


Abstract
Satellite DNA is one of the major classes of repetitive DNA, characterized by tandemly arranged repeat copies that form contiguous arrays up to megabases in length. This type of genomic organization makes satellite DNA difficult to assemble, which hampers characterization of satellite sequences by computational analysis of genomic contigs. Here, we present tandem repeat analyzer (TAREAN), a novel computational pipeline that circumvents this problem by detecting satellite repeats directly from unassembled short reads. The pipeline first employs graph-based sequence clustering to identify groups of reads that represent repetitive elements. Putative satellite repeats are subsequently detected by the presence of circular structures in their cluster graphs. Consensus sequences of repeat monomers are then reconstructed from the most frequent k-mers obtained by decomposing read sequences from corresponding clusters. The pipeline performance was successfully validated by analyzing low-pass genome sequencing data from five plant species where satellite DNA was previously experimentally characterized. Moreover, novel satellite repeats were predicted for the genome of Vicia faba and three of these repeats were verified by detecting their sequences on metaphase chromosomes using fluorescence in situ hybridization.
INTRODUCTION
Satellite DNA (satDNA) is a class of repetitive DNA that is characterized by its genomic organization into long arrays of tandemly arranged units called monomers. The monomer sequences are typically hundreds of nucleotides long and highly homogenized (1). Although monomer length is often used to classify genomic tandem repeats as microsatellites (2–7 bp), minisatellites (tens of bp) or satellites (hundreds of bp), it appears that satellites are best distinguished by forming longer arrays (tens of kilobases up to megabases) concentrated in relatively few genomic loci, while micro- and mini-satellite arrays are much shorter and scattered across the genome. These differences in genomic organization probably reflect different amplification and homogenization mechanisms acting on these repeats (2–5). In the majority of eukaryotic genomes studied to date, satDNA was predominantly located in subtelomeric and centromeric chromosome regions, and the role of satDNA in centromere determination and function is the subject of ongoing research (6). In some organisms, such as higher plants, satellite repeats are also located in interstitial chromosome regions, forming prominent heterochromatic bands (7,8). The overall patterns of satDNA distribution revealed by fluorescence in situ hybridization (FISH) are frequently used in karyotype studies because they can provide markers for distinguishing morphologically similar chromosomes (9,10).
Investigation of satDNA or its utilization as a cytogenetic marker requires a priori knowledge of the nucleotide sequences of satellite repeats in the species of interest. However, satDNA is among the most dynamic components of eukaryotic genomes and its high evolutionary rate results in considerable sequence diversification, therefore most satellite repeat families are species- or genus-specific (1). Consequently, identification of satDNA by its similarity to known repeats from phylogenetically distant taxa is not possible. For these reasons, there has been continuous demand for efficient ab initio methods for satDNA identification. Satellite DNA acquired its name from density gradient centrifugation experiments, where it was discovered as a constituent of satellite bands formed due to its different buoyant density compared to the bulk of genomic DNA (11). Thus, density centrifugation was the first method of satDNA isolation, followed by other experimental approaches based, for example, on the presence of specific restriction sites in monomer sequences (12) or on the self-priming of tandemly repeated sequences in a modified PCR protocol (8). Although these methods led to identification of numerous satellite repeats, they are mostly limited to isolation of highly amplified repeats and biased towards those that can be distinguished by some property of their sequences, such as the presence of conserved restriction site. However, the satellites lacking these features may remain unnoticed.
An alternative to experimental methods for satDNA isolation is to identify their presence in genomic sequence data. Due to the introduction of next generation sequencing technologies, generating such data is no longer a limiting factor for genome investigation. Bioinformatics tools, such as Tandem Repeats Finder (TRF) (13), can then be used to search genomic sequences for tandem repeats including satellite DNA. As reviewed by Glunčić and Paar (14), TRF is a representative of string matching algorithms, which are utilized in a number of computational tools for tandem repeat prediction, along with alternative approaches based on nucleotide autocorrelation functions (15,16) and Fourier transforms (17). However, a common limitation of these tools is their need for long input sequences, spanning more than one repeat monomer. Although such long contigs are routinely available from whole genome assemblies, they often lack or are severely underrepresented for satellite repeats. This is because satellite repeats are extremely difficult to assemble due to their structure and high sequence homogeneity (18). Thus, the search for satellite repeats should ideally be performed in unassembled reads but this approach is hampered by relatively short length of the reads produced by most of the currently used NGS technologies.
The task of repeat identification from unassembled NGS reads has been addressed by the introduction of a similarity-based clustering algorithm which evaluates all-to-all sequence comparisons between whole genome shotgun reads (19,20). When applied to low-coverage (0.01–0.50×) genome sequencing data, there are almost no similarities detected between reads derived from single-copy sequences. On the other hand, reads that originated from repetitive elements produce multiple similarity hits and can thus be identified as clusters of frequently overlapping sequences. The number of reads in each cluster is proportional to the genomic abundance of the corresponding repeat, thus enabling its quantification. This repeat clustering analysis is at the core of the RepeatExplorer pipeline (20), which was originally designed and used for repeat characterization in plants (reviewed in (21)), but also proved to be efficient in repeat identification in other organisms, including bats (22), fish (23) and insects (24).
The clustering algorithm employed by RepeatExplorer represents the reads and their sequence similarities as nodes and connecting edges, respectively, in a virtual graph, and identifies read clusters by examination of the graph topology (19). In addition to efficient partitioning of the graph into clusters, this approach has the benefit of providing graphical representation of individual clusters. The shapes of these graphs reflect the genomic organization and sequence variability of corresponding repeats, ranging from linear structures typical for dispersed transposable elements to circular or globular shapes of tandemly repeated sequences (19). It has been demonstrated for a number of species analyzed using graph-based read clustering that the graph shapes can be reliably used to discover novel satellite repeats (25–31). However, the need to visually inspect the graph shapes represented a limitation of this approach and prevented its full automation. Another problem with this approach concerned identification of the most abundant variants of monomer sequences which are then needed for downstream applications, including a design of hybridization probes or PCR primers. Inferring monomer consensus using traditional methods based on multiple sequence alignments is not feasible due to large numbers of analyzed reads and a principally similar approach employing sequence assembly results in multiple contigs which require further manual processing. However, it was shown that alignment-free approaches utilizing k-mer frequency statistics are more suitable for monomer reconstruction from unassembled sequence reads (29,32,33) and therefore could fill this last gap in the automated workflow once implemented into an efficient computational tool.
In this work, we present tandem repeat analyzer (TAREAN), a computational pipeline which was built on the principles of graph-based repeat clustering, enhanced and supplemented with additional tools facilitating unsupervised identification and characterization of satellite repeats from unassembled sequence reads. The pipeline uses low-pass whole genome sequence reads as its input and performs their graph-based clustering as the first step in the analysis. Resulting clusters, representing all types of repeats, are then examined for the presence of circular structures characteristic for tandem repeats. This is achieved by constructing directed graphs from read similarities and selecting clusters that contain strongly connected components in their graphs. In addition, paired-end read information is utilized to discriminate clusters representing potential satellite repeats from other types of tandemly repeated sequences. Reads from these clusters are then decomposed to k-mers and fractions of the most frequent k-mers are used for reconstructing representative monomer sequences for each satellite repeat. To test the efficiency and specificity of the pipeline, we first analyzed NGS data from five plant species with various numbers of previously characterized satellite repeat families which differ in their genomic abundance. Moreover, we demonstrated that TAREAN can also identify novel satellite repeats that were subsequently verified by their detection on metaphase chromosomes using FISH with probes designed according to reconstructed monomer sequences.
MATERIALS AND METHODS
The workflow of TAREAN
Input data. The analysis requires paired-end reads generated by whole genome shotgun sequencing provided as a single FASTA formatted file. Read length should be 100–200 nt and the number of analyzed reads should represent less than 1× genome equivalent (genome coverage of 0.01–0.50× is recommended). Illumina 2 × 100 nt reads were used in this work, however, paired-end reads generated by other NGS platforms should be also suitable for analysis, provided that the sequenced fragments are of sufficient length to avoid frequent overlaps of paired-end read sequences. Reads should be of uniform length, quality-filtered (quality score ≥10 over 95% of bases, no Ns allowed) and only complete read pairs should be submitted for analysis. The analysis workflow is schematically depicted in Figure 1.
Figure 1.

Schematic representation of the TAREAN analysis workflow.
Graph-based clustering
The read clustering algorithm is the same as described by Novák et al. (19). Briefly, reads are subjected to all-to-all sequence comparisons and their mutual similarities exceeding a specified threshold (90% similarity over at least 55% of the read length) are represented as a graph in which vertices correspond to sequence reads and overlapping reads are connected by edges. The resulting graph is then subjected to the Louvain method for community detection and partitioned into clusters (34) (Figure 1A). Although this method is computationally efficient and does not generate chimeric clusters, its drawback is that some families of repetitive elements frequently get split into multiple clusters rather than being represented as a single cluster (19,30). However, by utilizing paired-end read information, these split clusters can be identified and merged. Merging is performed for clusters that share significant proportions of broken read pairs (that is, when paired-end reads are present in different clusters), as determined with the formula:
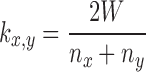 |
where W is the number of read pairs shared between clusters x and y, and nxand ny are the numbers of broken read pairs in clusters x and y, respectively. The cutoff for cluster merging used in our analysis was set to kx,y ≥ 0.2.
Automated detection of circular structures in cluster graphs
Following clustering, each cluster that represents abundant genomic repeat is examined for the presence of circular structures indicative of tandem repeats (clusters that contain at least 0.01% of input reads are analyzed by default). This is achieved by constructing a directed graph from the read similarities (Figure 1B) and testing if the graph is strongly connected, which means that it can be traversed from one read to any other read through a series of similarity overlaps (35). This is implemented by first constructing an edge signed graph Σ where vertices represent reads, edges connect overlapping reads and signs of the edges reflect orientation of the overlapping reads, being positive for forward to forward and negative for forward to reverse complement overlaps (36). The minimum spanning tree Σmsp of the graph Σ is then traversed using depth first search and each vertex which is connected to a previously visited vertex with a negative edge is switched (i.e. reverse-complemented). The resulting switching equivalent graph of Σmsp is used in the next iteration, finally leading to the directed graph G where all edges are positively signed. Next, the proportion of the largest strongly connected component in graph G is calculated as the connected component index C:
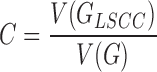 |
where V(G) is the number of vertices of the graph G and V(GLSCC) is the number of vertices in the graph GLSCC, which is a subgraph of G and corresponds to its largest strongly connected component (Figure 1C). For graphs derived from exact tandem repeats, C = 1.
Identification of putative satellite repeats
Although the parameter C facilitates the identification of clusters representing tandemly repeated genomic sequences, it does not efficiently discriminate clusters derived from satellite DNA from those representing other types of tandem repeats. Therefore, an additional cluster characteristic providing a proportion of broken read pairs is calculated. A typical feature of satellite repeats is that they occur in long contiguous arrays of monomers ranging up to megabases in length, whereas other tandem repeats form arrays in a range of hundreds to thousands of bp. Consequently, clusters of satDNA contain low proportions of broken read pairs, because most sequenced DNA fragments are entirely made of the same repeat. On the other hand, the proportions of broken pairs are much higher in tandem repeats scattered in the genome in a high number of short arrays, because many sequenced fragments span the junctions between a tandem repeat array and its neighboring genomic sequences. This is evaluated as the pair completeness index P using a formula:
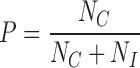 |
where NC is the number of complete read pairs in the cluster N and NI is the number of broken pairs. Both criteria, C and P, are then used simultaneously to detect putative satellite repeats, which have expected values close to 1 for both. Estimation of the threshold values of C and P suitable for sensitive yet reliable identification of putative satellite repeats was performed by re-analyzing 2968 manually annotated clusters from 11 plant species selected from the dataset published by Macas et al. (30). The estimation was done using discriminant analysis based on a Gaussian finite mixture model (37) as implemented in the R package mclust.
Reconstruction of monomer sequences from the most frequent k-mers
Reconstruction of prevailing sequence variants is performed by counting the occurrences of k-mers in a set of oriented reads obtained from the directed graph G. k-mers with lengths k = 11–27 are analyzed in parallel. The use of oriented reads ensures that the sequence reconstruction will be performed in one direction only, avoiding parallel reconstruction of its reverse complement. Identified k-mers are sorted based on their proportions in the analyzed sequence data and the resulting sorted list with the most frequent k-mers at the top is used in the subsequent analysis. The most frequent k-mers that represent 50% of the sequence data are used to construct a de Bruijn graph B and are removed from the k-mer list. k-mer frequencies are represented in the graph as weights of the corresponding vertices. The graph is then checked for the presence of cycles, and the subgraph BLSCC with the largest strongly connected component is identified. If there is no strongly connected component or if the sum of vertex weights in BLSCC is less than the threshold pkm, additional k-mers from the top of the list are iteratively added until the threshold is reached (the optimal value of pkm was tested empirically and set to 0.225). This process leads to graphs with reduced numbers of vertices yet containing cycles corresponding to prevalent monomers of tandem repeats (Figure 1D). Variants of monomer sequences are then extracted from the cycles by converting the sequences of k-mers making up de Bruijn graphs to nucleotide sequences, aligning sequences of the same length and calculating consensus and position probability matrices (PPM) from k-mer weights (Figure 1E). To limit the number of cycles used for monomer reconstruction, only the cycle with the highest weight (the sum of weights of all vertices in the cycle) is considered for each graph branch. In case of length variation in the reconstructed monomer sequences, multiple PPMs are produced and the one with the highest total weight is reported.
Identification of other types of repetitive sequences
Genes coding for 45S and 5S ribosomal RNAs are arranged as multi-copy tandem arrays in eukaryotic genomes and as such are detected as putative satellites by TAREAN. However, they are identified by similarity searches to a custom database of Viridiplantae rDNA sequences and reported separately in the program output. A specific type of repeats with potential for producing false-positive results are LTR-retrotransposons, which, due to the presence of direct terminal repeats, form circular graphs (19). To avoid this misclassification, we search for LTR retrotransposon-specific features in the reconstructed consensus sequences, including the presence of primer binding sites (PBS) complementary to some tRNAs (38) and retrotransposon protein-coding open reading frames longer than 300 bp.
Implementation
TAREAN is implemented using custom python and R scripts. Graph analysis was performed using igraph, a software collection for complex network research (39). All scripts and databases are available for download from http://w3lamc.umbr.cas.cz/lamc/resources.php. Additionally, TAREAN was implemented under Galaxy web-based environment (40) and made available as a tool in the public RepeatExplorer server (20) at http://www.repeatexplorer.org. The analyses presented in this paper were performed on Linux-based servers equipped with 16 GB RAM and 4–16 CPUs.
Pipeline testing and validation of the results
Data
The pipeline was tested using genomic shotgun Illumina reads from five species with previously characterized satellite repeats. The reads were downloaded from European Nucleotide Archive (http://www.ebi.ac.uk/ena) under accession numbers ERX379412 (Vicia faba L.), ERR063464 (Pisum sativum L.), ERP001569 (Luzula elegans Lowe), PRJEB9643 (Rhynchospora pubera (Vahl) Boeckeler) and SRX118541 (Zea mays L.).
Experimental validation of predicted satellite repeats
Reconstructed consensus sequences of satellites predicted by TAREAN in Vicia faba were used to design oligonucleotide probes for fluorescence in situ hybridization: Vf_TA11_H2, biotin-5΄-GGT TAC TTC ATC ACT AAG AAA CTA AGT TAA AAG ACT ATT AMT TAA TGA CAC-3΄; FokI_H1, fluorescein-5΄-CTA CCT TCC ATA ATG ACA AGG CTA CCA TCC ATT GGA GTA ACA AAA ATC TC-3΄. The oligo-probes were labeled with biotin or fluorescein at their 5΄ ends during synthesis. Alternatively, PCR primers were designed for amplification and cloning of satellites with longer monomers: Vf_TA39_1, 5΄-AGC ACG AAT AAA ACT AAA GTT C-3΄; Vf_TA39_2, 5΄-TAC TTT TGA AGT GAA ATG GAG-3΄; Vf_TA157_1, 5΄-GGT ATG AGA ATG GTG TAT CTT TTA TCA-3΄; Vf_TA157_2, 5΄-AGA AAA GAT ATT TGG TTT CGA ATG A-3΄. All oligonucleotides were synthesized by Integrated DNA Technologies (Leuven, Belgium). Probe amplification from total genomic DNA of V. faba and cloning was performed as described in Macas et al. (30). Probes were labeled with biotin-16-dUTP (Roche Diagnostics GmbH, Mannheim, Germany) or Alexa Fluor 568 (Thermo Fisher Scientific, Waltham, MA, USA) using nick translation (41) and FISH was performed according to Macas et al. (42). The oligo-probe FokI_H1 specific for FokI satellite (43) was used for simultaneous hybridization (two-color FISH) with the novel repeats to provide characteristic banding patterns allowing the discrimination of all chromosomes within the V. faba karyotype (44). Chromosomes were counterstained with DAPI and examined using a Nikon Eclipse 600 microscope. Images were captured using a DS-Qi1Mc cooled camera and NIS Elements 3.0 software (Laboratory Imaging, Praha, Czech Republic).
RESULTS
Major features of the pipeline and estimation of optimal parameters
The TAREAN pipeline takes paired-end NGS reads as input and outputs a list of clusters identified as putative satellite repeats, their genomic abundance and various cluster characteristics. The lengths and nucleotide sequences of reconstructed monomers are also provided and are accompanied by a detailed output from k-mer-based reconstruction including sequences and sequence logos of alternative variants of monomer sequences. A summary of this information is provided in HTML format and includes a table listing all analyzed clusters (an example of the HTML output is provided as Supplementary Data). More detailed information about clusters is provided in additional files. When the analysis is performed on a Galaxy server, all generated results are downloadable as a zip archive. Since read clustering results in thousands of clusters, the search for satellite repeats is limited to a subset of the largest clusters corresponding to the most abundant genomic repeats. The pipeline is set to analyze all clusters representing at least 0.01% of the input reads, but this size threshold can be changed in order to adjust the sensitivity of the analysis. Besides the satellite repeats, three other groups of clusters are reported in the output (i) LTR-retrotransposons, (ii) 45S and 5S rDNA and (iii) all remaining clusters passing the size threshold. As categories 1 and 2 contain sequences with circular graphs, their consensus is calculated in the same way as for the satellite repeats.
Since two cluster characteristics, the connected component index C and the pair completeness index P, are crucial for identification of satellite repeats, we searched for their optimal cutoff values by evaluating a pool of 2968 clusters from 11 species of legume plants. These clusters were manually annotated during our previous study (30) and included 174 satellites; the remaining clusters represented other kinds of genomic repeats. The C and P values of these clusters were used as training data for discriminant analysis to find the best model for satellite prediction (Figure 2A). Clusters identified as satellites according to this model were denoted as high-confidence satellites. Additionally, we also chose less strict criteria of P > 0.4 and C > 0.7 to be able to detect less typical satellite sequences which are then reported as low-confidence satellites. Examples of clusters with different P and C values with corresponding graph shapes are shown on Figure 2B E. In the model-based prediction using discriminant analysis, 143 (82%) of the reference satellites clusters were correctly classified as high-confidence satellites with a false positive rate of 1.4% (Table 1). Employing the low-confidence category criteria resulted in detection of 173 out of 174 control satellite clusters but the higher sensitivity led to an increased false positive rate (18%).
Figure 2.

Training dataset and examples of cluster graphs. (A) Scatter plot of C (connected component index) and P (pair completeness index) values for all reference clusters. Red dots mark clusters that were manually annotated as satellite repeats. Threshold for classification, based on the best discriminant analysis model, is shown as a blue line and defines the high-confidence satellite group. Green lines mark empirically selected thresholds for the low-confidence category. (B–D) Examples of repeat clusters visualized as graphs where nodes represent sequence reads and edges connect reads with sequence similarities. Nodes belonging to the largest strongly connected components of the graphs are red; corresponding C and P values are shown below each graph.
Table 1. Performance of automatic classification model. A confusion matrix of numbers of clusters annotated as satellite repeats compared to the reference obtained by manual annotation.
| Manual annotation (reference) | |||
|---|---|---|---|
| Non-satellite | Satellite | ||
| Automatic (model) | Non-satellite | 2756 | 31 |
| Satellite | 38 | 143 | |
Testing the pipeline performance using previously characterized satellite repeats
To validate the pipeline sensitivity and accuracy, we analyzed NGS data from five plant species in which satellite DNA was previously experimentally characterized (Table 2). The satellites were identified in these species using restriction digestion-based cloning and/or library screening (Z. mays, V. faba; (43,45–48)) or they were identified using bioinformatics tools, but subsequently verified by cloning, sequencing and FISH analysis (Rhynchospora pubera, Pisum sativum, Luzula elegans; (25–27, 42)). These control species were selected for carrying diverse satellites with different monomer lengths, sequence variability, abundance and location in the genome. Moreover, these species represented three different types of chromosome organization, including species with monocentric (Z. mays, V. faba), meta-polycentric (P. sativum) and holocentric chromosomes (L. elegans, R. pubera).
Table 2. Evaluation of TAREAN performance in species with previously characterized satellites. Successful detection is marked by ‘++’ (high-confidence) or ‘+’ (low-confidence).
| Species | TAREAN 500k | TAREAN max | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Satellite | Merge | Merge | ||||||||
| Monomer [bp] | Abundance [% genome] | Coverage | NO | 0.2 | Coverage | NO | 0.2 | Monomer [bp] | Notes | |
| Zea mays | ||||||||||
| Zea/Tripsacum | 180 | 2.10 | 5775 | – | ++ | 180 | (47) | |||
| CentC | 156 | 0.20 | 635 | ++ | ++ | 156 | (45) | |||
| TR-1(knobs) | 350/180 | 0.11 | 156/303 | ++ | ++ | 359 | (46) | |||
| Rhynchospora pubera | (26) | |||||||||
| Tyba-1 | 171 | 1.80 | 5211 | + | + | 172 | ||||
| Tyba-2 | 171 | 1.16 | 3358 | + | + | 172 | ||||
| Luzula elegans | (27) | |||||||||
| LeSAT4 | 190/220/360 | 2.40 | 6253/3300 | + | ++ | 25263/13333 | + | ++ | 170 | |
| LeSAT11 | 56 | 1.10 | 9723 | ++ | ++ | 39286 | ++ | ++ | 56 | |
| LeSAT7 | 75 | 0.97 | 6402 | ++ | ++ | 25867 | ++ | ++ | 75 | |
| LeSAT16 | 178/195 | 0.82 | 2280/2028 | ++ | ++ | 9213/8410 | ++ | + | 177/195 | |
| LeSAT18 | Variable | 0.55 | n.a. | ++ | ++ | n.a. | ++ | ++ | 56 | |
| LeSAT23 | 57 | 0.50 | 3397/1037 | ++ | ++ | 17544 | ++ | ++ | 57 | |
| LeSAT17 | 161 | 0.48 | 1476 | – | – | 5963 | + | – | 161 | |
| LeSAT38 | 137 | 0.37 | 1337 | – | – | 5401 | – | – | – | |
| LeSAT25 | 6 | 0.36 | 29700 | – | – | 120000 | – | – | – | SSR |
| LeSAT22 | 51/167 | 0.35 | 1037 | ++ | ++ | 13725/4192 | ++ | ++ | 51 | |
| LeSAT28 | 390/730 | 0.32 | 406/217 | ++ | ++ | 1730/877 | ++ | ++ | 392 | |
| LeSAT43 | 190 | 0.23 | 599 | ++ | ++ | 2421 | ++ | ++ | 189 | |
| LeSAT9 + 21 | 43 | 0.22 | 2533 | ++ | ++ | 10233 | ++ | ++ | 43 | |
| LeSAT63 | 90 | 0.13 | 715 | ++ | ++ | 2889 | ++ | ++ | 89 | |
| LeSAT72 | 4 | 0.13 | 16088 | – | – | 65000 | – | – | – | SSR |
| LeSAT36 | 6 | 0.12 | 9900 | – | – | 40000 | + | + | 24 | SSR |
| LeSAT99 | 180 | 0.11 | 303 | + | + | 1222 | ++ | ++ | 180 | |
| LeSAT109 | 33 | 0.08 | 1245 | ++ | ++ | 5030 | ++ | ++ | 33 | |
| LeSAT89 | 41 | 0.06 | 724 | – | – | 2927 | – | – | – | |
| LeSAT27 | 42 | 0.06 | 672 | ++ | ++ | 2714 | ++ | ++ | 84 | |
| Pisum sativum | (25,42) | |||||||||
| PisTR-B | 50 | 1.37 | 13740 | ++ | ++ | 39516 | ++ | ++ | 50 | |
| TR-5 | 54 | 0.51 | 4731 | ++ | ++ | 13608 | ++ | ++ | 54 | |
| TR-2 | 440 | 0.21 | 235 | – | – | 677 | – | – | – | |
| TR-4 | 172 | 0.20 | 581 | + | + | 1672 | + | + | 173 | |
| TR-7 | 164 | 0.14 | 412 | ++ | ++ | 1184 | ++ | ++ | 164 | |
| TR-11 | 510 | 0.10 | 101 | – | – | 290 | + | + | 459 | Low coverage |
| TR-3 | 81 | 0.06 | 358 | ++ | ++ | 1030 | ++ | ++ | 82 | |
| TR-19 | 2094 | 0.03 | 8 | – | – | 23 | – | – | – | Low coverage |
| TR-1 | 867 | 0.02 | 12 | – | – | 35 | + | + | 866 | Low coverage |
| TR-18 | 1644 | 0.01 | 4 | – | – | 11 | – | – | – | Low coverage |
| TR-17 | 191 | 0.01 | 31 | ++ | ++ | 90 | + | + | 191 | Low coverage |
| TR-6 | 245 | 0.01 | 22 | – | – | 65 | – | – | – | Low coverage |
| TR-10 | 659 | 0.01 | 8 | – | – | 22 | – | – | – | Low coverage |
| Vicia faba | ||||||||||
| FokI | 59 | 3.20 | 26847 | ++ | ++ | 53695 | ++ | ++ | 59 | (43) |
| pVf7 | 168 | 0.33 | 972 | + | + | 1945 | + | + | 169 | (48) |
The columns show (from left to right) previously reported monomer sizes and genome abundance of reference satellite repeats and results of their detection by TAREAN with 500 000 reads (‘TAREAN 500k’) and with maximal number of reads that could be analyzed (‘TAREAN max’). The last ‘Monomer’ column provides lengths of consensus monomer sequences reconstructed by TAREAN. Multiple values reflect several repeat variants differing in monomer length.
TAREAN runs were performed with 500 000 input reads which should provide sufficient sensitivity towards abundant satellites, yet keeping the computation time in the range of hours. Two analysis conditions were tested: the cluster merging option was either disabled or enabled at the cutoff value of 0.2. This analysis led to the successful detection of highly amplified satellites previously reported for Z. mays, V. faba and R. pubera (Table 2). In R. pubera, both previously characterized subfamilies of the same satellite (Tyba-1 and Tyba-2) sharing ∼70% similarity (26) were detected and distinguished. In the other two species, Luzula elegans and Pisum sativum, the analysis identified all highly abundant satellites with genome proportions exceeding 0.5%, but failed to detect some less-amplified satellites with estimated genomic proportions between 0.01 and 0.50% (Table 2). Thus, additional runs were performed with 2 million reads for L. elegans and 1.44 million for P. sativum, representing the maximal numbers of reads that could be processed at the given hardware configuration (the read numbers are different as they depend on the numbers of similarity hits between the reads, reflecting different proportions of repeats in each species). Although processing more reads improved the detection of four satellite repeats, 9 of 33 control satellites in these species remained unidentified. An investigation of the properties of these repeats provided an explanation of these results and enabled understanding of the sensitivity limits of TAREAN analysis, which were mostly determined by sequencing coverage, sequence homogeneity of monomers, genomic organization and similarities of satellites to other genomic repeats.
The sequencing coverage of a satellite was calculated as the total length of reads covering its sequences divided by monomer length. Thus, the calculation of coverage provided a normalization for genomic abundance (%) values, because satellites with the same genomic proportions but differing in monomer length have different coverages. For example, the P. sativum satellites, TR-17 and TR-18, both had genome proportions of 0.01%, but the former had higher coverage due to its shorter monomer length. The group of these less-amplified satellites (labeled as ‘low-coverage’ in Table 2) revealed that the sensitivity limit of the analysis was at the coverage range of 30–100×, probably depending on the sequence homogeneity of individual satellites. Thus, the failure to detect the P. sativum satellites TR-18, TR-6, TR-10 and TR-19 could be explained by their low abundance. Moreover, in the case of TR-19, the detection was also hampered by the fact that this repeat represents a longer monomer variant of TR-11 and thus the two repeats occurred in the same cluster. Since TR-19 has a lower genomic copy number compared to TR-11 (25) its monomer was not reconstructed and reported.
It has been reported that some satellite repeats originated by amplification of short tandem arrays present in other genomic repeats such as retrotransposons (49). Such satellites may be difficult to detect by TAREAN because their cluster graphs could contain substantial portions of non-circular components representing neighboring regions of repeats from which they originated. This was the case in the L. elegans satellite, LeSAT38, and in TR-2 in P. sativum. Another group of undetected satellites comprised three L. elegans repeats with extremely short monomers corresponding to simple sequence repeats of 4 bp (LeSAT72) or 6 bp (LeSAT25 and LeSAT36). These repeats failed to produce clusters due to active masking of low complexity regions during sequence similarity searches. The partial exception was LeSAT36 where the basic motif of 6 bp also occurred as a mutated higher order repeat of 24 bp which was reported by TAREAN (Table 2).
Identification and verification of novel satellite repeats
In addition to detecting previously described repeats, there were additional putative satellites reported for some of the analyzed species. The highest number was 12 novel satellites, which were identified for V. faba in the run analyzing a maximum number of 990 000 reads (Table 3 and Supplementary Data). One of them, Vf_TA70 was partially similar to the VicTR-B satellite described in several other Vicia species (8). No significant similarities were found for the other novel satellites.
Table 3. Putative novel satellite repeats identified in Vicia faba.
| Satellite | Monomer [bp] | Genome proportion [%] | Copy number /1C | Notes |
|---|---|---|---|---|
| Vf_TA_11 | 191 | 1.20 | 843 000 | Verified by FISH (Figure 3A) |
| Vf_TA_39 | 702 | 0.29 | 55 400 | Verified by FISH (Figure 3B) |
| Vf_TA_62 | 687 | 0.15 | 29 300 | |
| Vf_TA_70 | 38 | 0.12 | 423 000 | Similar to VicTR-B |
| Vf_TA_108 | 1482 | 0.05 | 4500 | |
| Vf_TA_109 | 870 | 0.05 | 7700 | |
| Vf_TA_123 | 878 | 0.03 | 4600 | |
| Vf_TA_137 | 603 | 0.03 | 6700 | |
| Vf_TA_143 | 352 | 0.02 | 7600 | |
| Vf_TA_154 | 560 | 0.02 | 4800 | |
| Vf_TA_157 | 781 | 0.02 | 3400 | Verified by FISH (Figure 3C) |
| Vf_TA_158 | 313 | 0.02 | 8600 |
Three of the novel repeats, Vf_TA11, Vf_TA39 and Vf_TA157, differing in monomer length and genomic abundance (Table 3) were chosen for experimental validation by detecting their hybridization patterns on V. faba metaphase chromosomes using FISH. In all three repeats, band or dot-like patterns of signals typical for satellite DNA were detected. The repeat Vf_TA11 represented a highly amplified satellite with a 191 bp monomer, which was estimated to occur in 843 000 copies per haploid genome. The oligonucleotide probe (49 nt), designed according to the most conserved part of its reconstructed monomer sequence, produced a strong signal on the satellite arm of metacentric chromosome 1, and a number of minor signals close to centromeres of all acrocentric chromosomes (Figure 3A). Since monomer sequences predicted for the other two satellites were too long to be covered by oligonucleotide probes (701 and 782 bp), FISH was performed using PCR-amplified and cloned genomic fragments. In both cases the PCR with primers designed according to reconstructed monomer sequences yielded specific bands of expected lengths and their sequences confirmed predictions made by TAREAN (95.5% and 93.6% similarity to reconstructed consensus of Vf_TA39 and Vf_TA157, respectively). The resulting FISH patterns of Vf_TA39 consisted of multiple intercalary bands on most chromosomes, in agreement with relatively high abundance (55 400 copies/1C) of this repeat in the genome (Figure 3B). As expected, the much less amplified satellite Vf_TA157 (3400 copies/1C) produced weaker labeling which was limited to a single locus on chromosome 3 (Figure 3C).
Figure 3.

FISH localization of novel satellite repeats on metaphase chromosomes of Vicia faba. The probes for the novel satellites, Vf_TA11 (panel A), Vf_TA39 (panel B) and Vf_TA157 (panel C), are green, FokI repeats used for chromosome discrimination are labeled red and chromosomes counterstained with DAPI are blue.
DISCUSSION
In this work, we have introduced and validated TAREAN, a computational pipeline for the automated identification of satellite repeats from unassembled NGS reads. Although there are a number of computational tools available for detecting tandem repeats in assembled genomic sequences (50,51), corresponding tools utilizing short sequence reads are scarce. To our best knowledge, only two algorithms, DExTaR and MixTaR (52,53) have been published to address this problem. The former was designed for the detection of tandem repeats from de Bruijn graphs constructed for the purpose of genome assembly. It uses parts of de Bruijn graphs that were omitted from assembly and detects potential tandem repeats in the form of cycles. The method requires previous global assembly by a de Bruijn assembler such as ABySS (54) and is limited to the identification of exact tandem repeats. MixTaR represents an improved approach allowing detection of approximate tandem repeats, however, it requires long PacBio reads in addition to short Illumina reads for its analysis. Moreover, the algorithm was tested for detecting repeats with monomers up to 100 bp only, while most satellite DNA families have longer monomers (1).
In our previous work, we demonstrated that an alternative approach based on graph representations of repeat populations in eukaryotic genomes can be utilized for the identification of satellite repeats (19,29,31). This method, employing the RepeatExplorer pipeline (20) for performing similarity-based repeat clustering and generating graph visualizations, allows identification of approximate tandem repeats of any length, provided they are sufficiently represented in the analyzed short reads to form recognizable circular structures in their cluster graphs. Consequently, satellite repeats with various degrees of sequence conservation, and monomer lengths up to 5 kb can be identified (25,30,55). Recently, a modification of this approach, employing iterative clustering in order to improve its sensitivity towards low-copy tandem repeats, has been published by Ruiz-Ruano et al. (28). Nevertheless, both setups require human intervention for graph shape examination and the former does not provide consensus sequences of identified satellite repeats; features that were fully addressed in TAREAN.
Testing TAREAN performance using short NGS reads from five control species revealed its excellent efficiency in detecting highly abundant satellite repeats and very good performance in identifying less-amplified satellites. In addition to the repeat identification, consensus monomer sequences were accurately reconstructed in most cases. On the other hand, a fraction of previously described repeat families was not identified in test runs. When evaluating TAREAN performance, it should be acknowledged that the tool was specifically designed for the detection of genuine satellite repeats, a category of tandemly repeated sequences characterized by the forming of long contiguous arrays of highly homogenized monomer sequences. However, this category is not always clearly separated from other genomic tandem repeats. For example, some satellite repeats originate through the amplification of short tandem repeat arrays present in mobile elements. Thus, the same monomer sequences occur in the genome as short, dispersed tandem arrays, as well as in a few long arrays typical for satellite repeats (49). Such repeats then produce cluster graphs with intermediate features combining circular and linear structures, thus hampering their identification. Satellite repeats derived from a large intergenic spacer (IGS) of 45S rDNA represent a similar case, being present as short arrays within IGS and as amplified satellites elsewhere in the genome (48,56). The repeat pVf7 from Vicia faba (48) represented IGS-derived satellites in our data; although it was successfully identified by TAREAN, it was only listed in the low-confidence category (Table 2) due to its complex graph structure. Another example of a satellite with intermediate features that was reported with lower confidence was the Tyba satellite of Rhynchospora pubera (Table 2). Tyba is the satellite associated with centromeric chromatin, which is dispersed along holocentric R. pubera chromosomes. Thus, Tyba is organized in multiple arrays only up to tens of kilobases long as revealed by FISH and sequence analysis of BAC clones (26).
Regarding the ability to detect less abundant satellite repeats, there is no simple rule that could be used to determine a TAREAN sensitivity threshold. This is because the successful identification of a particular repeat depends on multiple factors, including its copy number in the genome, sequence variability, genomic organization and number of reads that were analyzed. In principle, increasing the number of analyzed reads results in more efficient detection of less amplified satellites (Table 2) but the genome sequencing coverage should not exceed 0.5-1.0x in order to avoid similarity hits between single-copy sequences during clustering analysis. However, in species with large and repeat-rich genomes, such coverage might be hard to reach due to constraints imposed by computational resources. The limiting factor for read clustering analysis is the number of similarity hits between reads, which increases with increasing proportions of high copy number repeats in the genome. Therefore, smaller numbers of reads can be clustered in highly repetitive genomes compared to those with low proportions of highly repeated sequences (19). On the other hand, even the low coverage used in this study for the large, repeat-rich genome of V. faba (990 000 reads correspond to 0.007× genome equivalent) proved to be sufficient to identify relatively rare repeats like Vf_TA157 with only thousands of copies per haploid genome (Table 3, Figure 3C).
AVAILABILITY
Command-line version of TAREAN can be downloaded from http://w3lamc.umbr.cas.cz/lamc/resources.php. The pipeline can also be run via Galaxy web interface at our public RepeatExplorer server (http://www.repeatexplorer.org).
Supplementary Material
ACKNOWLEDGEMENTS
Access to computing and data storage facilities provided by the ELIXIR CZ infrastructure is greatly appreciated.
Author contributions: P.No. implemented the algorithms. P.No. and J.M. designed the algorithms and analyzed NGS data. L.A.R., A.K. and I.V. performed FISH experiments. P.Ne. participated in the data analysis and discussion of the results. J.M. supervised the work and drafted the manuscript. All authors contributed to preparation of the final manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Czech Ministry of Education, Youths and Sports [LM2015047]; Czech Science Foundation [BP501/12/G090]; Czech Academy of Sciences [RVO:60077344]. Funding for open access charge: Czech Ministry of Education, Youths and Sports [LM2015047].
Conflict of interest statement. None declared.
REFERENCES
- 1. Macas J., Mészáros T., Nouzová M.. PlantSat: a specialized database for plant satellite repeats. Bioinformatics. 2002; 18:28–35. [DOI] [PubMed] [Google Scholar]
- 2. Garrido-Ramos M.A. Satellite DNA in plants: More than just rubbish. Cytogenet. Genome Res. 2015; 146:153–170. [DOI] [PubMed] [Google Scholar]
- 3. Plohl M., Luchetti A., Meštrović N., Mantovani B.. Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene. 2008; 409:72–82. [DOI] [PubMed] [Google Scholar]
- 4. Ellegren H. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 2004; 5:435–445. [DOI] [PubMed] [Google Scholar]
- 5. Richard G.-F., Kerrest A., Dujon B.. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol. Mol. Biol. Rev. 2008; 72:686–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Plohl M., Meštrović N., Mravinac B.. Centromere identity from the DNA point of view. Chromosoma. 2014; 123:313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fuchs J., Strehl S., Brandes A., Schweizer D., Schubert I.. Molecular-cytogenetic characterization of the Vicia faba genome – heterochromatin differentiation, replication patterns and sequence localization. Chromosom. Res. 1998; 6:219–230. [DOI] [PubMed] [Google Scholar]
- 8. Macas J., Požárková D., Navrátilová A., Nouzová M., Neumann P.. Two new families of tandem repeats isolated from genus Vicia using genomic self-priming PCR. Mol. Gen. Genet. 2000; 263:741–751. [DOI] [PubMed] [Google Scholar]
- 9. Cai Z., Liu H., He Q., Pu M., Chen J., Lai J., Li X., Jin W.. Differential genome evolution and speciation of Coix lacryma-jobi L. and Coix aquatica Roxb. hybrid guangxi revealed by repetitive sequence analysis and fine karyotyping. BMC Genomics. 2014; 15:1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Navrátilová A., Neumann P., Macas J.. Karyotype analysis of four Vicia species using in situ hybridization with repetitive sequences. Ann. Bot. 2003; 91:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kit S. Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J. Mol. Biol. 1961; 3:711–716. [DOI] [PubMed] [Google Scholar]
- 12. Hemleben V., Kovařík A., Torres-Ruiz R.A., Volkov R.A., Beridze T.. Plant highly repeated satellite DNA: molecular evolution, distribution and use for identification of hybrids. Syst. Biodivers. 2007; 5:277–289. [Google Scholar]
- 13. Benson G. Tandem Repeats Finder: a program to analyse DNA sequences. Nucleic Acids Res. 1999; 27:573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glunčić M., Paar V.. Direct mapping of symbolic DNA sequence into frequency domain in global repeat map algorithm. Nucleic Acids Res. 2013; 41:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herzel H., Weiss O., Trifonov E.N.. 10-11 bp periodicities in complete genomes reflect protein structure and DNA folding. Bioinformatics. 1999; 15:187–193. [DOI] [PubMed] [Google Scholar]
- 16. Macas J., Navrátilová A., Koblížková A.. Sequence homogenization and chromosomal localization of VicTR-B satellites differ between closely related Vicia species. Chromosoma. 2006; 115:437–447. [DOI] [PubMed] [Google Scholar]
- 17. Sharma D., Issac B., Raghava G.P.S., Ramaswamy R.. Spectral repeat finders (SRF): identification of repetitive sequences using Fourier transformation. Bioinformatics. 2004; 20:1405–1412. [DOI] [PubMed] [Google Scholar]
- 18. Treangen T.J., Salzberg S.L.. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2012; 13:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Novák P., Neumann P., Macas J.. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics. 2010; 11:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Novák P., Neumann P., Pech J., Steinhaisl J., Macas J.. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics. 2013; 29:792–793. [DOI] [PubMed] [Google Scholar]
- 21. Weiss-Schneeweiss H., Leitch A.R., McCann J., Jang T.-S., Macas J.. Hörandl E, Appelhans M. Employing next generation sequencing to explore the repeat landscape of the plant genome. Next Generation Sequencing in Plant Systematics. Regnum Vegetabile 157. 2015; 158, Königstein: Koeltz Scientific Books; 155–179. [Google Scholar]
- 22. Pagan H.J.T., Macas J., Novák P., McCulloch E.S., Stevens R.D., Ray D.A.. Survey sequencing reveals elevated DNA transposon activity, novel elements, and variation in repetitive landscapes among vesper bats. Genome Biol. Evol. 2012; 4:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. García G., Ríos N., Gutiérrez V.. Next-generation sequencing detects repetitive elements expansion in giant genomes of annual killifish genus Austrolebias (Cyprinodontiformes, Rivulidae). Genetica. 2015; 143:353–360. [DOI] [PubMed] [Google Scholar]
- 24. Camacho J.P.M., Ruiz-Ruano F.J., Martín-Blázquez R., López-León M.D., Cabrero J., Lorite P., Cabral-de-Mello D.C., Bakkali M.. A step to the gigantic genome of the desert locust: chromosome sizes and repeated DNAs. Chromosoma. 2014; 124:263–275. [DOI] [PubMed] [Google Scholar]
- 25. Neumann P., Navrátilová A., Schroeder-Reiter E., Koblížková A., Steinbauerová V., Chocholová E., Novák P., Wanner G., Macas J.. Stretching the rules: monocentric chromosomes with multiple centromere domains. PLoS Genet. 2012; 8:e1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marques A., Ribeiro T., Neumann P., Macas J., Novák P., Schubert V., Pellino M., Fuchs J., Ma W., Kuhlmann M. et al. . Holocentromeres in Rhynchospora are associated with genome-wide centromere-specific repeat arrays interspersed among euchromatin. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:13633–13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heckmann S., Macas J., Kumke K., Fuchs J., Schubert V., Ma L., Novák P., Neumann P., Taudien S., Platzer M. et al. . The holocentric species Luzula elegans shows interplay between centromere and large-scale genome organization. Plant J. 2013; 73:555–565. [DOI] [PubMed] [Google Scholar]
- 28. Ruiz-Ruano F.J., López-León M.D., Cabrero J., Camacho J.P.M.. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci. Rep. 2016; 6:28333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Macas J., Kejnovský E., Neumann P., Novák P., Koblížková A., Vyskot B.. Next generation sequencing-based analysis of repetitive DNA in the model dioecious plant Silene latifolia. PLoS One. 2011; 6:e27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Macas J., Novák P., Pellicer J., Čížková J., Koblížková A., Neumann P., Fuková I., Doležel J., Kelly L.J., Leitch I.J.. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS One. 2015; 10:e0143424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Renny-Byfield S., Kovařík A., Chester M., Nichols R.A., Macas J., Novák P., Leitch A.R.. Independent, rapid and targeted loss of highly repetitive DNA in natural and synthetic allopolyploids of Nicotiana tabacum. PLoS One. 2012; 7:e36963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Macas J., Neumann P., Novák P., Jiang J.. Global sequence characterization of rice centromeric satellite based on oligomer frequency analysis in large-scale sequencing data. Bioinformatics. 2010; 26:2101–2108. [DOI] [PubMed] [Google Scholar]
- 33. Torres G.A., Gong Z., Iovene M., Hirsch C.D., Buell C.R., Bryan G.J., Novák P., Macas J., Jiang J.. Organization and evolution of subtelomeric satellite repeats in the potato genome. G3. 2011; 1:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blondel V.D., Guillaume J.-L., Lambiotte R., Lefebvre E.. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008; 10008:6. [Google Scholar]
- 35. Wilson R.J. Wilson R.J. Introduction to Graph Theory. 1996; 4th edn, Addison Wesley Longman Limited. [Google Scholar]
- 36. Zaslavsky T. Signed graphs. Discret. Appl. Math. 1982; 4:47–74. [Google Scholar]
- 37. Fraley C., Raftery A.E.. Model-based clustering, discriminant analysis, and densiy estimation. J. Am. Stat. Assoc. 2002; 97:611–631. [Google Scholar]
- 38. Havecker E.R., Gao X., Voytas D.F.. The diversity of LTR retrotransposons. Genome Biol. 2004; 5:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Csardi G., Nepusz T.. The igraph software package for complex network research. Inter J. Compex Syst. 2006. [Google Scholar]
- 40. Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Eberhard C. et al. . The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kato A., Albert P., Vega J., Birchler J.. Sensitive fluorescence in situ hybridization signal detection in maize using directly labeled probes produced by high concentration DNA polymerase nick translation. Biotech. Histochem. 2006; 81:71–78. [DOI] [PubMed] [Google Scholar]
- 42. Macas J., Neumann P., Navrátilová A.. Repetitive DNA in the pea (Pisum sativum L.) genome: comprehensive characterization using 454 sequencing and comparison to soybean and Medicago truncatula. BMC Genomics. 2007; 8:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kato A., Yakura K., Tanifuji S.. Sequence analysis of Vicia faba repeated DNA, the FokI repeat element. Nucleic Acids Res. 1984; 12:6415–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fuchs J., Pich U., Meister A., Schubert I.. Differentiation of field bean heterochromatin by in situ hybridization with a repeated FokI sequence. Chromosom. Res. 1994; 2:25–28. [DOI] [PubMed] [Google Scholar]
- 45. Ananiev E.V., Phillips R.L., Rines H.W.. Chromosome-specific molecular organization of maize (Zea mays L.) centromeric regions. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:13073–13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ananiev E.V., Phillips R.L., Rines H.W.. A knob-associated tandem repeat in maize capable of forming fold-back DNA segments: are chromosome knobs megatransposons. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:10785–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ananiev E.V., Phillips R.L., Rines H.W.. Complex structure of knob DNA on maize chromosome 9: retrotransposon invasion into heterochromatin. Genetics. 1998; 149:2025–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maggini F., Cremonini R., Zolfino C., Tucci G.F., D’Ovidio R., Delre V., DePace C., Scarascia Mugnozza G.T., Cionini P.G.. Structure and chromosomal localization of DNA sequences related to ribosomal subrepeats in Vicia faba. Chromosoma. 1991; 100:229–234. [DOI] [PubMed] [Google Scholar]
- 49. Macas J., Koblížková A., Navrátilová A., Neumann P.. Hypervariable 3΄ UTR region of plant LTR-retrotransposons as a source of novel satellite repeats. Gene. 2009; 448:198–206. [DOI] [PubMed] [Google Scholar]
- 50. Schaper E., Kajava A. V., Hauser A., Anisimova M.. Repeat or not repeat? Statistical validation of tandem repeat prediction in genomic sequences. Nucleic Acids Res. 2012; 40:10005–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lim K.G., Kwoh C.K., Hsu L.Y., Wirawan A.. Review of tandem repeat search tools: a systematic approach to evaluating algorithmic performance. Brief. Bioinform. 2013; 14:67–81. [DOI] [PubMed] [Google Scholar]
- 52. Fertin G., Jean G., Radulescu A., Rusu I.. DExTaR: detection of exact tandem repeats based on the de Bruijn graph. Proc. - 2014 IEEE Int. Conf. Bioinforma. Biomed. IEEE BIBM 2014. 2014; doi:10.1109/BIBM.2014.6999134. [Google Scholar]
- 53. Fertin G., Jean G., Radulescu A., Rusu I.. Hybrid de novo tandem repeat detection using short and long reads. BMC Med. Genomics. 2015; 8(Suppl. 3):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Simpson J.T., Wong K., Jackman S.D., Schein J.E., Jones S.J.M., Birol I.. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009; 19:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gong Z., Wu Y., Koblížková A., Torres G.A., Wang K., Iovene M., Neumann P., Zhang W., Novák P., Buell C.R. et al. . Repeatless and repeat-based centromeres in potato: implications for centromere evolution. Plant Cell. 2012; 24:3559–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Macas J., Navrátilová A., Mészáros T.. Sequence subfamilies of satellite repeats related to rDNA intergenic spacer are differentially amplified on Vicia sativa chromosomes. Chromosoma. 2003; 112:152–158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.