and RasGRP3 and noted that knockdown of GNAQ diminished not only p-RasGRP3 levels, but also decreased total RasGRP3 (Figure 4A). Conversely, expression of GNAQQ209L or GNA11Q209L, but not BRAFV600E, in human melanocytes significantly increased total RasGRP3 and p-RasGRP3 levels (Figure 4B). The upregulation of RasGRP3 expression depended on PKC activity. Treatment of OMM1.3 cells with PKC inhibitor AEB071 (500 nM) suppressed RasGRP3 levels in a time-dependent manner, starting after 10 hr, peaking at 24 hr after drug administration (Figure 4C). This time course contrasts with the suppression of RasGRP3 phosphorylation by AEB071, which was detectable already at 2 hr and peaked at 10 hr. Maximal pERK inhibition was observed at about 24 hr, probably reflecting the combined effect of inhibition of RasGRP3 phosphorylation and downregulation of its expression. Similar results were observed in the 92-1 cell line (Figure S4A). The effect of PKC inhibition on RasGRP3 expression levels and p-RasGRP3 levels was dose dependent in three different GNAQ mutant cell lines incubated with two different PKC inhibitors, AEB071 and AHT956 for 24 hr (Figures 4D and S4B), but had no effects on RasGRP3 expression while inhibiting p-RasGRP3 in melanoma cell lines with BrafV600E or NRAS mutations (Figure S4C), which have very weak expression of RasGRP3 compared with GNAQ mutant cells. TPA increased RasGRP3 expression levels and RasGRP3 phosphorylation in a time-dependent manner in human melanocytes, with no significant effects on RasGRP1 and SOS1 (Figures 4E and S4E). TPA also increased RasGRP3 expression in human melanoma cells with BRAF mutations (Figure S4D). These data indicate that activation of PKC in melanocytic cells results in upregulation of RasGRP3 expression as well as T133 phosphorylation of RasGRP3, and that the markedly elevated protein and phosphorylation levels found in GNAQ mutant melanoma cells are a consequence of Gαq-mediated upregulation of PKC activity. Among the four PKC isoforms, PKC α, δ, ε, and ζ, which we found to be invariably expressed in GNAQ or GNA11 mutant UM cell lines, only PKC δ and ε robustly phosphorylated T133 of RasGRP3, whereas PKC α and ζ had weaker effects with no significant consequences on MAPK signaling (Figures 4F and S4F). Combined knockdown of PKC δ and ε suppressed RasGRP3 expression and RasGRP3 phosphorylation, and resulted in maximal inhibition of pERK and pMEK compared with non-target siRNA control, while knockdown of individual PKC isoforms and combined knockdown of PKC α with PKC δ or ε had no significant effect (Figure 4G).
Figure 4. Oncogenic GNAQ Signaling Regulates RasGRP3 Expression and Phosphorylation in Uveal Melanoma Cells.

(A) Depletion of GNAQ with siRNAs decreased RasGRP3 expression and T133 phosphorylation in three GNAQ mutant uveal melanoma cells (OMM1.3, 92-1, and MEL202). Cells were transfected with mock (−), non-targeting siRNAs (NT), or GNAQ siRNAs for 72 hr and lysed.
(B) GNAQQ209L or GNA11Q209L, but not wild-type GNAQ, GNA11, or BrafV600E, induced increased RasGRP3 expression and phosphorylation in immortalized human melanocytes. Melanocytes were infected with lentivirus expressing indicated proteins for 48 hr and analyzed by immunoblot. Expression levels of GNAQ and GNA11 were monitored using Glu-Glu tags.
(C) The PKC inhibitor AEB071 decreased RasGRP3 expression and phosphorylation in OMM1.3 cells in a time-dependent manner. OMM1.3 cells were incubated with DMSO or 500 nM AEB071 for the times indicated.
(D) The PKC inhibitors AEB071 and AHT956 decreased RasGRP3 expression and phosphorylation in OMM1.3 cells in a dose-dependent manner. OMM1.3 cells were incubated with DMSO or AEB071 or AHT956 at indicated doses for 24 hr.
(E) TPA increased RasGRP3 expression and phosphorylation in human melanocytes. Immortalized human melanocytes were incubated with or without 200 nM TPA for the indicated times.
(F) PKC δ and ε increased T133 phosphorylation of RasGRP3 in 293FT cells. 293FT cells were transfected with indicated cDNAs for 24 hr.
(G) Knockdown of PKC δ and ε decreased RasGRP3 expression and phosphorylation in GNAQ mutant cells. Western blot of 92-1 cells transfected for 72 hr with mock (−), non-targeting siRNAs (NT), PKC α, δ, or ε siRNAs, singly or in indicated combination.
(H) The MEK inhibitor PD0325901 has no effect on RasGRP3 expression and phosphorylation in GNAQ mutant cells. MEL202 and 92-1 cells were incubated with PD0325901 at different doses for 24 hr. See also Figures S4S6.
Next we investigated the mechanism of RasGRP3 upregulation. We ruled out a positive feedback loop involving MAPK activation and RasGRP3 regulation by suppressing MAP-kinase signaling using the two different MEK inhibitors PD0325901 and Trametinib (Figures 4H and S4G). The YAP1 pathway has been shown to play an important role in GNAQ mutant melanoma development (Feng et al., 2014; Yu et al., 2014). Knockdown of YAP1 did not affect RasGRP3/MAPK regulation and activation (Figure S5A). Combined inhibition of the YAP1 and PKC/MAPK pathways neither synergistically suppressed MAPK signaling nor increased cleaved PARP levels, compared with combined inhibition of PKC and MEK (Figures S5B and S5C). The YAP1 inhibitor verteporfin, or the MET inhibitor INC280, did not increase the effects of the PKC inhibitor AEB or the MEK inhibitor 162 (Figure S5F). The oncogene tyrosine kinase receptor MET was found to be highly expressed in UM cell lines with GNAQ/11 mutation in our microarray experiment (Figure 3A). However, knockdown of MET or inhibition with the MET inhibitor INC280 had no effect on RasGRP3 and p-RasGRP3 levels or pERK in UM cell lines (Figures S5D and S5E). These data indicate that neither the YAP1 pathway nor MET signaling is involved in the upregulation of RasGRP3.
Some cancers have RasGRP3 amplifications (Figure S6D), but we found no incident of amplification of RasGRP3 or PKC δ and ε in UM cell lines (Figures S6A and S6B) and UM tissue samples (Figure S6C). To investigate whether the upregulation occurs at the transcription level, we knocked down GNAQ and found that it decreased RasGRP3 mRNA levels in the GNAQ-mutated UM cell lines OMM1.3 and 92-1 (Figure S6E). By contrast, the knockdown had no effect on the RASGEF SOS1. The PKC inhibitors AEB071 and AHT956 also both decreased RasGRP3 mRNA, also with no effect on SOS1 mRNAs (Figure S6G). Conversely, overexpression of GNAQQ209L or GNA11Q209L in melanocytes increased RasGRP3 mRNA (Figure S6F). Similarly, TPA increased RasGRP3 mRNA in both melanocytes and in the BRAF-mutant melanoma cell line UACC257 (Figure S6H). Together, these data suggest that RasGRP3 regulation in the context of GNAQ mutations occurs transcriptionally and involves PKC signaling.
RasGRP3 Mediates MAPK Activation in GNAQ Mutant Melanoma Cells
The consistently elevated expression levels implicated RasGRP3 as a candidate protein involved in activation of Ras signaling downstream of mutant GNAQ/11. Knockdown of RasGRP3 in GNAQ mutant melanoma cell lines significantly reduced levels of pMEK and pERK, but not pAKT (Figures 5A and S7A), but had no effect in other melanoma cell lines (Figure 5B). Similar results were obtained with individual siRNAs instead of siRNA pools (Figure S7B). Knockdown of other RasGRP isoforms had no effect on pMEK and pERK (Figure S7C), but the protein levels of RasGRP1, RasGRP2, and RasGRP4 were already too low to be detectable even before knockdown. Knockdown of SOS1 only slightly reduced pMEK and pERK levels (Figure S7D). Knockdown of either GNAQ or RasGRP3 also decreased Ras-GTP levels in GNAQ mutant UM cell lines (Figure S7E).
Figure 5. RasGRP3 Is Important for GNAQ-Mediated MAP-Kinase Pathway Activation and Proliferation.

(A) siRNA-mediated knockdown of RasGRP3 decreases pMEK and pERK levels in three GNAQ mutant melanoma cells (92-1, omm1.3, and mel270), but not in three GNAQ/11 wild-type melanoma cells.
(B) MUM2C is from uveal origin. Cells were transfected with mock(−), non-targeting, or RasGRP3 siRNAs for 72 hr and lysed for western blot analysis.
(C) RasGRP3 knockdown reduces proliferation of melanoma cells with GNAQ mutations (92-1, MEL270, and OMM1.3), but not of cell lines with other mutations (SK-MEL-5 and UACC257). Cells were transfected with non-targeting or RasGRP3 siRNAs; 48 hr after transfection, cells were counted and plated at equal density. Cells were counted at days 1, 3, and 5 after plating. Data represent the mean ± SEM.
(D) Western blot of stable knockdown of RasGRP3 using shRNA. 92-1 cells were infected with lentivirus expressing two different shRNAs for RasGRP3 and selected with puromycin. NT, non-targeting shRNA.
(E) shRNA-mediated knockdown of RasGRP3 in 92-1 cells from (D) decreased the number and size of colonies. Cells were stained with crystal violet after 18 days of culture.
(F) Nude mice were injected subcutaneously with 2 × 106 92-1 cells expressing RasGRP3 shRNA-1 (n = 5) or non-targeting shRNA (n = 5). Tumors sizes and weights for both experimental arms 13 weeks after injection. Data represent the mean ± SD.
(G) The effect of co-transfection of GNAQQ209L with four different RasGRP isoforms on MAPK signaling. 293FT cells were transfected with mock (−), GFP, GNAQQ209L, RasGRP1, RasGRP2, RasGRP3, or RasGRP4 plasmids either singly or in combination as indicated. After 24 hr, cells were lysed and blotted. Arrows indicate that pRasGRP1 antibodies recognized both RasGRP1 T184 phosphorylation and RasGRP3 T133 phosphorylation. See also Figure S7.
siRNA-mediated knockdown of RasGRP3 also significantly decreased proliferation of GNAQ mutant cells during a 5-day proliferation assay, whereas it had no significant effect on other melanoma cell lines (Figure 5C). We verified that RasGRP3 expression remained suppressed during the course of the experiment (Figure S7F). Stable small hairpin RNA (shRNA)-mediated knockdown of RasGRP3 also markedly reduced tumor growth in a mouse xenograft model of the GNAQ mutant cell line 92-1 (Figures 5D5F).
To further confirm that RasGRP3 modulates MAPK signaling in the setting of GNAQ mutation, we co-transfected 293FT cells with GNAQQ209L combined with individual RasGRP isoforms and examined their effects on MAPK signaling. As shown in Figure 5G, RasGRP1, RasGRP3, and RasGRP4 alone, but not RasGRP2, induced pERK, while only co-transfection GNAQQ209L with RasGRP1 or RasGRP3 potentiated MAPK signaling as demonstrated by markedly increased pMEK and pERK levels. In contrast, co-transfection GNAQQ209L with RasGRP2 had no synergistic effect on MAPK signaling, consistent with the notion that RasGRP2 activates Rap, but not Ras (Ksionda et al., 2013). We ruled out that these differences were due to variation in the expression levels of the transfection constructs using antibodies against the myc tag of RasGRP isoforms. GNAQQ209L increased p-RasGRP1 (T184) and p-RasGRP3 (T133) levels in RasGRP1-and RasGRP3-expressing cells, respectively, indicating that oncogenic GNAQ can in principle activate both RasGRP1 and RasGRP3. Our data indicate that in the context of UM cells RasGRP3 is the relevant effector and is upregulated at the mRNA level, whereas RasGRP1 is not expressed (Figure 3E).
RasGRP3 Is Activated by PKC Phosphorylation and DAG-Mediated Membrane Recruitment
Next we performed a series of experiments to examine the mechanisms by which RasGRP3 activates MAPK signaling in UM cells. In B cells, it has been shown that PKC can phosphorylate RasGRP3 at T133, located near the amino terminal region of CDC25 Ras activator domain (Zheng et al., 2005). A phosphorylation site mutant RasGRP3T133A substantially reduces but does not abrogate all RasGRP3 activity in B cells (Aiba et al., 2004). To determine the role of T133 phosphorylation in the context of mutant GNAQ/11, we introduced RasGRP3T133A into 293FT cells. RasGRP3 T133A also was less effective in synergizing with GNAQQ209L in activating the MAP-kinase pathway (Figure 6A) and Ras (Figures S7G) than wild-type RasGRP3. We confirmed that the PKC isoform expression patterns in 293FT cells and UM cells are comparable, with the exception of PKC θ, which is only expressed in 293FT cells (Figure S7H). These data indicate that PKC phosphorylation at T133 also contributes to RasGRP3-mediated Ras/MAPK signaling in the context of mutant GNAQ. However, the phosphorylation site mutant of RasGRP3 had a residual activating effect on the MAP-kinase pathway, indicating that some RasGRP3 activity may be PKC independent. To resolve this, we inhibited PKC activity using the pan-PKC inhibitor AEB071 in 293FT cells transfected with combinations of mutant or wild-type RasGRP3 with mutant GNAQ (Figure 6B). At a concentration of 1 μM AEB071 was able to completely abrogate T133 phosphorylation of RasGRP3, consistent with prior studies showing complete extinction of PKC activity at this concentration (Chen et al., 2014). By contrast, AEB071 had no significant effect on MAP-kinase pathway activation of wild-type RasGRP3 or RasGRP3T133A in the absence of mutant GNAQ, indicating that this residual activity was PKC independent. As a control, addition of a MEK inhibitor, PD0325901, in this experiment blocked all MAPK activation (Figure 6C). These data indicate that PKC phosphorylation at T133 contributes to RasGRP3-mediated MAPK signaling, but pinpoints a residual activating effect of RasGRP3 that does not rely on phosphorylation at T133 or PKC activity, which is consistent with the reported work in B cells (Aiba et al., 2004).
Figure 6. Both PKC-Mediated Phosphorylation and C1 Domain Are Required for RasGRP3-Mediated MAPK Activation.

(A) Mutating the PKC phosphorylation site of RasGRP3 (T133A) partially attenuated RasGRP3-mediated MAPK signaling in 293FT cells. 293FT cells were transfected with mock (−), GFP, GNAQQ209L, RasGRP3wt (GRP3wt), or RasGRP3T133A (GRP3T133A) either singly or in the indicated combination. After 24 hr, cells were lysed and subjected to western blot. RasGRP3 wild-type and mutant expression were detected with Myc tag antibody. GLU-GLU tag (EE) was used to detect GNAQQ209L. The top panel shows a schematic diagram of the RasGRP3 T133A construct.
(B) PKC inhibition fails to completely abrogate RasGRP3-mediated MAPK signaling. 293FT cells were transfected with GFP, GNAQQ209L, RasGRP3wt, or RasGRP3T133A, either singly or in the indicated combinations. After 24 hr, transfected cells were treated with 1 μM AEB071 (AEB) or vehicle for another 24 hr and then lysed.
(C) The MEK inhibitor PD0325901 (PD) completely suppressed RasGRP3-mediated MAPK signaling. 293 FT cells were co-transfected with GNAQQ209L and RasGRP3T133A or with GNAQQ209L and RasGRP3wt. After 24 hr, cells were treated for another 24 hr with DMSO, 1 μM AEB071 (AEB), 1 μm AHT956 (AHT), 100 nM PD0325901 (PD), 1 μM AEB + 100 nM PD, or 1 μM AHT956 + 100 nM PD, respectively.
(D) Both T133 phosphorylation and the C1 domain of RasGRP3 contribute to oncogenic GNAQ-mediated MAPK signaling. 293 FT cells were transfected with indicated constructs for 24 hr and lysed. The left panel shows a schematic diagram of the RasGRP3 C1 domain deletion constructs used.
(E) Restoration of MAPK activation by expression of membrane-targeted forms of RasGRP3 in 293FT cells. RasGRP3 constructs tagged with the membrane-targeting motifs of Fyn and Src protein as shown in the left panel. 293FT cells transfected with indicated RasGRP3 mutant constructs for 24 hr. See also Figure S7.
RasGRP3 is recruited to the inner leaflet of the plasma membrane by binding DAG via its C1 domain (Lorenzo et al., 2001). To determine the role of membrane recruitment of RasGRP3 on MAPK signaling, we generated C1 domain deletion constructs of RasGRP3. As shown in Figure 6D, deletion of the C1 domain of RasGRP3 significantly reduces the ability of mutant GNAQ to activate the MAP-kinase pathway, and the RasGRP3 activity could be further reduced by mutating the T133 phosphorylation site. The effect of C1 deletion could be completely rescued by adding Fyn or Src membrane binding motifs (Chen and Resh, 2001) to the RasGRP3 C1 deletion mutants (Figure 6E), indicating that C1-mediated membrane binding is essential for GNAQ-mediated activation of MAP-kinase signaling. Together, these data demonstrate that membrane binding via the C1 domain and T133 phosphorylation cooperate in RasGRP3-mediated activation of MAPK signaling in the setting of GNAQ mutations.
DISCUSSION
Constitutive activation of the MAPK pathway (RAS/RAF/MEK/ERK) is common in cutaneous melanoma and typically occurs through mutations and/or amplification of signaling modules such as BRAF or NRAS. The MAP-kinase pathway is also activated in UM, but mutations of these specific genes are conspicuously absent. Instead, this disease appears to be defined by mutations of the Gαq pathway. The mechanism of how these mutations result in activation of MAP-kinase signaling is incompletely understood. Prior studies have shown Gαq-mediated activation of MAPK through the kinases Pyk2 and Src-mediated activation of Ras in PC12 cells and HEK293 cells (Della Rocca et al., 1997; Dikic et al., 1996; Lev et al., 1995), an effect that is PKC independent. Gαq has also been shown to activate the MAP-kinase pathway independent of RAS through RasGRP2-and Rap1-mediated activation of B-Raf in PC12 cells (Guo et al., 2001). Another RAS-independent mechanism of MAP-kinase pathway activation by Gαq involving PKC-mediated phosphorylation of Raf-1 has been shown using NIH3T3cells (Kolch et al., 1993), COS-1 cells (Ueda et al., 1996), and COS-7 cells (Schonwasser et al., 1998) as models.
We show that, in the context of oncogenic GNAQ signaling found in UM, the activation of the MAP-kinase pathway occurs through a different mechanism. We find that Ras is required for GNAQ-mediated MAPK activation, and identify a signaling module that involves PKC δ and ε and RasGRP3, which is required and sufficient to activate Ras/MAPK signaling (Figure 7).
Figure 7. Model of Oncogenic GNAQ Activates MAPK Signaling in UM.
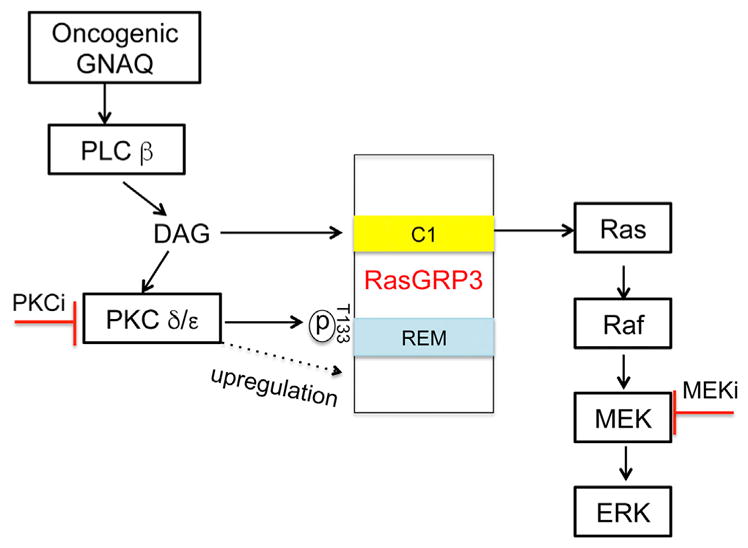
GTP-bound GNAQ directly activates PLC β, which generates the second messenger DAG. DAG activates PKC δ and ε by binding their C1 domains. The Ras activator RasGRP3 represents a critical signaling node linking oncogenic GNAQ to the RAS/RAF/MEK/ERK signaling pathway by three independent mechanisms: binding of membrane recruitment by DAG, phosphorylation and upregulation of expression by PKC δ and ε.
Among the more than ten different PKC isoforms we found that, irrespective of GNAQ or GNA11 mutation status, PKC α, δ, ε, or ζ were ubiquitously expressed in melanoma cells, while PKC ι was only weakly expressed, mostly consistent with published reports. Among the consistently expressed isoforms we identified PKC δ and ε as the main functionally important isoforms that mediate MAPK signaling in UM cells. Only knockdown of PKC δ or ε, but not other PKC isoforms significantly reduced MAPK signaling in GNAQ mutant melanoma cells. Only combined depletion of PKC δ and ε, but not combined knockdown of either PKC ζ with δ or ε, or PKC α with δ or ε extinguished MAP-kinase pathway signaling and inhibited proliferation of GNAQ mutant melanoma cell lines but had no such effect on melanoma cell lines with other mutations. Concordantly, co-transfection of GNAQQ209L with PKC δ or ε, but not PKC α, ζ, and ι potentiated MAPK signaling in 293FT cells.
We identified PKC δ and ε as the main effector isoforms downstream of oncogenic GNAQ/11 that activate MAPK signaling. The lack of MAPK activation by PKC α is of interest as it, like PKC δ and ε, also contains a DAG-binding domain. However, we found PKC α localized mainly in the cytoplasm in UM cell lines, while a considerable fraction of PKC δ and ε were localized at the membrane, consistent with their activation in this setting.
RasGRP3 belongs to family with four related RASGTP exchange factors (GEFs) RasGRP1-4, which all contain an REM (Ras exchange motif)-Cdc25 domain that mediates Ras activation and a C1 domain that mediates membrane localization by binding DAG (Ebinu et al., 1998; Ksionda et al., 2013; Stone, 2011). RasGRP proteins display tissue-specific expression patterns: RasGRP1 is primarily expressed in the cerebral cortex, cerebellum, T cells, and, to a lesser extent, in B cells; RasGRP2 is expressed in striatum and platelets and their precursors; RasGRP3 is highly expressed in B cells, macrophages, and endothelial cells; whereas RasGRP4 is restricted to mast cells, thymocytes, and neutrophils. Recent studies have shown that RasGRP3 plays an important functional role in the formation and progression of a variety of human cancers (Nagy et al., 2014; Yang et al., 2010, 2011).
We show that the significantly increased expression level of RasGRP3 in UM is a direct consequence of Gαq signaling, and that Gαq signaling not only increases RasGRP3 transcription in a PKC-dependent manner but activates RasGRP3 by two independent mechanisms: membrane recruitment and phosphorylation by PKC δ and ε.
RasGRP3 is reported to be expressed at elevated levels in some human cancers such as Burkitt’s lymphoma, metastatic prostate cancer, human breast cancer, and BRAF-mutant melanoma (Nagy et al., 2014; Teixeira et al., 2003; Yang et al., 2010, 2011). Here we show that the expression level of RasGRP3 in GNAQ or GNA11 mutant melanoma cell lines and human UM tissues by far exceeds the expression levels of the previously reported tissue types, indicating a specific role of this protein in UM. Our results indicate that GNAQ signaling and PKC δ and ε are involved in upregulating RasGRP3 expression, and that this effect is restricted to melanocytes, as mutant GNAQ did not increase RasGRP3 expression levels in 293FT (data not shown). Other RasGRP family members were not expressed at significant levels in UM cell lines. The mechanistic details behind this apparently RasGRP3- and cell type-specific regulation remain to be elucidated.
Phosphorylation of RasGRP3 at threonine 133 by PKC enhances the ability of RasGRP3 to maximally activate Ras signaling in antigen receptor-stimulated B cells, and in that context is suggested to be mediated by PKC θ (Aiba et al., 2004) and PKC β (Zheng et al., 2005). We confirm the critical role of RasGRP3 phosphorylation at T133 in the setting of GNAQ mutations with PKC δ and ε as the relevant PKC isoforms for this modification in UM. We show that mutating the PKC phosphorylation site of RasGRP3 (T133A) partially, but not completely, attenuated RasGRP3-mediated MAPK signaling. The residual RasGRP3 activity depended on its DAG-binding domain. These findings indicate the existence of two independent mechanisms that activate RasGRP3 in UM, one depends on PKC phosphorylation at T133 and the other requires DAG. DAG here most likely serves to recruit RasGRP3 to the plasma membrane, as the DAG-binding domain could be substituted by either Fyn or Src membrane target motifs. This PKC-independent activity may explain why PKC inhibition fails to induce sustained suppression of MAPK signaling in GNAQ mutant melanomas (Chen et al., 2014). In line with our findings, both the catalytic domain and the C1 domain of RasGRP3 are required in BCR-mediated Ras activation (Oh-hora et al., 2003).
Our finding that GNAQ mediates MAPK signaling via PKC δ and ε in UM may have important clinical implications and provide an opportunity for refined therapeutic targeting. To date, clinical studies on UM utilize pan-PKC inhibitors including AEB071. The combination of PKC and MEK inhibition with AEB071 and MEK162 was evaluated in a phase Ib/II clinical trial (NCT01801358), but the trial failed to progress to phase II due to toxicity. Thus, compounds specifically targeting PKC δ and ε hold the promise of an improved therapeutic index. The central role of RasGRP3 in mediating signals to the MAP-kinase pathway implicates RasGRP3 as a novel therapeutic target.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the lead contact, Xu Chen (xu.chen@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco. Nude mice were purchased from The Jackson Laboratory.
Cell Lines
The sources of uveal melanoma cell lines have been previously described(Griewank et al., 2012). Cutaneous melanoma cell lines SK-MEL-5, UACC257, MM415, MM485, SK-MEL-28 were purchased from UCSF cell culture facility. All melanoma cell lines were maintained in RPMI 1640 with 10%FBS. Human Burkitt’s lymphoma Ramos cells were obtained from ATCC (Manassas, VA) and were maintained in RPMI 1640 with 10%FBS. 293FT cells were obtained from Invitrogen (Grand Island, NY) and were cultured in DMEM with 10%FBS. Immortalized human melanocytes (a gift from Dr. David Fisher, Dana Farber Cancer Institute, Boston, MA) and normal human melanocytes from Invitrogen were cultured in 254 media supplemented by human melanocyte growth supplements (HMGS-2; Invitrogen). Melan-a cells (a gift from Dr Dorothy Bennett, St George University, London, UK) were maintained in RPMI with 10% FBS and 200 nM TPA.
Human Tumor Tissues
All pseudonymised human uveal melanoma tissue samples were provided by the University of Liverpool Ocular Oncology Biobank under the National Research Ethics Service approved study number NRES REC Ref 15/SC/0611. Informed consent was provided by all patients for the use of their biosamples for research and the study was conducted in accordance with the Declaration of Helsinki.
METHOD DETAILS
Plasmid and Reagents
The GNAQ and GNA11 constructs have been previously described (Chen et al., 2014). HA-tagged PKC isoforms full-length cDNA constructs (α, βII, δ, ε, γ, η, ζ, ι ), HA-tagged catalytic domains cDNAs (α, βII, δ, ε) and HA-tagged kinase dead cDNAs (δ, ε) are gifts from Bernard Weinstein(Soh and Weinstein, 2003) and purchased from Addgene (Cambridge, MA). Myc tagged human RasGRP1, RasGRP2, RasGRP3 and RasGRP4 cDNAs were cloned into pEF6 vector. RasGRP3-T133A construct was generated by site-directed mutagenesis using Quikchange II kit (Agilent Technologies, Santa Clara, CA) following the manufacturer’s instruction. C1 domain is deleted to create two C1 domain deletion mutant constructs (RasGRP3-ΔC1 and RasGRP3-T133A-ΔC1) by using overlap extension PCR. PCR products were inserted into pEF6 vector following EcoRI and XbaI digestion. RasGRP3-ΔC1 and RasGRP3-T133A-ΔC1 were fused at their N-terminus to 20 N-terminal Src or 16 N-terminal Fyn amino acids(Chen and Resh, 2001) by using PCR method, resulting in generation of Src-RasGRP3-ΔC1, Src-RasGRP3-T133A-ΔC1, Fyn-RasGRP3-ΔC1, and Fyn-RasGRP3-T133A-ΔC1. The primers used for Src fusion constructs are as follows: 5′-GGTGGAATTCATGGGGAGTAGCAAGAGCAAGCCTAAGGACCCCAGCCAGCGCCGGCGCAGCCTGGAGCCAGGATCAAGTGGCCTTGGG-3′ and 5′-ATCCTCTAGAGCCATCCTCACCATC-3′. The primers used for Fyn fusion constructs are: 5′-GGTGGAATTCATGGGGAGTAGCAAGAGCAAGCCTAAGGACCCCAGCCAGCGCCGGCGCAGCCTGGAGCCAGGATCAAGTGGCCTTGGG-3′ and 5′-ATCCTCTAGAGCCATCCTCACCATC-3′. All constructs were verified by DNA sequencing. AEB071, AHT956, MEK162 and INC280 were synthesized at Novartis Pharma AG (East Hanover, NJ, USA). Human HGF is from Peprotech( Rocky Hill, NJ). PD0325901 and Trametinib were obtained from Chemie TEK(Indianapolis, IN). TPA and Verteporfin were purchased from Sigma (St Louis, MO). All siRNAs were purchased from Dharmacon (Lafayette, CO)
Transient Transfection, Lentiviral Transduction and siRNA Mediated Gene Knockdown
For transient transfection, 293FT cells were transfected using lipofectamine 2000(Invitrogen) following the manufacturer’s instruction. Cells were processed 24 hr48 hr after transfection. Lentiviral generation and transduction have been described previously (Chen et al., 2014). All siRNA pools were obtained from Dharmacon (Rockford, IL). The list of siRNA for different genes is shown in the Supplemental Procedures. 30nM siRNAs for each gene were transfected using lipofectamine RNAimax reagent according to manufacturer’s instructions (Invitrogen). For combination knockdown, non-target siRNAs were used to keep each sample same amount of total siRNAs. After 48 to 72 hr transfection, cells were either lysed or used for cell proliferation analysis.
Western Blot Analysis and Cell Proliferation
Cell lysis and immunoblotting were performed as described(Chen et al., 2014). For antibody details, see Key Resources Table. Human RasGRP1 (JR-E160) was generated by the Roose lab together with Epitomics. P-RasGRP1(T184) was generated by the Roose lab through immunization with the peptide SRKL-pT-QRIKSNTC together with Eurogentech/AnaSpec (Fremont, CA). Western blot bands were subjected to densitometric analysis using image Studio software from Li-COR Biosciences (Lincoln, Nebraska). For short term proliferation assays (over one week), cells were transfected with indicated siRNAs. After 48 hr transfection, cells were counted using TC10 automated cell counter (Bio-Rad, Hercules, CA) and plated in triplicate into 6-well plates at 5x104 cells/well and cultured for the indicated times. Cells were collected by trypsinization and counted in a TC10 automated cell counter (Bio-Rad, Hercules, CA). For long term proliferation assays, cells were plated at 10 cm plates and then treated with or without inhibitors. After 12 to 18 days, cells were stained with Crystal Violet.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Rabbit PKC α | Cell Signaling Technology | Cat# 2056; RRID: AB_2284227 |
| Anti-Rabbit PKC δ | Cell Signaling Technology | Cat# 2058; RRID: AB_10694655 |
| Anti-Rabbit PKC ζ | Cell Signaling Technology | Cat# 9368; RRID: AB_10693777 |
| Anti-Rabbit PKC ι | Cell Signaling Technology | Cat# 2998; RRID: AB_2171737 |
| Anti-Mouse PKC θ | BD Bioscience | Cat# 610090; RRID: AB_397497 |
| Anti-Mouse PKC ε | BD Bioscience | Cat# 610085; RRID: AB_397492 |
| Anti-Mouse PKC γ | Invitrogen | Cat# 13-3800; RRID: AB_2533015 |
| Anti-Mouse PKC η | Abnova | Cat# H00005583-B02; RRID: AB_1018609 |
| Anti-Rabbit PKC βI | Santa Cruz | Cat# sc-209; RRID: AB_2168968 |
| Anti-Rabbit PKC βII | Santa Cruz | Cat# sc-210; RRID: AB_2252825 |
| Anti-Rabbit PKC δ | Santa Cruz | Cat# sc-937; RRID: AB_632229 |
| Anti- Rabbit PKC βII | Epitomics | Cat# 1514-1; RRID: AB_562197 |
| Anti-Rabbit pMEK | Cell Signaling Technology | Cat# 9121; RRID: AB_331648 |
| Anti-Rabbit pERK | Cell Signaling Technology | Cat# 4370; RRID: AB_2315112 |
| Anti-Rabbit SOS1 | Cell Signaling Technology | Cat# 12409 |
| Anti-Rabbit PARP | Cell Signaling Technology | Cat# 9542; RRID: AB_2160739 |
| Anti-Rabbit cleaved PARP | Cell Signaling Technology | Cat# 9541; RRID: AB_331426 |
| Anti-Rabbit YAP1 | Cell Signaling Technology | Cat# 14074 |
| Anti-Mouse MET | Cell Signaling Technology | Cat# 3127; RRID: AB_331361 |
| Anti-Rabbit phospho-MET | Cell Signaling Technology | Cat# 3077; RRID: AB_2143884 |
| Anti-Mouse Myc tag | Cell Signaling Technology | Cat# 2276; RRID: AB_331783 |
| Anti-Rabbit RasGRP3 | Cell Signaling Technology | Cat# 3334; RRID: AB_2269292 |
| Ant-Rabbit GNAQ (E-17) | Santa Cruz | Cat# sc-393; RRID: AB_631536 |
| Anti-Rabbit ERK2 (C-14) | Santa Cruz | Cat# sc-154; RRID: AB_2141292 |
| Anti-Mouse Nras (F155) | Santa Cruz | Cat# sc-31; RRID: AB_628041 |
| Anti-Mouse Kras | Santa Cruz | Cat# sc-30; RRID: AB_627865 |
| Anti-Rabbit Hras | Santa Cruz | Cat# sc-520; RRID: AB_631670 |
| Anti-Rabbit Braf (C-19) | Santa Cruz | Cat# sc-166; RRID: AB_630938 |
| Anti-Mouse β-actin | Sigma | Cat# A1978; RRID: AB_476692 |
| Anti-Mouse Glu-Glu tag | Covance Inc | Cat# MMS-115P-200; RRID: AB_291282 |
| Anti-Mouse HA tag | Covance Inc | Cat# MMS-101P; RRID: AB_2314672 |
| Anti-Rabbit p-p90RSK | Epitomics | Cat# 2006-1; RRID: AB_562342 |
| Anti-Rabbit RasGRP2 | Epitomics | Cat# S2451; RRID: AB_10640790 |
| Anti-Rabbit RasGRP1(JR-E160) | this paper | N/A |
| Anti-Rabbit RasGRP4 | Abcam | Cat# ab96293; RRID: AB_10697912 |
| Anti-Rabbit p-RasGRP3 (T133) | Abcam | Cat# ab124823; RRID: AB_11128345 |
| Anti-Mouse p-RasGRP1 (T184) | this paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human HGF | Peprotech | Cat# 100-39 |
| PD0325901 | Chemietek | Cat# CT-PD03 |
| Trametinib | Chemietek | Cat# CT-GSK212 |
| TPA | Sigma | Cat# P8139 |
| Verteporfin | Sigma | Cat# SML0534 |
| AEB071 | Novartis | N/A |
| AHT956 | Novartis | N/A |
| MEK162 | Novartis | N/A |
| INC280 | Novartis | N/A |
| Critical Commercial Assays | ||
| Ras activation assay kit | Millipore | Cat# 17-218 |
| RNeasy mini kit | Qiagen | Cat# 74104 |
| Human Transcriptome 2.0 microarray kit | Affmetrix | Cat# 902309 |
| PureLink RNA mini kit | Thermo Fisher Scientific | Cat# 12183018A |
| SuperScript III first-Strand Synthesis Super Mix | Thermo Fisher Scientific | Cat# 18080400 |
| Subcellular Protein Fractionation kit | Thermo Fisher Scientific | Cat#: 78840 |
| Deposited Data | ||
| Raw data files for microarray | NCBI Gene Expression Omnibus | GEO: GSE93666 |
| Experimental Models: Cell Lines | ||
| 293FT | Invitrogen | Cat#: R70007 |
| Ramos | ATCC | Cat#: CRL-1596 |
| Immortalized human melanocyte | Dr. David Fisher | N/A |
| Normal human melanocyte | Invitrogen | Cat#: C-002-5C |
| Normal human melanocyte | Invitrogen | Cat#: C-102-5C |
| Melan-a | Dr. Dorothy Bennett | N/A |
| SK-MEL-5 | UCSF cell culture facility | N/A |
| UACC257 | UCSF cell culture facility | N/A |
| MM415 | UCSF cell culture facility | N/A |
| MM485 | UCSF cell culture facility | N/A |
| SK-MEL-28 | UCSF cell culture facility | N/A |
| Uveal melanoma cell lines | Griewank et al., 2012 | N/A |
| Experimental Models: Organisms/Strains | ||
| Nude mice | Jackson Laboratory | Stock number: 007850 |
| Human uveal melanoma tissues | the University of Liverpool Ocular Oncology Biobank | N/A |
| Oligonucleotides | ||
| GNAQ siRNA | Dharmacon | Cat#: L-008562-00-0005 |
| PKC δ siPNA | Dharmacon | Cat#: L-003524-00-0005 |
| PKC ε siRNA | Dharmacon | Cat#: L-004653-00-0005 |
| PKC ζ siRNA | Dharmacon | Cat#: L-003526-00-0005 |
| RasGRP1 siRNA | Dharmacon | Cat#: L-008928-00-0005 |
| RasGRP2 siRNA | Dharmacon | Cat#: L-009365-00-0005 |
| RasGRP3 siRNA | Dharmacon | Cat#: L-008517-00-0005 |
| RasGRP3 siRNA | Dharmacon | Cat#: J-008517-07-0002 |
| RasGRP3 siRNA | Dharmacon | Cat#: J-008517-08-0002 |
| SOS1 siRNA | Dharmacon | Cat#: L-005194-00-0005 |
| HRAS siRNA | Dharmacon | Cat#: L-004142-00-0005 |
| KRAS siRNA | Dharmacon | Cat#: L-005069-00-0005 |
| NRAS siRNA | Dharmacon | Cat#: L-003919-00-0005 |
| Non-targeting control siRNA | Dharmacon | Cat#: D-001810-10-05 |
| MET siRNA | Dharmacon | Cat#: L-003156-00-0005 |
| YAP1 siRNA | Dharmacon | Cat#: L-012200-00-0005 |
| PKC α siRNA | Dharmacon | Cat#: L-003523-00-0005 |
| RasGRP3 primer forward: CTCTGCATGTATCGAAATGCCA | this paper | N/A |
| RasGRP3 primer reverse: CTACTTCCCGAAATTCCTCAGTC | this paper | N/A |
| SOS1 primer forward: GTAGGATGAACTTGCCCCTG | this paper | N/A |
| SOS1 primer reverse: GCTGCCCTACGAGTTTTTCA | this paper | N/A |
| Recombinant DNA | ||
| cDNA: GNAQQ209L | Chen et al., 2014 | N/A |
| cDNA: GNA11Q209L | Chen et al., 2014 | N/A |
| cDNA: HA-PKC α | addgene | 21232 |
| cDNA: HA-PKC βII (kinase dead) | addgene | 16385 |
| cDNA: HA-PKC δ | addgene | 16386 |
| cDNA: HA-PKC ε | addgene | 21240 |
| cDNA: HA-PKC γ | addgene | 21236 |
| cDNA: HA-PKC η | addgene | 21244 |
| cDNA: HA-PKC ζ | addgene | 21248 |
| cDNA: HA-PKC ι | addgene | 21252 |
| cDNA: HA-PKC α (catalytic domain) | addgene | 21234 |
| cDNA: HA-PKC βII (catalytic domain) | addgene | 16384 |
| cDNA: HA-PKC δ (catalytic domain) | addgene | 16388 |
| cDNA: HA-PKC ε (catalytic domain) | addgene | 21242 |
| cDNA: HA-PKC δ(kinase dead) | addgene | 16389 |
| cDNA: HA-PKC ε (kinase dead) | addgene | 21243 |
| cDNA: RasGRP1-Myc | this paper | N/A |
| cDNA: RasGRP2-Myc | this paper | N/A |
| cDNA: RasGRP3-Myc | this paper | N/A |
| cDNA: RasGRP4-Myc | this paper | N/A |
| cDNA: RasGRP3(T133A)-Myc | this paper | N/A |
| cDNA: RasGRP3-ΔC1-Myc | this paper | N/A |
| cDNA: RasGRP3-T133A-ΔC1-Myc | this paper | N/A |
| cDNA: Src-RasGRP3-ΔC1-Myc | this paper | N/A |
| cDNA: Src-RasGRP3-T133A-ΔC1-Myc | this paper | N/A |
| cDNA: Fyn-RasGRP3-ΔC1-Myc | this paper | N/A |
| cDNA: Fyn-RasGRP3-T133A-ΔC1-Myc | this paper | N/A |
| Other | ||
| Human primary uveal melanoma RNAseq and copy number data | TCGA | http://www.cbioportal.org |
Assay for Activated Ras
The Ras activation assay was performed using an agarose-bound GST-fused RBD of Raf-1 as described by the manufacturer (Millipore). Briefly, cells were transfected with indicated plasmids for 24 hr or indicated siRNAs for 72 hr and then were washed with cold PBS and lysed. The activated Ras was pulled down with agarose-conjugated Raf-1 RBD, followed by SDS/PAGE gel electrophoresis and immunoblotting with a pan-Ras antibody or a Ras isoform-specific antibody.
Taqman Real Time RT-PCR
RNA was isolated from cells by using RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacture’s instruction and reverse-transcribed with random primers (Invitrogen) and Moloney murine leukemia virus reverse transcriptase. Real-time PCR was performed in duplicate or triplicate using Eppendorf RealPlex2. Gene expression was evaluated using SensiMix II probe (Bioline) and specific probes described below. Each sample has been normalized to GAPDH and quantified with the comparative CT method according to the manufacturer’s instructions. The following combinations of primers and probes were used to analyze the expression of human RasGRP1: forward, AAGCTCCACCAACTACAGAACT; reverse, AGGGAGATGAGGTCCTTGAGAT; probe, FAM-CCACATGAAATCAATAAGGTTCTCGGTGAG-TAMRA and human GAPDH forward, GAAGGTGAAGGTCGGAGT; reverse, GAAGATGGTGA TGGGATTTC; probe, FAM-AGGCTGAGAACGGGAAGCTTGT-TAMRA. SOS1 (Hs00362308_m1), RasGRP2 (Hs00183378_m1), 3 (Hs00209808_m1) and 4 (Hs01073182_m1) probes and primers were obtained at Applied Bio System.
Microarray Experiments
Purified RNA was analyzed for quality using chip-based capillary electrophoresis (Bioanalyzer, Agilent, Inc) and quantity and purity was determined with a NanoDrop spectrometer. The GeneChip WT PLUS Reagent kit was used for amplification, fragmentation and biotin-labeling (Affymetrix, Santa Clara, CA). The labeled cDNA was hybridized to Human Transcriptome 2.0 microarrays (Affymetrix, Santa Clara, CA). The signal intensity fluorescent images produced during Affymetrix GeneChip hybridizations were read using the Affymetrix Model 3000 Scanner and converted into GeneChip probe results files (CEL) using Command and Expression Console software (Affymetrix). The array data were normalized at gene level and outliers were identified and removed if have by using Affymetrix expression console software. Gene level differential expression was compared between 5 GNAQ/11 mutant melanoma cells and 5 BRAF/Nras mutant melanoma cells by using Affymetrix transcriptome Analysis Console (TAC) software. Algorithm option is one-way between-subject Anova (Unpaired). Filter criteria: (1) fold change (linear)<−2 or fold change (linear)>2; (2) Anova p-value <0.05. Human Transcriptome Array 2.0 covered more than 285,000 full-length transcripts including >245,000 coding transcripts. Total 67528 genes are covered on this array including 44699 coding genes. Data are deposited in GEO (accession number: GSE93666).
Human Uveal Melanoma Cell Lines and Human Uveal Melanoma Tissues Immunohistochemistry
The 92.1, Omm1 and Mel270 UM cell lines were grown in RPMI (Life Technologies, Paisley, UK) plus 10% FCS. The CRMM1 and CRMM2 conjunctival melanoma cell lines were grown in Ham’s F-12K (Kaighn’s) Medium (Life Technologies) plus 10% FCS. At ~75% confluence the cells were removed from the bottom of a 75cm2 tissue culture flask using a cell scraper. The cells were then pelleted at 1500rpm for 2 minutes and fixed in 10% neutral buffered formalin for 10 minutes. Following fixation the cells were washed in PBS before transferring the cell pellet to a microfuge tube for a final wash with PBS and centrifugation at 8000rpm for 1 minute. The supernatant was removed and 100μl of 4% molten agar was added and mixed quickly into the cell pellet prior to centrifugation at 14,000rpm for 30 seconds. The agar cell pellet was then removed from the microfuge tube and placed into a tissue cassette for processing through a series of alcohols and xylenes. After dehydration the pellet was paraffin wax embedded such that the agar tip containing cells was at the surface of the block.
Sections (4 μm thick) cut from the cell pellet blocks, human uveal melanoma specimens from the University of Liverpool Ocular Oncology Biobank underwent immunohistochemistry as previously described (Lake et al., 2013). In brief, antigen retrieval and IHC were performed using the Dako PT Link and Autostainer Plus systems and Envision™ FLEX Kit according to standard manufacturers’ procedures (Dako UK Ltd, Cambridgeshire, UK). Positive staining was visualized either by use of 3-amino-9-ethylcarbazole (AEC, Vector Laboratories Ltd., Peterborough, UK, 30 min). The slides were counterstained with Mayer’s hematoxylin (VWR, Leicestershire, UK) and cover-slipped using an aqueous mountant (Aquatex™, VWR). The following RasGRP3 primary antibody was used: rabbit polyclonal, 13162-1-AP (ProteinTech, Manchester, UK) at 1:200. Omission of the primary antibody served as the negative control for the rabbit polyclonal. Human duodenal tissue served as a positive control. The cohort of patients included in this paper were as follows: GNAQ mutant: male 78 years, male 78 years, male 77 years, male 51 years; GNA11 mutant: female 73 years, female 73 years, female 53 years, female 43 years.
Mouse Xenograft
The constructs containing shRNA sequence targeting human RasGRP3 or non-target shRNAs were obtained from Genecopoeia (Rockville, MD). The RasGRP3 shRNA expressing stable 92-1 cell lines were generated following the manufacturer’s instructions. Nude mice (Jackson laboratory) were injected subcutaneously in the flanks with 2x106 cells suspended in matrigel. Mice were monitored for 13 weeks after injection and then were sacrificed. Tumors were collected and weighted.
Quantitative RT-PCR
Total RNA was extracted using PureLink RNA Mini kit (Thermo Fisher Scientific, Waltham, MA). For quantitative RT-PCR, mRNA was retrotranscribed using SuperScript III First-Strand Synthesis Super Mix (Thermo Fisher Scientific ). Real-time PCR was performed using standards and Power SYBR Green PCR Master Mix (Thermo Fisher Scientic) with Bio-rad C1000 Touch Thermal Cycler Samples were normalized to a ribosomal protein SB34 with standard comparative CT methods. Primer sequences are shown below: RasGRP3 forward-CTCTGCATGTATCGAAATGCCA; RasGRP3 reverse-CTACTTCCCGAAATTCCTCAGTC; SOS1 forward-GTAGGATGAACTTGCCCCTG; SOS1 reverse-GCTGCCCTACGAGTTTTTCA.
Genome-Wide SNP Genotyping
UM cell line DNAs were genotyped with the illumina Human Omni1-Quad V1.0 beadchip (Illumina, San Diego) according to the manufacture’s instructions. The beadchips were processed and imaged on an illumine Bead Array Reader. Bead intensity data obtained for each sample were loaded into Nexus Copy Number software from BioDiscovery (EI Segundo, CA) for gene copy number analysis.
Cell Fractionation
Subcellular fractionation was performed using the Subcellular Protein Fractionation Kit of Thermo Scientific (Waltham, MA, USA) following the manufacture’s instructions. Fractions from cytosolic, membrane and nuclear parts were subjected to western blotting with the indicated antibodies.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was assessed using a standard 2-tailed t test using prism 6.0 software (GraphPad software). p<0.05 was considered statistically significant.
DATA AND SOFTWARE AVAILABILITY
Data Resources
Raw data files for the Microarray experiment have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE93666.
Supplementary Material
Highlights.
PKC δ/ε mediate ERK activation in uveal melanoma with Gαq pathway mutations
The RAS exchange factor RasGRP3 is a critical module for ERK activation
PKC δ, PKC ε, and RasGRP3 are novel therapeutic targets for uveal melanoma
Significance.
Uveal melanoma (UM) is the most lethal form of melanoma with a 10-year survival rate of approximately 50%. Once meta-static, no effective treatment options exist as current targeted therapy approaches and immune checkpoint blockade regimens are less effective than in cutaneous melanomas. The identification of PKC δ and PKC ε as critical effectors downstream of an activated Gαq signaling pathway highlights them as specific targets for therapy. The critical role of RasGRP3 as an oncogenic effector and transcriptional target downstream of GNAQ/11 may offer additional opportunities for the development of treatments.
Acknowledgments
Our research was supported by an R01 grant CA142873 from the National Cancer Institute and a Stein Innovation Award from Research to Prevent Blindness and the Gerson and Barbara Baker Distinguished Professorship (to B.C.B.); a Young Investigator Award from the Melanoma Research Alliance (to X.C.); the Sandler Program in Basic Science (start-up), NIH-NCI Physical Science Oncology Center grant U54CA143874, NIH grant 1P01AI091580-01, a Gabrielle’s Angel Foundation grants, a UCSF ACS grant, and a UCSF Research Allocation Program (RAP) pilot grant (all to J.P.R.); the Eye Tumour Research Fund of the Royal Liverpool University Hospital (to S.T.).
Footnotes
Supplemental Information includes seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2017.04.002.
AUTHOR CONTRIBUTIONS
X.C. and B.C.B. designed the study and wrote the manuscript. P.D. and J.P.R. performed TaqMan real-time RT-PCR and provided RasGRP1-4 wild-type cDNA plasmids as well as p-RasGRP1 antibody. S.T., H.K., and S.E.C. performed RasGRP3 immunohistochemistry staining. X.C., Q.W., and P.C. performed the experiments except TaqMan real-time RT-PCR and RasGRP3 immunohistochemistry. X.C. analyzed microarray data as well as DNA copy number data. All authors reviewed the manuscript.
References
- Aiba Y, Oh-hora M, Kiyonaka S, Kimura Y, Hijikata A, Mori Y, Kurosaki T. Activation of RasGRP3 by phosphorylation of Thr-133 is required for B cell receptor-mediated Ras activation. Proc Natl Acad Sci USA. 2004;101:1661216617. doi: 10.1073/pnas.0407468101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Resh MD. Activation of mitogen-activated protein kinase by membrane-targeted Raf chimeras is independent of raft localization. J Biol Chem. 2001;276:3461734623. doi: 10.1074/jbc.M103995200. [DOI] [PubMed] [Google Scholar]
- Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, Bastian BC. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:47244734. doi: 10.1038/onc.2013.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Rocca GJ, van Biesen T, Daaka Y, Luttrell DK, Luttrell LM, Lefkowitz RJ. Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. Convergence of Gi- and Gq-mediated pathways on calcium/calmodulin, Pyk2, and Src kinase. J Biol Chem. 1997;272:1912519132. doi: 10.1074/jbc.272.31.19125. [DOI] [PubMed] [Google Scholar]
- Denning MF. Specifying protein kinase C functions in melanoma. Pigment Cell Melanoma Res. 2012;25:466476. doi: 10.1111/j.1755-148X.2012.01015.x. [DOI] [PubMed] [Google Scholar]
- Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- Dissanayake SK, Wade M, Johnson CE, O’Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal DT, Indig FE, et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem. 2007;282:1725917271. doi: 10.1074/jbc.M700075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280:10821086. doi: 10.1126/science.280.5366.1082. [DOI] [PubMed] [Google Scholar]
- Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25:831845. doi: 10.1016/j.ccr.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:16941703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilhooly EM, Morse-Gaudio M, Bianchi L, Reinhart L, Rose DP, Connolly JM, Reed JA, Albino AP. Loss of expression of protein kinase C beta is a common phenomenon in human malignant melanoma: a result of transformation or differentiation? Melanoma Res. 2001;11:355369. doi: 10.1097/00008390-200108000-00006. [DOI] [PubMed] [Google Scholar]
- Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, Wardell S, Bastian BC, Woodman SE. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012;25:182187. doi: 10.1111/j.1755-148X.2012.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- Guo FF, Kumahara E, Saffen D. A CalDAG-GEFI/Rap1/B-Raf cassette couples M(1) muscarinic acetylcholine receptors to the activation of ERK1/2. J Biol Chem. 2001;276:2556825581. doi: 10.1074/jbc.M101277200. [DOI] [PubMed] [Google Scholar]
- Hubbard KB, Hepler JR. Cell signalling diversity of the Gqalpha family of heterotrimeric G proteins. Cell Signal. 2006;18:135150. doi: 10.1016/j.cellsig.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Johansson P, Aoude LG, Wadt K, Glasson WJ, Warrier SK, Hewitt AW, Kiilgaard JF, Heegaard S, Isaacs T, Franchina M, et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7:46244631. doi: 10.18632/oncotarget.6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili JS, Yu X, Wang J, Hayes BC, Davies MA, Lizee G, Esmaeli B, Woodman SE. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012;18:43454355. doi: 10.1158/1078-0432.CCR-11-3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme D, Rapp UR. Protein kinase C alpha activates RAF-1 by direct phosphorylation. Nature. 1993;364:249252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- Ksionda O, Limnander A, Roose JP. RasGRP Ras guanine nucleotide exchange factors in cancer. Front Biol. 2013;8:508532. doi: 10.1007/s11515-013-1276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake SL, Damato BE, Kalirai H, Dodson AR, Taktak AF, Lloyd BH, Coupland SE. Single nucleotide polymorphism array analysis of uveal melanomas reveals that amplification of CNKSR3 is correlated with improved patient survival. Am J Pathol. 2013;182:678687. doi: 10.1016/j.ajpath.2012.11.036. [DOI] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- Lorenzo PS, Kung JW, Bottorff DA, Garfield SH, Stone JC, Blumberg PM. Phorbol esters modulate the Ras exchange factor RasGRP3. Cancer Res. 2001;61:943949. [PubMed] [Google Scholar]
- Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, Zhang JQ, Walczak EG, Kazmi MA, Taylor BS, et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48:675680. doi: 10.1038/ng.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z, Kovacs I, Torok M, Toth D, Vereb G, Buzas K, Juhasz I, Blumberg PM, Biro T, Czifra G. Function of RasGRP3 in the formation and progression of human breast cancer. Mol Cancer. 2014;13:96. doi: 10.1186/1476-4598-13-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-hora M, Johmura S, Hashimoto A, Hikida M, Kurosaki T. Requirement for Ras guanine nucleotide releasing protein 3 in coupling phospholipase C-gamma2 to Ras in B cell receptor signaling. J Exp Med. 2003;198:18411851. doi: 10.1084/jem.20031547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Kikkawa U. Protein kinase C in melanoma. Cancer Metastasis Rev. 2005;24:287300. doi: 10.1007/s10555-005-1578-8. [DOI] [PubMed] [Google Scholar]
- Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selzer E, Okamoto I, Lucas T, Kodym R, Pehamberger H, Jansen B. Protein kinase C isoforms in normal and transformed cells of the melanocytic lineage. Melanoma Res. 2002;12:201209. doi: 10.1097/00008390-200206000-00003. [DOI] [PubMed] [Google Scholar]
- Sheldahl LC, Park M, Malbon CC, Moon RT. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr Biol. 1999;9:695698. doi: 10.1016/s0960-9822(99)80310-8. [DOI] [PubMed] [Google Scholar]
- Shin MK, Levorse JM, Ingram RS, Tilghman SM. The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature. 1999;402:496501. doi: 10.1038/990040. [DOI] [PubMed] [Google Scholar]
- Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18:7584. doi: 10.1016/j.ohc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Soh JW, Weinstein IB. Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem. 2003;278:3470934716. doi: 10.1074/jbc.M302016200. [DOI] [PubMed] [Google Scholar]
- Stone JC. Regulation and function of the RasGRP family of Ras activators in blood cells. Genes Cancer. 2011;2:320334. doi: 10.1177/1947601911408082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira C, Stang SL, Zheng Y, Beswick NS, Stone JC. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood. 2003;102:14141420. doi: 10.1182/blood-2002-11-3621. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem. 1996;271:2351223519. doi: 10.1074/jbc.271.38.23512. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:21912199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Li J, Zhu M, Fletcher JA, Hodi FS. Protein kinase C inhibitor AEB071 targets ocular melanoma harboring GNAQ mutations via effects on the PKC/Erk1/2 and PKC/NF-kappaB pathways. Mol Cancer Ther. 2012;11:19051914. doi: 10.1158/1535-7163.MCT-12-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Kedei N, Li L, Tao J, Velasquez JF, Michalowski AM, Toth BI, Marincsak R, Varga A, Biro T, et al. RasGRP3 contributes to formation and maintenance of the prostate cancer phenotype. Cancer Res. 2010;70:79057917. doi: 10.1158/0008-5472.CAN-09-4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Tao J, Li L, Kedei N, Toth ZE, Czap A, Velasquez JF, Mihova D, Michalowski AM, Yuspa SH, et al. RasGRP3, a Ras activator, contributes to signaling and the tumorigenic phenotype in human melanoma. Oncogene. 2011;30:45904600. doi: 10.1038/onc.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, Zhao L, Peyman G, Ouyang H, Jiang W, et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25:822830. doi: 10.1016/j.ccr.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Liu H, Coughlin J, Zheng J, Li L, Stone JC. Phosphorylation of RasGRP3 on threonine 133 provides a mechanistic link between PKC and Ras signaling systems in B cells. Blood. 2005;105:36483654. doi: 10.1182/blood-2004-10-3916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.