


Abstract
Insertional mutagenesis using engineered transposons is a potent forward genetic screening technique used to identify cancer genes in mouse model systems. In the analysis of these screens, transposon insertion sites are typically identified by targeted DNA-sequencing and subsequently assigned to predicted target genes using heuristics. As such, these approaches provide no direct evidence that insertions actually affect their predicted targets or how transcripts of these genes are affected. To address this, we developed IM-Fusion, an approach that identifies insertion sites from gene-transposon fusions in standard single- and paired-end RNA-sequencing data. We demonstrate IM-Fusion on two separate transposon screens of 123 mammary tumors and 20 B-cell acute lymphoblastic leukemias, respectively. We show that IM-Fusion accurately identifies transposon insertions and their true target genes. Furthermore, by combining the identified insertion sites with expression quantification, we show that we can determine the effect of a transposon insertion on its target gene(s) and prioritize insertions that have a significant effect on expression. We expect that IM-Fusion will significantly enhance the accuracy of cancer gene discovery in forward genetic screens and provide initial insight into the biological effects of insertions on candidate cancer genes.
INTRODUCTION
Transposon-based insertional mutagenesis (TIM) is a high-throughput method for cancer gene discovery in mice (1). In TIM, discrete DNA elements called transposons can migrate throughout the genome by a cut-and-paste mechanism, in which they are excised from their original location in the genome and randomly reintegrated elsewhere (2). Depending on the location and orientation of their reintegration, these integrations can activate oncogenes or inactivate tumor suppressors, thereby inducing tumor development and progression (3). By identifying genomic loci that are recurrently affected by transposon insertions in multiple independent tumors, this approach can be used to identify candidate cancer genes (1,3,4).
Transposon insertion sites are typically identified using targeted DNA-sequencing approaches, in which junction fragments containing transposon and flanking genomic sequences are selectively amplified and sequenced (5). The genomic parts of these sequences are mapped to the reference genome to identify insertion sites and their genomic locations (6). These insertions are then assigned to their putative target gene(s) using heuristics, typically picking genes in the direct vicinity of the insertion. Examples of such heuristics are nearest gene (6), fixed window (7) and rule-based mapping approaches (8).
A significant drawback of DNA-sequencing approaches is that they do not provide any direct evidence that an insertion actually affects a gene. In ambiguous cases with multiple genes in the vicinity of an insertion, heuristic approaches are frequently unable to identify the true target(s) of the insertion. This typically leads to an arbitrary selection of a single gene (nearest gene), potentially selecting the wrong gene or missing other targets (false negatives). Alternatively, heuristics may select many genes in the direct vicinity of the insertion (fixed window, rule-based mapping), resulting in the selection of many non-target genes (false positives).
Additionally, DNA-sequencing approaches provide limited insight into how the expression of a target gene is affected by a transposon insertion and which novel transcripts may result from the insertion. This has two main drawbacks. First, it prevents prioritizing insertions that have a strong effect on gene expression and are therefore likely of more importance than insertions without an effect on expression. This limits effective discrimination between driver and passenger insertions, resulting in long lists of candidate loci which are likely to include a substantial fraction of false positives that do not affect expression. Second, it limits our understanding of how gene expression or the expression of (novel) gene transcripts is affected by insertions. These insights may be key to ultimately understanding the biological effect of insertions and how they may contribute to tumorigenesis.
In previous work, Temiz et al. have demonstrated that insertions can be identified in paired-end RNA-sequencing data using their tool Fusion Finder (9). In Fusion Finder, insertions are detected from discordant mate pair alignments, in which one mate aligns to a genomic sequence and the other to part of the transposon sequence. A drawback of this approach is that it does not use information from chimeric reads overlapping the fusion boundary between the gene and the transposon (split reads), limiting the accuracy and sensitivity of insertion detection. Additionally, the dependency on mate pair information prevents its use for analyzing datasets based on single-end RNA-sequencing.
In this work, we present an approach called IM-Fusion, which uses fusion-aware RNA-seq alignment to identify transposon insertions from splicing events between endogenous genes and the transposon. Key advantages of this approach are that it identifies exactly which gene(s) are affected by a transposon insertion and how the transposon is incorporated into the resulting gene transcript. Additionally, by using both split reads and discordant mate pairs to identify insertions, IM-Fusion is more sensitive than existing approaches and can be used to analyze single-end RNA-sequencing datasets. Finally, by combining insertions with exon-level expression data, we are able to accurately predict the consequences of integrations on gene transcripts.
MATERIALS AND METHODS
IM-Fusion
Identifying insertion sites
First, we create an augmented reference genome by adding the transposon as an extra sequence in the reference genome. Then, for each sample, we align sequence reads to the augmented reference genome using a fusion-aware RNA-seq aligner such as STAR (10) or Tophat-Fusion (11). By default, STAR is used for alignment, with the argument ‘–chimSegmentMin’ to ensure that chimeric read alignments are produced. Chimeric alignments from STAR are filtered to select alignments that represent fusions between the transposon and genomic sequences. Alignments that overlap with the fusion junction (represented by split-read alignments) are grouped by the position of their breakpoints, as these reads precisely identify the location of a fusion. Each such group is considered to represent a single gene-transposon fusion. For paired-end sequencing data, alignments that do not overlap with the fusion boundary are grouped if their mate positions fall within a pre-defined distance, which depends on the insert size of the dataset. Where possible, these ‘spanning’ read groups are assigned as additional support for fusions identified from split-reads. For cases where no such fusion is found, approximate locations for the corresponding fusions are predicted based on the bounds provided by the spanning reads.
The identified fusions are annotated to identify which gene(s) and which transposon feature(s) are involved in each fusion. Fusions that do not involve splice acceptor (SA) or splice donor (SD) features of the transposon or fusions that represent biologically implausible situations (such as fusions between transposon features and gene exons in opposite orientations) are considered artifacts and removed from the list of fusions. Optionally, fusions supported by less than a pre-defined number of reads can be removed to avoid fusions with low support. For this filtering, we provide two distinct measures: a support score and an FFPM (fusion fragments per million) score. The support score simply indicates the number of reads/mates that supports the corresponding fusion. The FFPM score is a scaled version of the support score, which is normalized for differences in sequencing depth between samples. This score is analogous to the FFPM score used by STAR-Fusion (12). The list of filtered fusions is used to predict approximate locations of the corresponding insertion sites, based on the breakpoints of the fusions.
Transcript assembly
To identify cases in which insertions lead to the expression of non-canonical transcripts, IM-Fusion provides an optional step which uses StringTie (13) to perform a reference-guided assembly of novel transcripts using the read alignment from STAR. The produced transcript annotation is used to assign any previously unannotated insertions to any novel transcripts that overlap with the insertion. If such a novel transcript overlaps with any known genes, the corresponding insertion is also assigned to these known genes, as the transcript likely represents an alternative transcript of these existing genes.
Selecting commonly targeted genes
Commonly targeted genes (CTGs) are selected by testing if genes are affected by insertions more frequently than would be expected by chance according to the Poisson distribution. The Poisson distribution expresses the probability of a given number of events occurring in a fixed interval of time or space, as long as the expected number of events in a fixed window is known and events occur independently. Specifically,
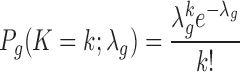 |
where k is the number of events and λg is the expected number of events in a fixed window. Here, each insertion represents an independent event and the fixed window is the genomic region of the gene of interest, optionally expanded to include a window around the gene. The expected number of insertions is calculated based on the size of the gene window, the size of the transcriptome (the union of windows for all genes) and the total number of insertions within the transcriptome windows.
In detail, we first count the number of insertions that were identified for a given gene g (by the insertion identification step) and were located within a pre-defined window (by default 20 kb) around the gene. This count is denoted as Ng. Second, we calculate the expected number of insertions in gene g (λg) based on its window size and the total number of insertions within the transcriptome as follows:
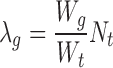 |
in which Wg corresponds to the size of the window around gene g, Wt the size of the transcriptome windows (the sum of windows for all genes in the genome, corrected for overlap between gene windows) and Nt represents the total number of insertions within the transcriptome windows. Using λg, we then calculate the probability of observing Ng or more insertions in gene g as:
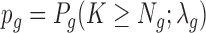 |
After testing all genes of interest (by default all genes with at least one insertion in the gene), calculated P-values are corrected for multiple testing using Bonferroni correction.
If the transposon employed in the screen is known to be biased toward integrating at specific nucleotide sequences, λg can be calculated differently to take this integration bias into account. In this case, instead of using the size of the gene windows, we use the number of occurrences of the nucleotide sequence with the gene window (Sg) and within the transcriptome windows (St) to calculate λg:
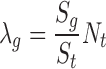 |
To account for a potential bias in integrations on the chromosome on which the transposon concatemer is located, insertions and genes on the donor chromosome can be excluded from the analysis. In this case, genes on the donor chromosome are also excluded when calculating the transcriptome size (Wt/St) and the number of insertions (Nt).
Differential expression analysis
To test for differential expression, we first generate exon expression counts from the read alignments using featureCounts (14). For this count summarization, we use a flattened version of the reference GTF file, which is similar to the flattened GTF files produced by DEXSeq (15). This flattened GTF is required to ensure that overlapping exons from different transcripts of the same gene are only counted once by featureCounts.
Next, to test a given gene g for differential expression, we divide the exons of gene g into two groups: those before the transposon insertions in the gene (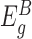 ) and those after the insertions (
) and those after the insertions (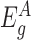 ). We assume that the expression counts of exons before the insertions (
). We assume that the expression counts of exons before the insertions (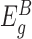 ) are not directly affected by the presence of an insertion and therefore reflect differences in the overall expression of the gene between samples. Based on this assumption, we normalize the counts of each sample for differences in overall expression of the gene by dividing the counts by a sample-specific normalization factor, which is calculated from the counts of the exons in
) are not directly affected by the presence of an insertion and therefore reflect differences in the overall expression of the gene between samples. Based on this assumption, we normalize the counts of each sample for differences in overall expression of the gene by dividing the counts by a sample-specific normalization factor, which is calculated from the counts of the exons in 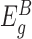 using DESeq2's median-of-ratios approach (16). We then sum the normalized counts of exons in
using DESeq2's median-of-ratios approach (16). We then sum the normalized counts of exons in 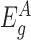 per sample, to get a single (normalized) count of expression after the insertion site for each sample. Finally, to actually test for differential expression in the presence of an insertion in gene g, we use a two-tailed Mann–Whitney-U test to compare the distribution of these counts between samples with an insertion in gene g and samples without an insertion in the gene.
per sample, to get a single (normalized) count of expression after the insertion site for each sample. Finally, to actually test for differential expression in the presence of an insertion in gene g, we use a two-tailed Mann–Whitney-U test to compare the distribution of these counts between samples with an insertion in gene g and samples without an insertion in the gene.
In some cases, the above test is not possible because some samples do not have at least one exon before and after their insertion sites. This mostly occurs when insertions are located upstream of the first exon of the gene. To handle these cases, we first try to remove these problematic samples and repeat the test using the remaining samples. For cases where this does not leave us with any samples to test, we provide an additional gene-level test, which compares the expression of the overall gene between samples with/without insertions after normalizing for overall differences in sequencing depth.
By default, we do not use multiple testing correction for the differential expression test, as we primarily select CTGs using the Poisson-based test and use the differential expression test as an extra test to determine whether to keep the CTG. Additionally, not all CTGs may be subjected to the same test, as some genes may be tested using the gene-level test if the exon-level version is not applicable.
Single-sample differential expression
To test for differential expression in a single sample (as opposed to the group-wise test described above), we provide an alternative approach that uses the same normalization procedure, but uses a negative binomial distribution to compare the expression of the sample of interest to samples without an insertion. In this approach, a negative binomial is fitted using the after insertion counts of samples without an insertion in the gene. The after count of the sample of interest is then compared to this distribution using a two-tailed test to determine if the gene is differentially expressed.
Implementation
For convenience and reusability, we implemented the different steps of IM-Fusion in a Python package called imfusion, which is freely available on GitHub (https://github.com/nki-ccb/imfusion). Jupyter notebooks containing the code and results of the various computational analyses are also available on GitHub (https://github.com/jrderuiter/imfusion-analyses).
The Python package provides commands for each main step of IM-Fusion, including the construction of the custom reference genome, identification of insertions from RNA-seq reads, selection of CTGs and analysis of differential expression. The current implementation supports the use of STAR or Tophat-Fusion to detect fusions, although support for additional fusion-aware aligners may be added in the future. For full functionality, working installations of STAR/Tophat2, StringTie and featureCounts are required; as STAR or Tophat2 (which implements Tophat-Fusion) are used to align reads and detect fusions, StringTie is used to detect novel transcripts and featureCounts is used to generate the expression counts. Optionally, STAR-Fusion (12) can also be used to detect endogenous gene fusions as part of the STAR insertion detection pipeline.
Datasets
ILC dataset (RNA-seq)
Single-end RNA-sequencing data from 123 tumors were obtained from a dataset of a Sleeping Beauty (SB) transposon screen in a mouse model of invasive lobular breast carcinoma (ILC) (17). The RNA-seq data were downloaded from ENA in fastq format (accession number PRJEB14134) and analyzed using IM-Fusion (version 0.3.1) to detect SB insertion sites in each sample, as well as subsequently identify CTGs and their effects. For this analysis, we created an augmented reference genome using the mm10 version of the mouse genome and the T2/Onc transposon sequence (18). STAR (version 2.5.2b) was used to perform the alignment, StringTie (version 1.3.0) was used for transcript assembly and featureCounts (version 1.5.0-post3) was used to generate expression counts. Reference genome features were downloaded from Ensembl 76.
ILC dataset (ShearSplink)
DNA-sequencing data prepared using the ShearSplink protocol (19) for the same tumors as the ILC RNA-seq dataset were downloaded from Figshare (DOI: 10.6084/m9.figshare.4765111) and analyzed using the ShearSplink pipeline in PyIM (version 0.2.0, https://github.com/jrderuiter/pyim) to identify SB insertion sites. In essence, this pipeline first extracts genomic DNA from reads by removing the transposon and linker sequences. The genomic sequences are then aligned to the reference genome using Bowtie2 (version 2.2.8) (20), and the resulting alignments are grouped by sample and position to identify the location of insertion sites. Finally, identified insertions are assigned to their predicted target genes using the windows outlined in KC-RBM (8). To reduce the number of identified target genes for each insertion, we selected a single target gene for each insertion by picking the closest gene identified by KC-RBM. In cases where this was not possible, e.g. due to overlapping genes, we retained multiple target genes.
B-ALL dataset
Insertion data and paired-end RNA-seq data from 20 B-cell acute lymphoblastic leukemias (B-ALLs) were obtained from a previously published dataset of a SB screen performed in a mouse model of B-ALL (21). The RNA-seq data were downloaded from ENA in fastq format (study ID: ERP005291, array expression ID: E-ERAD-264). The insertion data were obtained from the Supplementary Materials of the publication or through personal communication. Control samples were omitted from the performed analyses.
Methods—ILC dataset
Gene-transposon fusion validation in RNA
Tumor RNA was extracted as previously described (22) and 300 ng was converted to complementary DNA (cDNA) with a Moloney murine leukemia virus reverse transcriptase using random hexamer primers according to manufacturer's protocol (Tetro cDNA synthesis kit, Bioline). Gene-transposon fusions were detected by standard polymerase chain reaction (PCR) with an annealing temperature of 58°C. The following primer sequences were used:
-
SA reverse
5΄-TTCCCGCGAATCCATCTTTC-3΄
-
En2SA reverse
5΄-GTCGACTGCAGAATTCGATGA-3΄
-
SD forward
5΄-GCCCATCAAGCTTGCTACTA-3΄
-
Myh9 forward
5΄-CTGTGTGGTCATCAACCCTTAT-3΄
-
Trp53bp2 reverse
5΄-ATCGCTCTGGTTTCGATAAGG-3΄
-
Ctnnd1 forward 1
5΄-GCTACATGCCTTGACAGATGA-3΄
-
Ctnnd1 forward 2
5΄-GAGAGGAGAAAGGCAGGAAAG-3΄
-
Hprt forward
5΄-CTGGTGAAAAGGACCTCTCG-3΄
-
Hprt reverse
5΄-TGAAGTACTCATTATAGTCAAGGGCA-3΄
Effects of insertions
To study the effects of individual SB insertions on expression, we visualized single insertions together with the expression of each of their targets in the affected sample and tested for differential expression over the insertion site in the sample. The visualization was generated using the Python package geneviz, which is freely available on GitHub (https://github.com/jrderuiter/geneviz). Gene annotations for the plot were obtained from Ensembl 76. Expression profiles were generated from the RNA-seq alignment of the sample using pysam (23), by counting the number of reads overlapping each nucleotide position in the plotted range. Junction strengths were derived from the junction files (SJ.out.tab) generated by STAR during the alignment. To test for differential expression, we used the single-sample exon-level test implemented by IM-Fusion.
Effects on CTGs
To identify biases in SA/SD insertions for the various CTGs, we counted the number of times each transposon feature (SD, SA, En2SA) was involved in the insertions affecting each CTG. The results were visualized to show the different distributions across CTGs. To test for differential expression, we applied IM-Fusions group-wise DE test for each CTG.
Insertion comparison
To compare the overlap in insertions between IM-Fusion and ShearSplink, we matched two insertions between IM-Fusion and ShearSplink under the following conditions: both insertions were identified in the same sample, had the same predicted target gene and their relative location and orientation was compatible. The latter restriction was used to ensure that a ShearSplink insertion was in the correct location to generate the fusion observed by IM-Fusion in the RNA-seq data. Insertions matched between the two approaches were marked as ‘Shared’, unmatched insertions were designated ‘IM-Fusion only’ or ‘ShearSplink only’ depending on the approach that identified them.
To identify features distinguishing shared insertions from insertions that were unique to either approach, we compared the set of shared insertions to the IM-Fusion- and ShearSplink-specific insertions. For both comparisons (Shared/ShearSplink and Shared/IM-Fusion), we first defined a set of features that could potentially affect insertion detection by either method. We then trained a logistic regression model on these features to predict whether an insertion was matched or unique to the corresponding approach. This model was used to determine the significance of each feature. Finally, we visualized the distributions of significant features for both the matched/unmatched insertions using kernel density estimation (KDE) plots for interpretation.
Candidate gene comparison
To compare the candidate genes identified by ShearSplink and IM-Fusion, we first identified significant common insertion sites (CISs) and differentially expressed CTGs (DE CTGs) separately using the respective approaches. We then visualized the resulting gene rankings, linking genes that were identified as candidate genes by both approaches. Candidate genes were colored to distinguish whether they were (i) shared between both approaches (black), (ii) were identified to have insertions but were not selected as a CTG/CIS by the other approach (blue), (iii) were selected as a CTG/CIS but were not differential expressed (green), (iv) were not selected as a CTG/CIS and were not differentially expressed (purple) and (v) were omitted entirely by the other approach (red).
ShearSplink insertion validation in DNA
Tumor DNA was isolated using a phenol–chloroform extraction. Transposon insertions were detected in 500 ng DNA by standard PCR with an annealing temperature of 58°C. The following primer sequences were used:
-
En2SA forward
5΄-GCTTGTGGAAGGCTACTCGAA-3΄
-
Nf1 11KOU029-R5.INS_12 reverse
5΄-CTCACGTGAAGTGGGAAAGACA-3΄
-
Nf1 12SKA029-R3.INS_15 reverse
5΄-GGCGCACACCTTTAATCCTAAC-3΄
-
Nf1 12SKA033-R3.INS_10 reverse
5΄-TAGCTCCCTGTGTGTTCCTTTG-3΄
-
Nf1 12SKA068-L3.INS_15 reverse
5΄-AAGGGTGAAGCAGGAGGATTAC-3΄
-
Nf1 12SKA092-L2.INS_10 reverse
5΄-ACGGAGAAGGAGAGAGGGAAA-3΄
-
Nf1 12SKA104-R3.INS_1 reverse
5΄-CCAACATCCCTGTTGTGTGTATG-3΄
-
Hprt forward
5΄-CTGGTGAAAAGGACCTCTCG-3΄
-
Hprt reverse
5΄-TGAAGTACTCATTATAGTCAAGGGCA-3΄
Endogenous fusion identification
Endogenous gene fusions were identified by applying STAR-Fusion (version 0.5.4) (12) to the raw RNA-seq data (fastq files) using recommended settings. The resulting list of fusions were combined across samples and filtered for fusions with breakpoints at known splice junctions, as these are most likely to reflect proper gene fusions. The filtered fusions were prioritized by grouping fusions on the involved genes and ranking by the recurrence of these gene pairs across samples. The fusions involving Fgfr2 were validated using the same approach as for the gene-transposon fusions, with the following additional primers:
Fgfr2 forward 5΄-TGGCCAGGGATATCAACAAC-3΄
Kif16b reverse 5΄-CTTTCCTGAGGGCTAGAGTTTG-3΄
Myh9 reverse 5΄-GATAGCGCCTTTGTCTCCTT-3΄
Tbc1d1 reverse 5΄-CCAGGCTGTGAGAAGGATTT-3΄
Methods—B-ALL dataset
Candidate gene comparison
To compare IM-Fusion with the DNA-seq results from the original publication, we applied IM-Fusion to the paired-end RNA-seq data and compared the identified DE CTGs with the published candidate genes (DE CISs). To avoid selecting CTGs with very low support in this relatively deeply sequenced dataset (as these are more likely to represent false positives), we filtered insertions with fewer than 10 supporting reads or mates from the CTG analysis.
Effect of sequencing depth
The B-ALL samples were downsampled to depths of 15, 30, 50 and 70 million reads using Seqtk (https://github.com/lh3/seqtk). IM-Fusion was applied to each of these downsampled datasets to identify DE CTGs, using the same settings as were used for the full dataset. The number of insertions and DE CTGs were compared between the different depths, as well as the overlap in DE CTGs between depths.
Single- versus paired-end comparison
A single-end version of the dataset was simulated by supplying only the first pair as input to IM-Fusion. The results from the paired-end and single-end analyses were compared by juxtaposing DE CTGs and insertions in these genes between the two analyses.
Fusion Finder comparison
We created an augmented version of the mm10 reference genome containing the T2/Onc transposon sequence in the same manner as described by Temiz et al. (9). This reference was modified to mask the En2 and Foxf2 gene loci, which contain sequences homologous to parts of the transposon sequence. Tophat2 (version 2.1.0) (24) was used to align reads to this augmented reference, after which the Fusion Finder script (version 3.1) was used to identify insertions in each sample. The results were compared with IM-Fusions DE CTGs and published candidate genes by analyzing the overlap between the identified insertions and the CTGs/CISs. To determine why certain CTGs/candidates were not identified by Fusion Finder, we visualized the distribution of the used transposon features and compared the alignments of reads supporting insertions unique to IM-Fusion between the Tophat2 and STAR alignments using pysam (23).
Endogenous fusion identification
Endogenous gene fusions were identified in the same manner as for the ILC dataset.
RESULTS
Identifying insertion sites from gene-transposon fusions
Transposon insertions can affect the expression of nearby genes, potentially leading to the activation of oncogenes or the inactivation of tumor suppressors. For example, consider the T2/Onc transposon (Figure 1A) that is used in this work. When integrated in the vicinity of a gene, this transposon can induce (over)expression of nearby genes by initiating transcription from its promoter sequence (MSCV) and then splicing into the gene using the SD sequence (Figure 1B). Alternatively, the transposon can truncate transcripts using either of its SA sites (SA/En2SA) and their corresponding polyA (pA) sites (Figure 1C). Depending on the gene and the location of the transposon, these truncations can inactivate the gene by resulting in an unstable transcript or inactive protein, or activate the gene by removing inhibitory protein domains.
Figure 1.

Overview of the T2/Onc transposon and its effects on gene expression. (A) The transposon sequence contains two splice acceptor sequences (SA and En2SA) with corresponding polyA sequences (pA), and a single promoter sequence (MSCV) combined with a splice donor (SD) sequence. (B) Sense insertions of the transposon either within or upstream of a gene may drive overexpression of the downstream gene sequence by initiating expression from the transposons promoter and SD sequence. (C) Insertions within genes (in either orientation) may truncate gene transcripts by splicing to either of the SA sites (SA or En2SA). The resulting truncations may inactive tumor suppressor genes, but can also activate oncogenes by removing inhibitory domains from the resulting protein.
In both of these cases, part of the transposon sequence is incorporated into the resulting mRNA transcript(s) via splicing between the affected gene and the transposon. As such, these transcripts effectively represent fusions between the transposon sequence and the affected gene. We therefore hypothesized that it should be possible to detect transposon insertion sites from RNA-sequencing by identifying gene-transposon fusions using existing gene fusion detection tools. By further analyzing the breakpoints of each fusion, we could determine exactly which gene and which feature of the transposon are involved in the fusion, and use this information to predict the location of the corresponding insertion site.
IM-Fusion
In this work, we developed a tool called IM-Fusion, that uses a three-step approach to (i) identify insertions from gene-transposon fusions in RNA-sequencing data, (ii) select genes that are more frequently affected by insertions than would be expected by chance and (iii) test if the expression of these genes is significantly changed by their insertions (Figure 2A). A brief description of each of the steps is provided below, more details are available in the ‘Materials and Methods’ section.
Figure 2.

Overview of IM-Fusion. (A) The IM-Fusion pipeline. Samples are initially processed individually to identify insertions and generate gene and exon expression counts for each sample separately. The per-sample results are then combined to identify genes that are recurrently affected across samples. For these genes, we then combine the expression and insertion data to test for differential expression over the insertion site. The results of this analysis are used to determine if insertions have a significant effect on the expression of their target genes and exactly how the insertions affect the resulting gene transcript. (B) Transposons that affect gene expression are included in gene transcripts and are therefore detectable as fusion transcripts between genes and the transposon. These fusions are detected by reads or mate-pairs that bridge the fusion site. The breakpoints of the identified gene-transposon fusions are analyzed to identify the involved gene(s) and predict an approximate location for the corresponding insertion(s). (C) Insertion and expression data are combined to test if an insertion significantly affects the expression of exons downstream of the insertion site. Expression counts are calculated both before/after the insertion site for a sample with an insertion and a set of background samples without an insertion. The ‘before’ count is then used to normalize the sample counts, after which the normalized ‘after’ counts are compared to the ‘before’ counts to test for differential expression. Samples with a truncating insertion are expected to show a lower level of expression after the insertion relative to the background, whilst samples with an activating insertion are expected to show increased expression after the insertion.
Identifying insertion sites
IM-Fusion identifies transposon insertion sites from gene-transposon fusions in the RNA-seq data. To identify these fusions, IM-Fusion first creates an augmented version of the host reference genome by adding the sequence of the transposon as an extra sequence to the original reference sequence. Then, for each sample, IM-Fusion uses a fusion-aware RNA-seq aligner to align RNA-seq reads to the augmented reference and identify gene fusions. By default, STAR (10) is used for this purpose, although Tophat-Fusion (11) is also supported. The identified fusions are filtered to only select fusions between genes and the transposon sequence. These gene-transposon fusions are then analyzed to identify the involved genes and transposon features, and to infer the approximate locations of the insertions (Figure 2B). Optionally, the RNA-seq alignment can be used to perform a reference-guided transcript assembly, which allows IM-Fusion to detect insertions that result in the expression of novel (unannotated) transcripts.
An important advantage of IM-Fusion over DNA-sequencing based approaches is that, instead of focusing on deriving the exact location of insertion sites, it focuses on determining which genes are affected by insertions. This gene-centric approach allows us to select only those insertions that affect expressed genes and are therefore most likely to have an actual biological effect. By doing so, IM-Fusion provides an important filter that strongly enriches for biologically relevant insertions and avoids selecting many extraneous insertions that are unlikely to affect gene expression. This greatly increases the specificity of our results, providing more confidence in the identified hits.
Selecting commonly targeted genes
To identify genes that are commonly targeted by insertions, we use the Poisson distribution to test whether a given gene has more insertions than may be expected by chance (see ‘Materials and Methods’ section). To correct for cases in which a single insertion is detected multiple times in the same gene, either due to its involvement in multiple gene isoforms or due to local hopping within the gene, insertions are by default collapsed into a single insertion per gene per sample (taking the average location of the insertions) before testing for enrichment. This ensures that selected CTGs indeed represent recurrent insertions across multiple samples, and not just multiple insertions within a single or few samples.
Testing for significant effects on expression
To establish whether the expression of a CTG is significantly altered by its insertions, we test for differential expression over the insertion sites in the gene. The main goal of this analysis is to determine if we see a significant increase in expression after the insertion site, indicating that (partial) gene transcripts are (over)expressed by the insertions, or observe a significant decrease in expression, indicating that gene transcript(s) are truncated by the insertions.
To perform the test, we first normalize for differences in overall expression of the gene across all samples based on the expression of exons before the insertion site, which we assume are not directly affected by the presence of an insertion. After this normalization, we compare the normalized expression levels after the insertion site between samples with and without an insertion in the gene to test for differential expression (Figure 2C). By default the test performs a group-wise comparison using the Mann–Whitney U test, in which the expression of samples with an insertion is compared to samples without insertions in the gene. Alternatively, we also provide a single-sample test based on the negative binomial distribution, which determines whether the gene is differentially expressed in a specific sample.
For cases without exons before the insertion site(s), which can occur if insertions are located upstream of the gene, an additional gene-level test is provided. This test compares the expression of the overall gene between samples with/without insertions, after normalizing for overall differences in sequencing depth.
Applying IM-Fusion to a mouse model of breast cancer
We tested our approach by using IM-Fusion to identify SB transposon insertions in 123 tumors from a mouse model of invasive lobular breast cancer (ILC) (17). On average, 0.1% of the reads in each sample were chimeric reads supporting a potential fusion, of which 0.42% represented a putative gene-transposon fusion (Supplementary Table S1). From these fusions, IM-Fusion identified a total of 2057 transposon insertion sites across all tumors, with a median of 12 insertions per tumor (Supplementary Table S2). A total of 1043 genes were affected by at least one insertion, 14 of which were selected as differentially expressed (DE) CTGs (Supplementary Table S3). To confirm the existence of the identified insertions, a subset of insertions was validated using PCRs targeting the predicted gene-transposon fusion transcripts (Supplementary Figure S1).
Effects of individual insertions
To evaluate the effect of individual insertions, we visualized single insertions together with the expression of their target genes in the corresponding sample. An example is shown in Figure 3A, which shows an antisense insertion in the Trps1 gene. This insertion was identified from a fusion between the transposons En2SA site and the fourth exon of the gene, indicating that the insertion truncated the gene after this exon. This hypothesis was supported by the expression profile of the gene in this sample, which showed a marked reduction in expression after the insertion site. Using the single sample DE test, we confirmed that this reduction in expression was indeed significant compared to background samples without an insertion in the gene (Figure 3B).
Figure 3.

Examples of identified insertions, CTGs and their effect on gene expression. (A) An example of an antisense insertion in Trps1 that results in truncation of the gene transcript. The insertion (red arrow) is shown above the main transcript of the gene, together with expression levels of the gene. The expression of the exons is shown along the top in blue, which reflects the number of reads covering the various exons. Similarly, the black arches below indicate the strength of the splicing junctions between the different exons, with the height of the arch indicating the number of reads supporting the splice junction. Taken together, these expression profiles show a strong decrease in expression after the insertion site, supporting the hypothesized truncation. (B) Quantified expression levels before/after the insertion site for the Trps1 insertion shown in (A). Compared to the samples without an insertion (gray), the sample with this insertion (blue) shows a significant decrease in expression after the insertion. (C and D) An example of a sense insertion in Trp53bp2 (blue arrow). This insertion results in both truncation of the gene and overexpression of a partial transcript. This overexpression is clearly reflected by the increase in expression after the insertion site. (E) Frequencies of the transposon features involved in the insertions of the top six identified CTGs. A bias toward SA/En2SA favors truncation of the gene, whereas a bias toward SD favors overexpression. (F) Differential expression across the insertion sites for each of the CTGs. An increase in the presence of an insertion indicates overexpression, a decrease indicates truncation.
A second example (Figure 3C) shows a sense insertion in the Trp53bp2 gene, which was identified from two distinct gene-transposon fusions. The first fusion, between the SA site of the transposon and exon 12 of the gene, indicated that the insertion truncated gene transcription after this exon. However, the second fusion, between the SD site and exon 13, indicated that the insertion also drove overexpression of a partial gene transcript downstream of the insertion. Taken together, this showed that the insertion simultaneously resulted in both the truncation of the original gene transcript and overexpression of a C-terminal transcript containing exons 11–18. This overexpression was clearly reflected in the expression levels of the gene, which were significantly increased after the insertion site (Figure 3D). Finally, from the shown splice junctions we saw that the full-length transcript of Trp53bp2 (and/or the truncated N-terminal transcript) was still expressed in this sample, though at lower levels than the partial transcript.
General effects of insertions on CTGs
To determine how each identified CTG was affected by its insertions, we first analyzed the insertions in each CTG to identify if the gene was biased to SD or SA insertions. In this analysis, a bias to SD insertions would indicate the gene is mainly overexpressed by insertions in the gene (Figure 1B). Conversely, a bias toward the SA/En2SA sites would indicate the gene is mainly truncated by its insertions (Figure 1C). Second, we used IM-Fusion to test for differential expression across the insertion site to determine if the insertions affect the expression of the gene and whether the observed effect points to truncation or overexpression of the gene. For clarity we limited our analysis here to the top six candidate genes; similar results for the other candidates are available in Supplementary Figure S2.
This analysis showed that most top CTGs (Ppp1r12a, Trps1, Myh9, Tgfbr2 and Runx1) were clearly biased toward SA/En2SA insertions (Figure 3E), indicating that transcripts of these genes were being truncated by the transposon insertions. This hypothesis was further supported by the DE tests (Figure 3F), which confirmed that each of these genes showed a significant decrease in expression after the insertion site, indicating that genes are indeed truncated. Conversely, for one top CTG, Trp53bp2, we saw a clear bias toward SD insertions, indicating that this gene is overexpressed by its insertions. This was again supported by the DE analysis, which determined that Trp53bp2 showed a significant increase in expression after its insertion sites.
Comparison with targeted DNA-sequencing
To assess if IM-Fusion identifies similar insertions to targeted DNA-sequencing approaches, we compared our results to those obtained by targeted DNA-sequencing of insertions using the ShearSplink protocol (19). For this comparison, we matched insertions between the two approaches (IM-Fusion and ShearSplink) if they identified the same target gene and had compatible genomic locations and orientations. Note that, using this approach, an insertion is counted multiple times if it is assigned to multiple genes, thereby increasing the apparent total number of insertions.
Matched insertions were considered to be shared by both approaches, whereas unmatched insertions were categorized as ‘ShearSplink-specific’ or ‘IM-Fusion-specific’ depending on their source. This analysis showed that the majority of the insertions identified by IM-Fusion (578/818) were shared with ShearSplink (Figure 4A). However, a substantial number of insertions were unique to either IM-Fusion (240) or ShearSplink (2838), indicating a considerable disparity between the two approaches.
Figure 4.

Comparison of insertions identified by IM-Fusion and ShearSplink. (A) Venn diagram of the insertions identified by ShearSplink (red) and IM-Fusion (blue). Many IM-Fusion insertions are shared with ShearSplink (green), but a considerable number of insertions are unique to either approach. (B and C) Distribution of features reflecting biases of RNA-sequencing that affect the detection of insertions by IM-Fusion. ShearSplink-specific insertions (red) typically have low expression compared to shared insertions (green) and are therefore more difficult to detect by RNA-seq. Similarly, insertions toward the start of the gene are more frequently missed by IM-Fusion due to the 3΄ bias of the polyA tail selection used in the RNA-sequencing. (D and E) Distributions of support of DNA-seq insertions and support of RNA-seq insertions. Insertions with low DNA-seq support are more often missed by IM-Fusion, whilst insertions with low IM-Fusion support are often not detected by ShearSplink. These differences likely reflect heterogeneity of subclonal insertions present in the tumor tissue samples used for DNA-seq and RNA-seq, respectively. (F) Comparison of the frequency-based ranking of candidate genes identified by IM-Fusion and ShearSplink. Gray lines indicate the relative rankings of genes that were identified by both approaches. Genes missed by the other approach are marked red. Genes that were identified to have insertions but not selected as CISs/CTGs by the other approach are colored blue or purple, depending on their differential expression status. Genes that were identified as CISs/CTGs but were not differentially expressed are marked green.
ShearSplink-specific insertions
To investigate why certain insertions were not identified by IM-Fusion, we compared the ShearSplink-specific insertions to the insertions identified by both approaches. The goal of this comparison was to identify features that distinguished the two sets of insertions (see ‘Materials and Methods’ section) and might therefore provide insight into the underlying reasons for the observed differences. Of the considered features, the following were determined to be significantly predictive: the expression level of the predicted target gene, the relative location of the insertion within its target, the distance of an insertion to its target and the support of the ShearSplink insertion.
The first two of these features point toward biases in the sequencing coverage of the RNA-seq data that affect the detection of insertions. The first feature, the expression level of the target gene, indicates that IM-Fusion had trouble identifying insertions in genes with no or low expression (Figure 4B). The lack of insertions in non-expressed genes was expected, as these insertions are not represented in the RNA-seq data. As these insertions are unlikely to have any biological effect, their omission is expected to increase the specificity of IM-Fusion with regard to biologically relevant insertions. The lack of insertions in genes with low expression reflects an inherent bias of RNA-seq toward highly expressed genes, which results in less sequencing coverage for genes with low expression.
Similarly, the second feature, the relative position of an insertion within the gene, showed that IM-Fusion misses more insertions at the 5΄ end of genes (Figure 4C). This is due to a well documented 3΄ bias of the polyA-tail selection used to enrich for mRNAs in RNA-sequencing, which results in decreasing coverage toward the 5΄ end of gene transcripts. Together, these two biases limit the ability of RNA-seq-based approaches such as IM-Fusion to detect insertions in lowly expressed genes, particularly at their 5΄ end. This effect can be mitigated by deeper sequencing and by using a different approach to enrich for mRNAs in the preparation for RNA-sequencing (such as rRNA depletion).
Another significant feature, the support of an insertion (Figure 4D), showed that IM-Fusion mainly missed ShearSplink insertions with a low support score. This bias may be due to one or more of the following reasons. First, our RNA-seq data may not be deep enough to detect very subclonal insertions that are only present in a very small fraction of the tumor cells. Second, the observed differences may reflect intratumoral heterogeneity, as we did not use the same tumor fragments for RNA and DNA extraction and sequencing, but instead used two separate pieces of the same tumor. For clonal insertions this is not an issue, but subclonal insertions might be present in only one of the tumor pieces, therefore leading to some of the observed differences between IM-Fusion and ShearSplink.
Finally, we found that the heuristic assignment of target genes by ShearSplink also introduced biases. Even after restricting the assignment of target genes to the closest gene, ShearSplink was unable to identify a unique target gene for some insertions. For example, insertions within the Arfip1/Fbxw7 locus were frequently assigned by ShearSplink to both Fbxw7 and Arfip1. Closer inspection of these insertions indicated that these insertions are in fact closely clustered in Fbxw7 and are therefore unlikely to affect the Arfip1 transcript that overlaps with Fbxw7. This hypothesis was supported by the IM-Fusion results, which only identified insertions in Fbxw7, indicating that Arfip1 is a false positive of the heuristic assignment by ShearSplink. Similarly, the distance to target gene feature indicated that insertions further away from their target genes are rarely matched by IM-Fusion. These hits are also likely to be artifacts of the heuristic assignment of target genes by Shearsplink.
IM-Fusion-specific insertions
To determine why some insertions were only identified by IM-Fusion, we also compared the set of insertions unique to IM-Fusion to the shared insertions. This comparison identified the support score of an insertion as the most predictive feature of IM-Fusion-specific insertions (Figure 4E). This feature, which reflects the number of reads supporting the corresponding gene-transposon fusion, showed that ShearSplink mainly misses insertions with a low IM-Fusion support score. As these insertions are only supported by a few reads in the RNA-seq data, they are likely either false positives of IM-Fusion or subclonal insertions that are present in a small fraction of tumor cells or in specific parts of the tumor. In the latter case, the missed insertions are again likely attributable to heterogeneity between the DNA- and RNA-seq samples, as previously explained for the ShearSplink support feature.
Comparison of identified candidate genes
To assess if IM-Fusion identified different candidate genes than ShearSplink, we compared the DE CTGs from IM-Fusion to the genes associated with CISs from the ShearSplink analysis. This comparison showed that IM-Fusion and ShearSplink identified 14 and 32 candidate genes respectively, of which 12 were shared between both approaches. From a comparison of the rankings of the candidate genes (Figure 4F), we saw the strongest concordance between the most frequently recurring candidate genes, with more discrepancy among the less frequent candidates.
To determine why some ShearSplink candidate genes were not identified by IM-Fusion, we examined them in more detail. Five genes (Arfip1, Gm26836, Gm14798, Ppp2r2a and Bach2) were not identified at all by IM-Fusion, suggesting that these are either false positives of the ShearSplink analysis, as we have already argued for Arfip1, or are weak/subclonal insertions that were not picked up by IM-Fusion. For Nf1, IM-Fusion did detect several weak insertions, which were only supported by single reads and were therefore filtered from the CTG analysis. These insertions, together with additional validation of several ShearSplink insertions (Supplementary Figure S3), demonstrated that Nf1 was not a false positive of the ShearSplink analysis. However, closer inspection showed that Nf1 insertions were generally supported by few reads in the ShearSplink data, thereby explaining their omission by IM-Fusion.
Several other genes (Setd5, Gab1, Ppp1r12b, e.g.) were identified to have insertions by IM-Fusion, but were not detected in enough samples to be selected as a CTG. Further analysis showed that insertions in missing samples were supported by few ShearSplink reads, indicating that these insertions are missed due to their low clonality. This also explains why several of these genes (Ppp1r12b, Nfix, Rmb47, etc.) are not differentially expressed in the presence of an insertion, as we are less likely to pick up expression differences if the signal is weak due to subclonality.
A few candidate genes, including Fgfr2—the top hit from the ShearSplink analysis, were not selected as DE CTGs due to a lack of differential expression. Closer analysis showed that Fgfr2 is affected by a mix of sense and antisense insertions. Whilst the antisense insertions merely truncate the gene, the sense insertions both truncate the gene and induce the overexpression of a partial C-terminal transcript (Supplementary Figure S4). Together, this results in a mix of samples with increased and decreased expression, thereby representing a more complex pattern of expression changes than the overall changes that the DE test was designed to detect. This indicates that, although the DE test is useful for prioritizing candidate genes, frequently recurring CTGs that are not differentially expressed should be investigated in more detail to avoid filtering out more complex cases of differential expression. This can, for example, be done by grouping samples based on the orientation of their insertions (as done here) or on the involved SD/SA sites if these are expected to have different effects on expression.
Finally, besides the known candidates, IM-Fusion identified two novel candidates that were not identified by ShearSplink. Interestingly, both of these genes were identified in similar numbers of samples (two-three samples) by both ShearSplink and IM-Fusion, indicating that IM-Fusion may have more power to identify rare CTGs.
Application of IM-Fusion to paired-end RNA-sequencing data from B-ALL tumors
To test IM-Fusion on paired-end RNA-sequencing data, we used an additional dataset of SB-induced B-cell acute lymphoblastic leukemias (B-ALL) for which both targeted DNA-sequencing and relatively deeply sequenced paired-end RNA-sequencing (70–90 million reads) was available (21). In the original analysis of this dataset, Van der Weyden et al. first identified CISs from targeted DNA-sequencing data, and then selected predicted target genes that showed significant differential expression in the presence of an insertion (DE CISs). For our comparison, we applied IM-Fusion using only the RNA-sequencing data and compared the identified insertions and CTGs to the results of the DNA-seq analysis. In light of the higher sequencing depth of the B-ALL dataset (relative to the ILC dataset), we removed insertions with fewer than 10 supporting reads in the CTG analysis to avoid selecting genes that are recurrently detected but have low support, as these are likely to represent false positives (Supplementary Figure S5).
CTG comparison
On average, 0.72% of the mate pairs in each sample reflected chimeric alignments, of which 0.45% supported potential gene-transposon fusions (Supplementary Table S4). From these fusions (Supplementary Table S5), IM-Fusion identified six CTGs (Jak1, Stat5b, Cblb, Zfp423, Dlx3 and Bmi1), of which all except Bmi1 and Dlx3 coincided with the six DE CISs identified by the DNA-seq analysis (Figure 5A and B). Two genes were only identified by the DNA-seq analysis (Foxp1 and Il2rb). Closer inspection of the original DNA-seq data showed that insertions in these genes were generally supported by <10 reads (Supplementary Table S6), suggesting that these insertions are subclonal and are therefore not represented in the RNA-seq sample due to the afore-mentioned issues with sample heterogeneity. Interestingly, both of the novel CTGs (Bmi1, Dlx3) have been reported to play a role in the development of B-ALL (25,26), suggesting that these are true hits that were missed by the DNA-based analysis.
Figure 5.

Comparison of candidate genes identified by IM-Fusion and the original DNA-sequencing analysis in the B-ALL dataset. (A) Comparison of candidate gene rankings between IM-Fusion (left) and the original DNA-seq-based analysis (right). Colors are coded as in Figure 4F. (B) Overlap of IM-Fusions DE CTGs and the published DE CISs. (C) Comparison of DE CTG rankings by IM-Fusion on the single- and paired-end versions of the B-ALL dataset.
Effect of sequencing depth
To determine how sequencing depth affects the detection of insertions and CTGs, we made use of the high sequencing depth of the B-ALL dataset to repeat the analysis at reduced depth by downsampling the original dataset to 15, 30, 50 and 70 million reads per sample. These analyses showed that the number of detected insertions increases linearly with the sequencing depth (Supplementary Figure S6a), indicating that additional sequencing depth provides more power to detect insertions. In contrast, only one extra DE CTG (Dlx3) was detected at higher sequencing depths (Supplementary Figure S6b and c), suggesting that deep sequencing may provide limited returns when screening for candidate genes. However, less insertions were detected in some of these CTGs at the lower depths (Supplementary Table S7), demonstrating that a higher sequencing depth will provide more accuracy in the detection of weak insertions.
Single- versus paired-end sequencing
To study the added value of paired-end sequencing, we simulated a single-end version of the dataset by applying IM-Fusion to only one of the paired-ends. Although the analysis of the single-end dataset identified the same DE CTGs as the paired-end analysis (Figure 5C), the paired-end data yielded on average two times higher support scores for insertions due to the higher effective depth of paired-end sequencing, and identified a number of insertions that were not detected in the single-end sequencing data (Supplementary Table S8). Overall, this suggests that paired-end sequencing data is not strictly necessary for detecting insertions, but is beneficial for the detection of weak insertions.
Comparison with Fusion Finder
Finally, to compare IM-Fusion with existing approaches, we analyzed the B-ALL dataset using Fusion Finder (9), which uses Tophat2 (24) to identify transposon insertions from discordant mate pairs in paired-end RNA-sequencing data. Comparison of the identified insertions showed that Fusion Finder identified recurrent insertions in Cblb and Dlx3, but was only able to identify insertions in a single sample for Jak1 and Bmi1, and was unable to detect insertions in any of the other DE CTGs identified by IM-Fusion (Supplementary Table S9).
More detailed analyses of the results showed that the insertions in CTGs missed by Fusion Finder are (i) biased toward SD insertions and (ii) mainly supported by chimeric reads overlapping the fusion boundary, rather than mate pairs that span the fusion (with one mate on either side of the fusion). The latter explains why the majority of these insertions were not detected by Fusion Finder, as Fusion Finder does not incorporate split read information into its insertion detection. This highlights an important advantage of using fusion-aware aligners such as STAR and Tophat-Fusion, as these aligners explicitly account for chimeric fusion reads in their alignment, resulting in increased sensitivity for the detection of these insertions.
Although Fusion Finder failed to detect insertions involving the SD site of the transposon in this dataset, it did identify SD insertions in the original study by Temiz et al. (9). We expect that the differences between our result and theirs are due to differences in read lengths, as the B-ALL dataset uses 100 bp reads compared to the 50 bp read length used in their dataset. The longer read length makes it more likely that reads overlap the fusion boundary, making an approach that uses chimeric reads preferable with longer read lengths.
DISCUSSION
We have presented IM-Fusion, a novel approach for identifying transposon insertion sites from gene-transposon fusions in RNA-sequencing data. A key advantage of this approach is that it focuses on identifying insertions that affect gene expression. As such, IM-Fusion provides a significant filter that strongly enriches for insertions that actually affect the expression of their target genes and are therefore most likely to be biologically relevant. This greatly increases the specificity of the approach, providing more confidence in detected insertions and genes and increasing our power to identify rare candidate genes. Furthermore, by combining the insertions with a differential expression analysis, IM-Fusion provides valuable insight into the effect of insertions on the affected target genes.
An important advantage of using RNA-sequencing rather than targeted DNA-sequencing for identification of transposon insertions, is that RNA-sequencing provides much more information than just the location of insertion sites. For example, IM-Fusion uses RNA-expression information to determine how a gene is affected by the presence of an insertion. The same expression data may also be used to identify more global changes in gene expression associated with tumor subtypes or with specific insertions (17), or be used to detect single nucleotide variants and somatic gene fusions that contribute to tumorigenesis. As an example of the latter, we have identified several endogenous fusions in the ILC and B-ALL datasets (Supplementary Figure S7 and Table S10), including several Fgfr2 fusions that reflect known oncogenic fusions previously identified in human cancers (27). Most importantly, these extra analyses can be performed on the same RNA-seq sample, thereby inherently avoiding potential discrepancies resulting from the use of different tumor material for DNA- and RNA-sequencing, an issue that we encountered in the analyses of both the ILC and the B-ALL datasets.
A potential limitation of IM-Fusion is that it requires splicing between the transposon and the affected genes to identify the corresponding insertions. As a result, it will not detect transposon insertions that affect expression via enhancer sequences, as the effects of these insertions are not mediated via splicing. In our analyses, this does not seem to be an issue, as DNA-sequencing approaches did not identify any candidate genes that were perturbed via enhancer effects. This suggests that the MSCV enhancer sequence present in the T2/Onc transposon is not particularly active and that the transposon therefore mainly affects expression via splicing. This notion is in agreement with previous studies reporting preferential intragenic insertion of the T2/Onc transposon (8), making it less likely to act as an enhancer. Enhancer effects may however play a more important role in case other transposons are employed. Similarly, IM-Fusion may be unable to detect insertions that result in transcript instability or degradation, as these will be under-represented in the RNA-seq data. Although we do not observe evidence pointing to transcript degradation in the presence of (clonal) SB insertions (Supplementary Figure S8), other transposons might have different effects on transcript stability.
A strategy to identify both insertions whose effects are mediated by transcriptional enhancement and insertions that affect expression via splicing, would be to combine DNA- and RNA-sequencing methods, ideally using RNA and DNA isolated from the same sample. In such a combined approach, RNA-sequencing could be used to identify and characterize insertions that are mediated via splicing. For insertions that are uniquely identified by DNA-sequencing, the RNA-seq data could be used to analyze their effects on expression of the predicted target genes. Such a strategy would effectively unite the advantages of both approaches, by combining the unbiased identification of insertion sites by DNA-sequencing with the additional biological information provided by RNA-sequencing in a single analysis.
Although Temiz et al. (9) have provided a proof-of-concept showing that transposon insertions can be identified via paired-end RNA-sequencing, our analysis was performed on a much larger dataset (123 versus 20 samples), allowing us to determine biases that affect insertion detection in DNA- and RNA-sequencing data and identify potential limitations of either approach. Furthermore, IM-Fusion improves on Fusion Finder by using a fusion-aware RNA-seq aligner to identify transposon insertions, which enables the use of single-end RNA-sequencing data and increases the sensitivity and the accuracy of insertion detection by also using chimeric reads to identify gene-transposon fusions. Finally, IM-Fusion is provided as comprehensive software package that enables users to perform the entire analysis from start to finish, including the generation of augmented reference genomes, identification of CTGs and testing for differential expression.
In summary, IM-Fusion provides a convenient approach for the identification of insertion sites and their effects on target gene expression from standard single- and paired-end RNA-sequencing data. By combining the identification of insertion sites with expression data, our approach provides valuable insight into the effect of an insertion on its target gene(s) and helps prioritize insertions that are biologically relevant. We expect that this approach will significantly enhance the accuracy of cancer gene discovery in forward genetic screens and prioritization of the identified candidate cancer genes for functional validation studies.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the animal housing, animal pathology and genomics core facilities for their help in generating the mice and the genomics data used in this work. We would also like to thank Sander Canisius for his critical reading of the draft manuscript and for his help in testing the software.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Worldwide Cancer Research grant 07-0585; Netherlands Organization for Scientific Research [NWO-NGI Zenith grant 93512009, NWO-ZonMW Vici grant 91814643, Cancer Genomics Netherlands (CGCNL), Cancer Systems Biology Center (CSBC)]; European Research Council (ERC Synergy grant 319661: CombatCancer). Funding for open access charge: ERC.
Conflict of interest statement. None declared.
REFERENCES
- 1. Copeland N.G., Jenkins N.A.. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer. 2010; 10:696–706. [DOI] [PubMed] [Google Scholar]
- 2. Ivics Z., Li M.A., Mátés L., Boeke J.D., Nagy A., Bradley A., Izsvák Z.. Transposon-mediated genome manipulation in vertebrates. Nat. Methods. 2009; 6:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kool J., Berns A.. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat. Rev. Cancer. 2009; 9:389–399. [DOI] [PubMed] [Google Scholar]
- 4. Collier L.S., Largaespada D.A.. Hopping around the tumor genome: transposons for cancer gene discovery. Cancer Res. 2005; 65:9607–9610. [DOI] [PubMed] [Google Scholar]
- 5. Largaespada D.A., Collier L.S.. Transposon-mediated mutagenesis in somatic cells. Methods in Molecular Biology. 2008; 435:95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarver A.L., Erdman J., Starr T., Largaespada D.A., Silverstein K.A.. TAPDANCE: An automated tool to identify and annotate transposon insertion CISs and associations between CISs from next generation sequence data. BMC Bioinformatics. 2012; 13:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Ridder J., Uren A., Kool J., Reinders M., Wessels L.. Detecting statistically significant common insertion sites in retroviral insertional mutagenesis screens. PLOS Comput. Biol. 2006; 2:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Jong J., de Ridder J., van der Weyden L., Sun N., van Uitert M., Berns A., van Lohuizen M., Jonkers J., Adams D.J., Wessels L.F.. Computational identification of insertional mutagenesis targets for cancer gene discovery. Nucleic Acids Res. 2011; 39:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Temiz N.A., Moriarity B.S., Wolf N.K., Riordan J.D., Dupuy A.J., Largaespada D.A., Sarver A.L.. RNA sequencing of Sleeping Beauty transposon-induced tumors detects transposon-RNA fusions in forward genetic cancer screens. Genome Res. 2016; 26:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim D., Salzberg S.L.. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011; 12:R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stransky N., Cerami E., Schalm S., Kim J.L., Lengauer C.. The landscape of kinase fusions in cancer. Nat. Commun. 2014; 5:4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 15. Anders S., Reyes A., Huber W.. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012; 22:2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kas S.M., de Ruiter J.R., Schipper K., Annunziato S., Schut E., Klarenbeek S., Drenth A.P., van der Burg E., Klijn C., ten Hoeve J.J. et al. . Insertional mutagenesis identifies drivers of a novel oncogenic pathway in invasive lobular breast carcinoma. Nature Genetics. 2017; in press. [DOI] [PubMed] [Google Scholar]
- 18. Collier L.S., Carlson C.M., Ravimohan S., Dupuy A.J., Largaespada D.A.. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005; 436:272–276. [DOI] [PubMed] [Google Scholar]
- 19. Koudijs M.J., Klijn C., van der Weyden L., Kool J., ten Hoeve J., Sie D., Prasetyanti P.R., Schut E., Kas S., Whipp T. et al. . High-throughput semiquantitative analysis of insertional mutations in heterogeneous tumors. Genome Res. 2011; 21:2181–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Weyden L., Giotopoulos G., Wong K., Rust A.G., Robles-Espinoza C.D., Osaki H., Huntly B.J., Adams D.J.. Somatic drivers of B-ALL in a model of ETV6-RUNX1; Pax5(+/-) leukemia. BMC Cancer. 2015; 15:585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boelens M.C., Nethe M., Klarenbeek S., de Ruiter J.R., Schut E., Bonzanni N., Zeeman A.L., Wientjens E., van der Burg E., Wessels L. et al. . PTEN loss in E-cadherin-deficient mouse mammary epithelial cells rescues apoptosis and results in development of classical invasive lobular carcinoma. Cell Rep. 2016; 16:2087–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. et al. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. et al. . TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heltemes-Harris L., Larson J., Starr T., Hubbard G., Sarver A., Largaespada D., Farrar M.. Sleeping Beauty transposon screen identifies signaling modules that cooperate with STAT5 activation to induce B-cell acute lymphoblastic leukemia. Oncogene. 2016; 35:3454–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dell’Orto M.C., Banelli B., Giarin E., Accordi B., Trentin L., Romani M., Kronnie G.T., Basso G.. Down-regulation of DLX3 expression in MLL-AF4 childhood lymphoblastic leukemias is mediated by promoter region hypermethylation. Oncol. Rep. 2007; 18:417–423. [PubMed] [Google Scholar]
- 27. Wu Y.-M., Su F., Kalyana-Sundaram S., Khazanov N., Ateeq B., Cao X., Lonigro R.J., Vats P., Wang R., Lin S.-F. et al. . Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013; 3:636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.