


Abstract
Fusion proteins, comprising peptides deriving from the translation of two parental genes, are produced in cancer by chromosomal aberrations. The expressed fusion protein incorporates domains of both parental proteins. Using a methodology that treats discrete protein domains as binding sites for specific domains of interacting proteins, we have cataloged the protein interaction networks for 11 528 cancer fusions (ChiTaRS-3.1). Here, we present our novel method, chimeric protein–protein interactions (ChiPPI) that uses the domain–domain co-occurrence scores in order to identify preserved interactors of chimeric proteins. Mapping the influence of fusion proteins on cell metabolism and pathways reveals that ChiPPI networks often lose tumor suppressor proteins and gain oncoproteins. Furthermore, fusions often induce novel connections between non-interactors skewing interaction networks and signaling pathways. We compared fusion protein PPI networks in leukemia/lymphoma, sarcoma and solid tumors finding distinct enrichment patterns for each disease type. While certain pathways are enriched in all three diseases (Wnt, Notch and TGF β), there are distinct patterns for leukemia (EGFR signaling, DNA replication and CCKR signaling), for sarcoma (p53 pathway and CCKR signaling) and solid tumors (FGFR and EGFR signaling). Thus, the ChiPPI method represents a comprehensive tool for studying the anomaly of skewed cellular networks produced by fusion proteins in cancer.
INTRODUCTION
Fusion proteins or chimeras in cancers are usually produced by chromosomal translocations and incorporate parts of two different parental proteins (1). Fusions often function as oncoproteins or cancer drivers (2,3). One of the best-known examples is the BCR/ABL fusion protein, considered to be the primary oncogenic driver of chronic myelogenous leukemia (4). The identification of this specific fusion event led to the development of a drug (Imatinib/Gleevec) that is highly specific for inhibiting the fusion kinase, resulting in a breakthrough treatment for a poorly responsive disease (5). More recently, gene fusions in solid tumors and in prostate cancer in particular have been shown to drive carcinogenic processes such as invasiveness (e.g. the TMPRSS2/ERG fusion) (6–8). Thus, gene fusions are being recognized as important diagnostic and prognostic biomarkers in malignant hematological disorders and childhood sarcomas (2,3,9,10).
The organization of proteins into groups and networks due to physical interactions with each other has led to the discovery of numerous functions of otherwise anonymous proteins (11). For example, the physical interaction between transcription factors and non-DNA binding cofactors led to the understanding that the latter may often be assigned the role of co-activator or co-repressor (12). In an attempt to provide a comprehensive list of interactions that govern the biological processes in a cell, high-throughput protein interaction assays have been developed with great success. However, the large-scale sets of interactions, represented as protein–protein interaction (PPI) networks that are often analyzed by mathematical methods for comprehensive biological interpretation, have only been applied to the normal form of a protein (11,13–24). Furthermore, the question of the straightforward prediction of the PPIs of fusion proteins has not been yet systematically addressed.
PPI network analysis has been effectively used to highlight the role of cancer driver genes in disrupting the normal regulation of cellular processes (25–28). These analytical methods score the tendency of the fusion parental genes in their cancer-mutant form to induce changes in protein networks. However, only a few attempts have been made to study the PPI network properties of fusion genes (as a hybrid of two genes) (27,29–31). In a recent most comprehensive study, Latysheva et al. (32) used a fusion network approach to reveal that certain parental proteins of fusions occupy central positions in PPI networks losing their functional activities following fusion events in cancer as well as that fusions escape regulation by losing post-translational modification sites.
To demonstrate the systematic identification of PPI of fusions, uncovering their influence on networks and thus on cancer phenotypes, we present here chimeric protein–protein interaction (ChiPPI) method. It uses a ‘domain–domain co-occurrence’ (DDCOS) score to calculate PPI likelihood. The DDCOS score is based on the previous observations about the preference of domain–domain interactions and the co-occurrence methods (33–36). The final score for a fusion protein and its interactors combines all the preserved domains of two parental proteins of the fusion. As the result, our method predicts the differences in networks of the fusion protein in the context to its two parental proteins (and their preserved protein domains). ChiPPI differs from current methods in that most methods utilize a simple ‘unification’ of the PPI networks of the parental proteins, but are not able to accurately evaluate ‘missing’ and ‘preserved’ interactions that result from the domains of a fusion (27,32). Nevertheless, ChiPPI finds ‘missing’ and ‘preserved’ interactors of a fusion based on the preserved protein domains of the parental proteins. We applied ChiPPI to the analysis of 11 528 fusions from ChiTaRS-3.1 (37), accurately mapping alterations in network properties that delineate the fusion protein network from parental networks.
Thus, using ChiPPI, we found a loss of tumor suppressors from fusion protein networks and the novel inclusion of oncoproteins. We further tested the power of ChiPPI to resolve differences in cellular metabolism and pathways (enrichment) in different cancer types, i.e. leukemia/lymphoma, sarcoma and solid tumors. While certain pathways are enriched in all three cancer types (Wnt, Notch, and Transforming-Growth-Factor beta (TGFB) signaling pathway), there are distinct patterns for leukemia (p53 pathway, Epidermal Growth Factor Receptor (EGFR) signaling, DNA replication and Cholecystokinin Receptor (CCKR) signaling), for sarcoma (p53 pathway and CCKR signaling) and solid tumors (Fibroblast Growth Factor Receptor (FGFR) and EGFR signaling). Therefore, ChiPPI is a comprehensive method for analyzing protein fusion networks and our results show that it is particularly useful to uncover remodeling of the PPI networks in different cancer phenotypes.
MATERIALS AND METHODS
Datasets of cancer fusions
In order to study the fusion networks we used a dataset of 358 cancer fusions of 337 genes from the study of Mitelman et al. (10) as a training set and the Breakpoints Collection of 11 528 fusions of the ChiTaRS-3.1 database (37) as a test set. In addition, we downloaded the COSMIC datasets of all point mutations in human genes in cancers (38). We used cBioPortal (http://www.cbioportal.org/public-portal/index.do) to study the number of patients where cancer-associated mutations and mutated genes have been detected (Supplementary Table S1). We downloaded the BioGrid database (http://thebiogrid.org/download.php) of PPIs (39), consisting of 16 102 genes and 218 979 molecular interactions, for the DDCOS analysis of human genes (Supplementary Table S1 and Supplementary Data). For every new release of the BioGrid database (39), we update the ChiPPI collection automatically.
Enrichment analysis
The enrichment analysis was performed to determine which pathways are over-represented, rendering a significant P-value < 1E-4. This analysis was carried out using GSEA (40) and PANTHER (41) methods for every set of 50 fusion networks found in leukemia/lymphoma, sarcoma or solid tumors (totally 150 networks). Finally, the pathways enrichment analysis was performed using the hypergeometric distribution as follows: given an input list of proteins, a comparison is done considering a number of proteins involved in a certain pathway or PPI network using the Benjamini–Hochberg false discovery rate (FDR) adjustment that gives us a list of pathways for an FDR of 1% (Supplementary Table S2).
Domain–domain co-occurrence table
We took all human proteins and determined their protein domains (from the set of more than 1000 different known protein domains) in PFAM (42,43), STRING (44) and ELM (45,46) and other resources (Supplementary Table S3 and Supplementary Data). We then used the BioGrid database of PPIs (39) to ascribe specific domains to known PPI events (Figure 1). Thus, we built the domain-domain co-occurence (DDCOS) table, T(x, y), using the PPI networks of all the proteins, where x and y are protein domains (Figure 1). It combines frequencies, F(x, y), to identify how often domain, x, co-occurred with domain, y. To normalize for domain over- or under-representation across the proteome, we calculated F(x) and F(y) as frequencies of domain x and domain y correspondingly throughout all human proteins. Thus, to determine whether two domains are frequently found in pairs of interacting proteins in different PPI events, we calculated a log-odds score as follows:
 |
(1) |
Figure 1.

The domain–domain co-occurrence (DDCOS) table. Generation of a proteome-wide database of interactions between discrete protein domains, followed by determining their protein domains and finally building the DDCOS table using all the interactors in the BioGrid database (39).
where Fobserved(domain(x)) is an observed frequency of a particular domain x in all human proteins, Fobserved(domain(x) and domain(y)) is an observed frequency of the DDCOS of domain(x) and domain(y) in two interactors as previously described in (48–50).
Scoring chimeric protein–protein interaction (ChiPPI) events
To evaluate an interaction between two proteins we used the following interaction score (Int):
 |
(2) |
where DDCOS(domain(x), domain(y)) is the DDCOS score for domains x of protein1 and domain y of protein2 calculated by formula [1].
To set a threshold for Int values that represent bona fide data-supported interactions, we studied the distribution of interaction scores for 100 000 randomly selected pairs of real interactors and 100 000 random pairs of non-interactors (Supplementary Table S5 and 6; Supplementary Data). These random datasets have been utilized to verify the score threshold at 1% FDR. We selected PPIs that occur in well-known cancer pathways, using different score thresholds. For each fusion protein of interest, we predicted all the protein domains associated with the corresponding fusion protein sequence, using a Hidden Markov Model-based method, HMMer (51). To build PPI networks for the fusion protein we first ‘unified’ all the interactors of the parent proteins from BioGrid (39). Next, using the DDCOS scores we calculated the Int score for every protein interactor and the fusion. Thus, for every PPI event we determined whether it is maintained or lost upon fusion, using the Int score threshold at 1% FDR (see examples in Supplementary Table S4 and Supplementary Data). Finally, we also used the clustering coefficient as a measure of centrality that calculates a number of triangles in the ChiPPI network of a fusion. The clustering coefficient was calculated as an average of the local clustering coefficients of all the nodes in the ChiPPI network, based on the local clustering coefficient for each node that was calculated as follows:
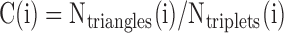 |
(3) |
where Ntriangles(i) is a number of connected triangles around node i and Ntriplets(i) is a number of triplets centered around node i in the ChiPPI network.
RESULTS
ChiPPI is based on the assumption that PPIs can be approximated by calculating the propensity of discreet domain–domain interactions. Accordingly, we posed that each PPI can be converted into a corresponding set of pairwise–domain interactions. To generate a proteome-wide scoring system for interactions using discrete protein domains, we devised the process described on Figure 2 and explained below in details.
Figure 2.

The computing steps that have been done to generate a proteome-wide scoring system for all the ChiPPI interactions using discrete protein domains.
Fusion network construction
Having built a table of the biologically feasible DDCOS between protein domains throughout the human proteome (Figure 1), we then built a network that best predicts the PPIs for each particular fusion protein (Figure 2), as follows:
We unified all the interactors of both parental proteins;
We removed ‘missing’ interactors, lost presumably due to deletion of parental protein domains (having an Int score less than the threshold value);
To match this theoretical prediction with experimental data, we tested how many interactors were missed in any computed pair (from the 358 fusions in the training set), as compared to the published data. To do this, we generated using the DDCOS table four sets of interacting proteins for each of the 358 fusions, using the Int score thresholds: 0, 0.5, 1.0 and 5.0. While threshold score 5.0 lost many bona fide interactors, a 0.5 threshold resulted in 99% of the real cases being included (Supplementary Table S6 and Supplementary Data).
We manually verified an existence of every interactor in the 50 fusions' networks using previously published information on the experimentally verified interactors (2,3,9,10,52,53). In this way, the correct threshold for every fusion network was fixed as the percentage of correctly verified interactors at 1% FDR (P-values adjusted using the Benjamini–Hochberg procedure) (Supplementary Table S6 and Supplementary Data). For more than 95% of fusions, the correct interactors were found for Int scores > 0.5. Therefore, this threshold was used for our fusion network analysis (Supplementary Table S7 and Supplementary Data). As a result, to construct any fusion network, we removed nodes that were characterized by Int ≤ 0.5, which we defined as the ‘missing’ interactors (consistent with the missing domains of the parental proteins).
To study the fusion network properties and their highly connected parts, we transformed the protein interaction network to a graph-based format. For example, the highly connected PPI sub-networks of BCR and ABL1 are distinct from each other and from that of the BCR/ABL1 fusion protein (Figure 3) based on the predicted domains in the interacting proteins, our DDCOS analysis and sub-networks (Int score > 0.5, FDR <1%). While ChiPPI was primarily designed to identify interactors that are lost from networks upon protein fusion, we found that ChiPPI also identified five ‘preserved’ interactors in the networks (in comparison to the network of ABL1 protein): CRKL, JAK2, RASA1, CD2AP and INPPL1 (Figure 3). Inclusion of new interactors into the BCR/ABL network is likely to promote the disease phenotype, since CRKL and RASA1 regulate tumorigenic signaling in chronic myeloid leukemia (4,54), JAK2 mutation and fusion is tumorigenic in myeloproliferative disorders and in acute myeloid leukemia (AML) (52,54) and CD2AP and INPPL1 are suspected oncogenes involved in segmental glomerulosclerosis type 3 and Metabolic Syndrome (52–54), all of which potentially represent drug targets in disease therapy (Figure 3). In general, all interactors in the fusion networks were extended (‘preserved’) set of all interactors from both parental proteins (with the exception of missing interactors), as well as new interactors at occurrence of new protein fusion domains. For example, we can observe the new domains in the fusions as follows: ESTid=L22179.1 (the KMT2A/AFF1 fusion in ChiTaRS-3.1 (37)) has seven new domains (Actin, Vfa1, Ery_res_leader2, Filament_head, YL1, DUF3446 and HPS3_Mid), which were not in the parental proteins KMT2A and AFF1. Thus, DDCOS analysis by ChiPPI can be used to identify novel suspect oncogenes and potential drug targets, as ‘preserved’ interactors in the cancer fusion PPI networks (comparing to the parental PPI networks).
Figure 3.

The protein–protein interaction (PPI) networks of the BCR/ABL fusion and two parental proteins, which show alterations. (A) The ABL1 highly connected disease module. (B) The BCR highly connected disease module. (Note only the closest interactors are shown here). (C) The BCR/ABL1 PPI disease module contains five additional nodes: CRKL, JAK2, RASA1, CD2AP and INPPL1, where CRKL and RASA1 oncogenes are involved in the developing of CML, JAK2 is associated with acute myeloid leukemia (AML), and CD2AP and INPPL1 are suspected oncogenes, involved in segmental glomerulosclerosis type 3 and Metabolic Syndrome, respectively. Moreover, some genes are involved in mental diseases (colored in yellow), immunodeficiency (green), suspected oncogenes in few cancers (blue) and oncogenes (red).
Comparison between the parental PPI networks and the fusion PPI networks
The underlying question in studying cancer fusion proteins is: what is their carcinogenic function that is selected for? We applied ChiPPI to study how fusions can distort parental protein PPI networks, leading to cell deregulation. Compared to point mutations, insertions and deletions, it is clear that fusion events can delete functions from the protein (e.g. loss of a regulatory domain or interaction with an oncoprotein), as well as generate a partial fusion of the parental PPI networks, bringing together proteins that normally never meet (Figure 4).
Figure 4.

Cancer fusions make interactors of gene1 and gene2 to be ‘close’ interactors with shorter pathways. (A) Interactors of gene1 are depicted in blue and interactors of gene2 are in green. (B) Interactors of a fusion of gene1 and gene2. The red colored line shows the fusion of gene1 and gene2. (C) A shortest distance between interactors of gene1 and gene2 may have different distributions. (D) After a fusion event, the interactors of gene1 and gene2 have a shortest distance of 1.
To study how networks change between the parental and fusion networks, we used graph theory to represent the biological networks of the fusions and the following graph properties: the node degree, closeness and betweenness. We compared the networks of the 337 genes that participate in the 358 fusions (10) with those of 300 genes frequently mutated in cancer and, used as a control, a set of 300 randomly selected genes. Centrality analysis showed that fusion parental proteins tend to have much higher total degree centrality (P-value < 6.3E-6, the Wilcoxon signed-rank test) and higher betweenness centrality (P-value < 1.3E-4) than all genes in the control sets (Supplementary Table S8 and Supplementary Data). Thus, Figure 5 shows that parental genes found in different cancer phenotypes, e.g. leukemia, sarcoma and solid tumors, in ChiPPI networks have higher clustering coefficient than their corresponding fusions. Therefore, ChiPPI uncovers that parental genes in fusions are hubs in biological networks prior to the fusion event, as has also been suggested in a recent study by Latysheva et al. (32). Consequently, fusions reduce the radius of the protein interaction networks by connecting previously non-connected parts and reduce the network betweenness centrality.
Figure 5.

The centrality analysis for 337 parental genes in the 358 fusions in the training set (10). The analysis showed that the parental proteins of fusions tend to have much higher clustering coefficient (P-value < 2.7E-4, the Wilcoxon signed-rank test) than all other genes in the network. Moreover, parental genes found in leukemia, sarcoma and solid tumors in ChiPPI networks show a similar property, namely, parental genes are central nodes in the PPI networks.
We then asked whether fusion events in cancer act to deregulate the general organization of the cellular PPI network. We found that by connecting all 337 proteins involved in the 358 fusions in our training dataset, three large connected clusters of genes are formed, with each cluster being typically involved in a morphologically distinct tumor type (10). Interestingly, we found that these three clusters are connected in one highly joined cancer network with some nodes that are frequently found in many distinct PPI networks (Figure 6). Particularly, the Human DNA topoisomerase I (Top1) that is a validated target for the treatment of human cancers (colored in red, Figure 6) is the most connected gene in the network. Other most connected genes are, for example, found in AML (CREBBP, EP300, NPM1, Figure 6), in sarcoma (FUS, Figure 6), in renal cell carcinoma (NONO, Figure 6). As it can be seen from Figure 6, a large sub-network (left) includes hematological ETV6, IGH and NUP98 fusions, with the mainly lymphoma-associated BCL6 fusions as well as epithelial RET fusions (56). The upper right network (Figure 6) includes the lymphoma-associated ALK fusions, the carcinoma-associated transcription factor for IGHM, the enhancer 3 (TFE3) fusions and the sarcoma-associated EWSR1 fusions. The lower right network (Figure 6) contains mainly hematological MLL fusions connected to HMGA2 fusions that are typically found in solid tumors (9,10,56–59). Taken together, these results indicate that parental proteins in fusions are indeed central nodes in the PPI networks (32) and they produce together one highly connected large cancer PPI network that unify different cancer-associated nodes within different cancer phenotype.
Figure 6.

Among the 358 fusions, 90% of proteins form three clusters of proteins involved in morphologically distinct tumor types. Highly frequent 158 genes found in highly connected sub-networks of the 358 fusions. The dash line means the score is ‘NA’ because the involved genes are not characterized by well-known protein domains. The frequency is presented as a heatmap from a red color (highly frequent), to green (less frequent).
If a fusion event occurs between two proteins in non-connected PPI networks then we might expect that the two networks will now be brought close together (Figure 4). However, if both proteins are in the same PPI network (e.g. BCR and ABL), then the effect of a fusion event could be 2-fold: loss of interactions through deletion of protein domains in the fusion and enhanced activity of the fusion. Particularly, we used ChiPPI to systematically analyze network changes in known cancer fusion events. Specifically, we analyzed the parental proteins of 2632 fusions from the ChiTaRS-2.1 database (57) finding that 886 fusions (34%) incorporate parental genes which are already closely associated in the same PPI network, while the majority (66%) link previously non-associated networks (Supplementary Table S9 and 10 and Supplementary Data). We studied PPI networks of these fusions and identified potential ‘missing’ proteins from the networks (Supplementary Table S9 and Supplementary Data). Thus, we found that all fusions incorporating such parental proteins which are interactors, change PPI networks of parental proteins in order to reduce a number of interactors (e.g. radius) in the ChiPPI network in order to enhance activity of the fusions (Supplementary Table S10 and Supplementary Data).
Fusion networks frequently lose tumor suppressors
We speculated that the fusion of two proteins within the same PPI network has a dual function: the loss of interactors in the same PPI (e.g. removal of a tumor suppressor) (Supplementary Table S10 and 11 and Supplementary Data), while bringing closer other interactions (for example, enhancing the activity of a signaling pathway). However, when two non-connected PPI networks are joined by a fusion event it is not clear whether there will be loss of tumor suppressors from the fusion network. To test this, we used ChiPPI to rebuild the PPI networks of 11 528 fusion proteins (ChiTaRS-3.1 (37)) and analyzed the proteins frequently missing from the networks (using our previously described procedure). In most networks, we found that more than five tumor suppressors are missing from the interactors because of a loss of the functional domains in the fusions (Supplementary Table S1 and Supplementary Data). The most frequently lost tumor suppressors in all cancers are TP53, HDAC3, PML, SMAD3/SMAD2, RB1 and BRCA1 (Supplementary Table S1 and Supplementary Data) and in fusion networks this is also the case, indicating that fusion events might be specifically selected for due to the loss of the tumor suppressors. To directly compare the association of fusion events or other types of mutation to loss of tumor suppressors from PPI networks, we took 572 cancer genes from the COSMIC database (38) that are causally linked to cancer mutations and asked what changes occur to tumor suppressors in their corresponding PPI networks upon gene mutation (Supplementary Table S1 and 2 and Supplementary Data). We first mapped the PPI network for each wild-type protein, followed by mapping the cataloged mutations to specific protein domains within the 572 proteins. Finally, we built a new PPI network for the mutant protein by subtracting the mutant domain from the interaction map of the wild-type protein. The resultant maps for 572 mutant PPI networks revealed a loss of at least 10 interactors per protein due to mutated domains (Supplementary Table S9 and 10 and Supplementary Data). While analyzing the predicted or known function of these lost interactors we found that 25% percent were classified as tumor suppressors, significantly different from the random controls (FDR< 1%, P-value ≤ 3E-4, the Wilcoxon signed-rank test). Further, the rate of loss of tumor suppressors from mutant gene PPI networks was found to be similar to that of fusion gene PPI networks (25% compared to 20%, respectively). Taken together, these results indicate that while the mechanistic aspect of fusion events as well as other forms of deleterious mutation in cancer appears to be complex, the same final result is observed, namely, the loss of tumor suppressors from the PPI networks.
Pathway enrichment of ChiPPI networks delineates between leukemia, sarcoma and solid tumor
Having shown that fusions dramatically skew PPI networks, we next asked if these network aberrations are associated with particular signaling pathways in different cancer types. First, we classified all 358 fusions (10) into three disease types: leukemia/lymphoma, sarcoma or solid tumors. This resulted in 238 characterized fusions for leukemia, 51 for sarcoma and 69 for solid tumors (in our training set). Then we performed a pathway enrichment analysis of all the interactors for every ChiPPI network, in order to identify over-represented (cancer-associated) pathways. Next, we analyzed the remaining fusions in ChiPPI (∼11 000) and their PPI network members, for pathway enrichment, finding that once again, three distinct groups are formed. While certain pathways are enriched in all three diseases (Wnt, Notch and TGF β), there are distinct patterns for leukemia (p53 pathway, EGFR signaling, DNA replication and CCKR signaling), for sarcoma (p53 pathway and CCKR signaling) and solid tumors (FGFR and EGFR signaling) (P-values are all < 1.0E-6) (Figure 7, Supplementary Figure S1–8 and Supplementary Data), leading us to hypothesize that (i) fusion events are selected for based on specific pathway activation, and (ii) different cancer types rely on specific combinations of altered pathways. Reassuringly, pathway enrichment analyzes of fusions using ChiPPI closely matches previous pathway enrichment studies (33). Thus, in leukemia KEGG analysis revealed Notch, TGF-β, ErbB, MAPK, Jak- STAT, Wnt, T-cell receptor and B-cell receptor pathway activation (58,59) (Figure 7). Particularly, Supplementary Figure S9 represents the enrichment results for fusion transcripts from different leukemia and lymphoma types that shows unique profiles for the leukemia and lymphoma sub-types. Taken together, these results indicate that pathway enrichment specific for fusions follows a similar pattern to total pathway activity in the studied cancer types.
Figure 7.

The comparison between the pathways enrichment in leukemia/lymphoma, sarcoma and solid tumors. There is a different pattern of the enrichment. The pathways enriched in leukemia are: Wnt Signaling (P-value = 2.24E-16), p53 pathway (P-value = 1.95E-10), Notch signaling (P-value = 2.95E-09), EGFR signaling (P-value = 2.31E-08), DNA replication (P-value = 9.81E-07), TGF-β signaling (P-value = 1.95E-06) and CCKR signaling (P-value = 2.30E-06). The pathways enriched in sarcoma are: Notch signaling (P- value = 8.48E-14), TGF-β signaling (P-value = 3.01E-10), p53 pathway (P-value = 7.31E-09), Wnt signaling (P-value = 2.32E-08) and CCKR signaling (P-value = 8.48E-07). The pathways enriched in solid tumors are: FGFR signaling (P-value = 6.32E-15), EGFR signaling (P-value = 2.13E-11), TGF-β signaling (P- value = 2.20E-07), Wnt signaling (P-value = 2.24E-06) and Notch signaling (P-value = 8.48E-06).
DISCUSSION AND CONCLUSION
Overview
In this paper we present ChiPPI, (Figure 8, The ChiPPI interface), a novel approach implemented in an openly accessible web-server, for performing a global analysis of changes to PPI networks upon protein fusion events in cancer. We have analyzed the largest number of fusions known today (11 528 fusions, ChiTaRS-3.1) (37). We have compared the PPI networks of fusion proteins with the protein PPI networks of both parental proteins, and have mapped additional (non-fusion) cancer mutations to parental proteins or to other proteins that coincide in the fusion protein PPI network. This computational methodology successfully identifies the changes induced to the known chimeric PPI networks. For the specific set of protein fusion events unique to cancer cells the methodology described here appears to account for most of the known protein interaction perturbations upon gene fusion. While it is possible, and even likely, that we have ruled out certain real changes to PPI networks due to the presence or absence of promiscuous domains in the fusion proteins. This model can provide novel insight on cancer cell physiology. For example, using ChiPPI analysis of over 2000 fusions we were able to identify missing tumor suppressors from the fusion networks. Thus, ChiPPI provides a predictive model for demonstrating how fusions may act as drivers of cancer by reducing or losing interactions with tumor suppressors in PPI networks.
Figure 8.

The interface of ChiPPI with querying and analysis features. The fusion networks can be searched using whatever two protein names or fusion IDs. The interactions can be visualized in the form of different layouts like Tree, Circle, Radial and Force Directed. Various network properties can be studied, such as a number of interacting pairs, oncoproteins, affected interactors, missing interactors, the diameter, clustering coefficient and centrality measures. Finally, for each interaction, cross-links are present for databases like ChiTaRS, Pfam, InterPro and SMART. The statistics part and the network structure information also appears on the ChiPPI website at the bottom.
Comparison to other methods
To verify the DDCOS scoring system of ChiPPI, we compared it with IDDI (60), Domine (61) and iPFAM (62). The difference between our ChiPPI method and other methods is that we predict interactors of fusions while the other methods predict interactors of normal proteins only (63). We found that ChiPPI includes ten times more DDCOS cases than other methods. The reason being, that IDDI has ∼204 705 domain–domain interactions (last updated in May, 2011) while Domine has 20 513 domain–domain interactions (last updated in September, 2010). In ChiPPI, we used the DDCOS table of size equals to 9380 * (9380−1)/2 = 43 987 510 domain–domain interactions, where 9380 is amount of various unique domains for all interactors from BioGrid (39). Thus, by comparison, ChiPPI has more domain–domain interactions (∼200-fold more than IDDI), giving it its distinct advantage over other methods (Table 1). Taking into consideration normal proteins, we found that our scoring system is in good agreement with these three published methods (Pearson correlation coefficient is 0.57, 0.56 and 0.64 for the top 100 scores of IDDI) (60), Domine (61) and iPFAM (62) respectively (Table 1, Supplementary Table S12 and Supplementary Data). Those results indicate that ChiPPI is a reliable tool for studying PPI events for normal as well as for fusion networks.
Table 1. The methods comparison with ChiPPI.
| ChiPPI (last updated September, 2016) | IDDI (last updated May, 2011) | DOMINE (last updated September, 2010) | iPFAM (last updated August, 2013) | Latysheva et al. (27) | |
|---|---|---|---|---|---|
| Domain–domain Interactions | 43 987 510 | 204 705 | 20 513 | 9500 | <1000 |
| Number of domains | 16 953 | 7351 | 5410 | 16 640 | <2000 |
| Number of fusions | 23 141 | NO | NO | NO | 2699 |
Mapping PPI networks of cancer fusions
We applied ChiPPI to the analysis of 11 528 fusions from ChiTaRS-3.1 (37), accurately mapping alterations in network properties that delineate the fusion protein network from parental networks. We ranked the PPI-pathway associations with respect to a distribution obtained for the real interactions versus random datasets. Based on these data, we compared cellular pathways ‘enrichment’ in leukemia/lymphoma, sarcoma and solid tumors, for each cancer fusion protein. While a set of core pathways is enriched in all three cancer types, i.e. Wnt, Notch and TGF β, there is also a set of pathways specifically affected by fusions, in leukemia, i.e. p53 pathway, EGFR signaling, DNA replication and CCKR signaling; sarcoma, i.e. p53 pathway and CCKR signaling; and solid tumors, i.e. FGFR and EGFR signaling. Our findings fit well with previous observations on pathways enrichment in cancer based on the analysis of mutation load or altered gene expression (33,47,55,59,64) leading us to speculate that both fusion events and other types of alterations, including mutation and gene expression alterations, are similarly selected for in cancer and they might be mediated by network aberrations affecting similar pathways. Finally, using ChiPPI, we found that tumor suppressors are lost from fusion networks, while oncoproteins are maintained and even brought into closer proximity with other network members. Therefore, ChiPPI provides a comprehensive method for analyzing protein fusion networks and can be successfully utilized to uncover dramatic changes in the cancer cellular PPI networks.
Detailed investigation of individual fusion events are essential to distinguish other effects such as changes in tissue specificity (65) that may give rise to new functions that are not present in the individual parental proteins, or to determine whether the fusion is simply an elaborate way of introducing a truncating stop codon. To conclude, the ChiPPI results suggest a new role for fusions proteins as hubs in cancer interaction networks, confirming previous studies (27,66). Identifying drugable targets in the cancer networks skewed by the introduction of fusion proteins may lead to the discovery of new fusion-specific anti-cancer drugs in the future.
AVAILABILITY
Supplementary Material
Footnotes
Present addresses:
Vaishnovi Sekar, Department of Biology, Lund University, Lund, 22362, Sweden.
Alfonso Valencia, Barcelona Supercomputing Center (BSC), Institució Catalana de Recerca i Estudis Avançats (ICREA), 08010 Barcelona, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work is funded by the Project Retos BFU2015-71241-R of the Spanish Ministry of Economy, Industry and Competitiveness (MEIC), co-funded by European Regional Development Fund (ERDF) and by the Project PT13/0001/0030, Instituto de Salud Carlos III (ISCIII), Strategic Action in Health, co-funded by European Regional Development Fund (ERDF). The work of MFM is supported by the Israel Cancer Association (ICA) fund, the work of ST is supported by the VaTaT Postdoctoral Fellowship for excellent students [22351, 20027, 26912]. AV is supported by the Joint BSC-CRG-IRB Programme in Computational Biology. Funding for open access charge: ICA [e-cancer-diagnosis].
Conflict of interest statement. None declared.
REFERENCES
- 1. Taki T., Taniwaki M.. Chromosomal translocations in cancer and their relevance for therapy. Curr. Opin. Oncol. 2006; 18:62–68. [DOI] [PubMed] [Google Scholar]
- 2. Mertens F., Johansson B., Fioretos T., Mitelman F.. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015; 15:371–381. [DOI] [PubMed] [Google Scholar]
- 3. Mertens F., Antonescu C.R., Mitelman F.. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer. 2016; 55:291–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bentz M., Cabot G., Moos M., Speicher M.R., Ganser A., Lichter P., Döhner H.. Detection of chimeric BCR-ABL genes on bone marrow samples and blood smears in chronic myeloid and acute lymphoblastic leukemia by in situ hybridization. Blood. 1994; 83:1922–1928. [PubMed] [Google Scholar]
- 5. Roeder I., Horn M., Glauche I., Hochhaus A., Mueller M.C., Loeffler M.. Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implications. Nat. Med. 2006; 12:1181–1184. [DOI] [PubMed] [Google Scholar]
- 6. Maher C.A., Palanisamy N., Brenner J.C., Cao X., Kalyana-Sundaram S., Luo S., Khrebtukova I., Barrette T.R., Grasso C., Yu J. et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:12353–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M.. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009; 458:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kannan K., Wang L., Wang J., Ittmann M.M., Li W., Yen L.. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:9172–9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mitelman F., Mertens F., Johansson B.. Prevalence estimates of recurrent balanced cytogenetic aberrations and gene fusions in unselected patients with neoplastic disorders. Genes Chromosomes Cancer. 2005; 43:350–366. [DOI] [PubMed] [Google Scholar]
- 10. Mitelman F., Johansson B., Mertens F.. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007; 7:233–245. [DOI] [PubMed] [Google Scholar]
- 11. Shih Y.K., Parthasarathy S.. Identifying functional modules in interaction networks through overlapping Markov clustering. Bioinformatics. 2012; 28:i473–i479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brayer K.J., Segal D.J.. Keep your fingers off my DNA: protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem. Biophys. 2008; 50:111–131. [DOI] [PubMed] [Google Scholar]
- 13. Buljan M., Chalancon G., Eustermann S., Wagner G.P., Fuxreiter M., Bateman A., Babu M.M.. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell. 2012; 46:871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ellis J.D., Barrios-Rodiles M., Colak R., Irimia M., Kim T., Calarco J.A., Wang X., Pan Q., O’Hanlon D., Kim P.M. et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell. 2012; 46:884–892. [DOI] [PubMed] [Google Scholar]
- 15. Goldberg D.S., Roth F.P.. Assessing experimentally derived interactions in a small world. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:4372–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoffmann R., Krallinger M., Andres E., Tamames J., Blaschke C., Valencia A.. Text mining for metabolic pathways, signaling cascades, and protein networks. Sci. STKE. 2005; 2005:pe21. [DOI] [PubMed] [Google Scholar]
- 17. Valencia A., Pazos F.. Computational methods for the prediction of protein interactions. Curr. Opin. Struct. Biol. 2002; 12:368–373. [DOI] [PubMed] [Google Scholar]
- 18. Yeger-Lotem E., Sattath S., Kashtan N., Itzkovitz S., Milo R., Pinter R.Y., Alon U., Margalit H.. Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:5934–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang L.V., King O.D., Wong S.L., Goldberg D.S., Tong A.H., Lesage G., Andrews B., Bussey H., Boone C., Roth F.P.. Motifs, themes and thematic maps of an integrated Saccharomyces cerevisiae interaction network. J. Biol. 2005; 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beltrao P., Kiel C., Serrano L.. Structures in systems biology. Curr. Opin. Struct. Biol. 2007; 17:378–384. [DOI] [PubMed] [Google Scholar]
- 21. Juan D., Pazos F., Valencia A.. High-confidence prediction of global interactomes based on genome-wide coevolutionary networks. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mazurie A., Bottani S., Vergassola M.. An evolutionary and functional assessment of regulatory network motifs. Genome Biol. 2005; 6:R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stirnimann C.U., Petsalaki E., Russell R.B., Müller C.W.. WD40 proteins propel cellular networks. Trends Biochem. Sci. 2010; 35:565–574. [DOI] [PubMed] [Google Scholar]
- 24. Frenkel-Morgenstern M., Valencia A.. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 2012; 28:i67–i74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Akavia U.D., Litvin O., Kim J., Sanchez-Garcia F., Kotliar D., Causton H.C., Pochanard P., Mozes E., Garraway L.A., Pe’er D.. An integrated approach to uncover drivers of cancer. Cell. 2010; 143:1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang X.S., Prensner J.R., Chen G., Cao Q., Han B., Dhanasekaran S.M., Ponnala R., Cao X., Varambally S., Thomas D.G. et al. An integrative approach to reveal driver gene fusions from paired-end sequencing data in cancer. Nat. Biotechnol. 2009; 27:1005–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Latysheva N.S., Babu M.M.. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016; 44:4487–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abate F., Zairis S., Ficarra E., Acquaviva A., Wiggins C.H., Frattini V., Lasorella A., Iavarone A., Inghirami G., Rabadan R.. Pegasus: a comprehensive annotation and prediction tool for detection of driver gene fusions in cancer. BMC Syst. Biol. 2014; 8:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Höglund M., Frigyesi A., Mitelman F.. A gene fusion network in human neoplasia. Oncogene. 2006; 25:2674–2678. [DOI] [PubMed] [Google Scholar]
- 30. Vandin F., Upfal E., Raphael B.J.. Algorithms for detecting significantly mutated pathways in cancer. J. Comput. Biol. 2011; 18:507–522. [DOI] [PubMed] [Google Scholar]
- 31. Wu C.C., Kannan K., Lin S., Yen L., Milosavljevic A.. Identification of cancer fusion drivers using network fusion centrality. Bioinformatics. 2013; 29:1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Latysheva N.S., Oates M.E., Maddox L., Flock T., Gough J., Buljan M., Weatheritt R.J., Babu M.M.. Molecular principles of gene fusion mediated rewiring of protein interaction networks in cancer. Mol. Cell. 2016; 63:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Memišević V., Wallqvist A., Reifman J.. Reconstituting protein interaction networks using parameter-dependent domain-domain interactions. BMC Bioinformatics. 2013; 14:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gupta S., Wallqvist A., Bondugula R., Ivanic J., Reifman J.. Unraveling the conundrum of seemingly discordant protein-protein interaction datasets. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2010; 2010:783–786. [DOI] [PubMed] [Google Scholar]
- 35. Apic G., Gough J., Teichmann S.A.. Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J. Mol. Biol. 2001; 310:311–325. [DOI] [PubMed] [Google Scholar]
- 36. Itzhaki Z., Akiva E., Altuvia Y., Margalit H.. Evolutionary conservation of domain-domain interactions. Genome Biol. 2006; 7:R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gorohovski A., Tagore S., Palande V., Malka A., Raviv-Shay D., Frenkel-Morgenstern M.. ChiTaRS-3.1—the enhanced chimeric transcripts and RNA-seq database matched with protein–protein interactions. Nucleic Acids Res. 2017; 45:D790–D795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Forbes S.A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C.G., Ward S., Dawson E., Ponting L. 2017 et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017; 45:D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chatr-Aryamontri A., Breitkreutz B.J., Oughtred R., Boucher L., Heinicke S., Chen D., Stark C., Breitkreutz A., Kolas N., O’Donnell L. et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015; 43:D470–D478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mi H., Muruganujan A., Casagrande J.T., Thomas P.D.. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013; 8:1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bateman A., Coin L., Durbin R., Finn R., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E. et al. The Pfam protein families database. Nucleic Acids Res. 2004; 32:D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Finn R.D., Tate J., Mistry J., Coggill P.C., Sammut S.J., Hotz H.R., Ceric G., Forslund K., Eddy S.R., Sonnhammer E.L. et al. The Pfam protein families database. Nucleic Acids Res. 2008; 36:D281–D288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dinkel H., Van Roey K., Michael S., Kumar M., Uyar B., Altenberg B., Milchevskaya V., Schneider M., Kühn H., Behrendt A. et al. ELM 2016–data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 2016; 44:D294–D300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gould C.M., Diella F., Via A., Puntervoll P., Gemünd C., Chabanis-Davidson S., Michael S., Sayadi A., Bryne J.C., Chica C. et al. ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010; 38:D167–D180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ben-Hamo R., Efroni S.. MicroRNA regulation of molecular pathways as a generic mechanism and as a core disease phenotype. Oncotarget. 2015; 6:1594–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sprinzak E., Margalit H.. Correlated sequence-signatures as markers of protein-protein interaction. J. Mol. Biol. 2001; 311:681–692. [DOI] [PubMed] [Google Scholar]
- 49. Sprinzak E., Sattath S., Margalit H.. How reliable are experimental protein-protein interaction data. J. Mol. Biol. 2003; 327:919–923. [DOI] [PubMed] [Google Scholar]
- 50. Sprinzak E., Altuvia Y., Margalit H.. Characterization and prediction of protein-protein interactions within and between complexes. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:14718–14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Finn R.D., Clements J., Arndt W., Miller B.L., Wheeler T.J., Schreiber F., Bateman A., Eddy S.R.. HMMER web server: 2015 update. Nucleic Acids Res. 2015; 43:W30–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Panagopoulos I., Isaksson M., Lindvall C., Hagemeijer A., Mitelman F., Johansson B.. Genomic characterization of MOZ/CBP and CBP/MOZ chimeras in acute myeloid leukemia suggests the involvement of a damage-repair mechanism in the origin of the t(8;16) (p11;p13). Genes Chromosomes Cancer. 2003; 36:90–98. [DOI] [PubMed] [Google Scholar]
- 53. Panagopoulos I., Kitagawa A., Isaksson M., Mörse H., Mitelman F., Johansson B.. MLL/GRAF fusion in an infant acute monocytic leukemia (AML M5b) with a cytogenetically cryptic ins (5;11) (q31;q23q23). Genes Chromosomes Cancer. 2004; 41:400–404. [DOI] [PubMed] [Google Scholar]
- 54. Brehme M., Hantschel O., Colinge J., Kaupe I., Planyavsky M., Köcher T., Mechtler K., Bennett K.L., Superti-Furga G.. Charting the molecular network of the drug target Bcr-Abl. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:7414–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ben-Hamo R., Gidoni M., Efroni S.. PhenoNet: identification of key networks associated with disease phenotype. Bioinformatics. 2014; 30:2399–2405. [DOI] [PubMed] [Google Scholar]
- 56. Cierpicki T., Grembecka J.. Targeting protein-protein interactions in hematologic malignancies: still a challenge or a great opportunity for future therapies. Immunol. Rev. 2015; 263:279–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frenkel-Morgenstern M., Gorohovski A., Vucenovic D., Maestre L., Valencia A.. ChiTaRS 2.1-an improved database of the chimeric transcripts and RNA-seq data with novel sense-antisense chimeric RNA transcripts. Nucleic Acids Res. 2015; 43:D68–D75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jamil K., Jayaraman A., Rao R., Raju S.. In silico evidence of signaling pathways of notch mediated networks in leukemia. Comput. Struct. Biotechnol. J. 2012; 1:e201207005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Platzer A., Perco P., Lukas A., Mayer B.. Characterization of protein-interaction networks in tumors. BMC Bioinformatic. 2007; 8:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim Y., Min B., Yi G.S.. IDDI: integrated domain-domain interaction and protein interaction analysis system. Proteome Sci. 2012; 10:S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yellaboina S., Tasneem A., Zaykin D.V., Raghavachari B., Jothi R.. DOMINE: a comprehensive collection of known and predicted domain-domain interactions. Nucleic Acids Res. 2011; 39:D730–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Finn R.D., Miller B.L., Clements J., Bateman A.. iPfam: a database of protein family and domain interactions found in the Protein Data Bank. Nucleic Acids Res. 2014; 42:D364–D373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee H., Deng M., Sun F., Chen T.. An integrated approach to the prediction of domain-domain interactions. BMC Bioinformatics. 2006; 7:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Efroni S., Ben-Hamo R., Edmonson M., Greenblum S., Schaefer C.F., Buetow K.H.. Detecting cancer gene networks characterized by recurrent genomic alterations in a population. PLoS ONE. 2011; 6:e14437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bossi A., Lehner B.. Tissue specificity and the human protein interaction network. Mol. Syst. Biol. 2009; 5:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Latysheva N.S., Flock T., Weatheritt R.J., Chavali S., Babu M.M.. How do disordered regions achieve comparable functions to structured domains. Protein Sci. 2015; 24:909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.