


Abstract
Serine integrases are bacteriophage enzymes that carry out site-specific integration and excision of their viral genomes. The integration reaction is highly directional; recombination between the phage attachment site attP and the host attachment site attB to form the hybrid sites attL and attR is essentially irreversible. In a recent model, extended coiled-coil (CC) domains in the integrase subunits are proposed to interact in a way that favors the attPxattB reaction but inhibits the attLxattR reaction. Here, we show for the Listeria innocua integrase (LI Int) system that the CC domain promotes self-interaction in isolated Int and when Int is bound to attachment sites. Three independent crystal structures of the CC domain reveal the molecular nature of the CC dimer interface. Alanine substitutions of key residues in the interface support the functional significance of the structural model and indicate that the same interaction is responsible for promoting integration and for inhibiting excision. An updated model of a LI Int•attL complex that incorporates the high resolution CC dimer structure provides insights that help to explain the unusual CC dimer structure and potential sources of stability in Int•attL and Int•attR complexes. Together, the data provide a molecular basis for understanding serine integrase directionality.
INTRODUCTION
Serine integrases are bacteriophage enzymes that carry out site-specific integration and excision of their viral genomes using a serine recombinase (SR) mechanism (1,2). These enzymes are currently used in a variety of applications ranging from genome engineering to construction of biocomputers (3–5). The integration and excision reactions are remarkable because of their simplicity and high level of directionality. The serine integrases recombine substrates containing a ∼50 bp phage attachment site (attP) and a ∼40 bp bacterial site (attB) in a variety of topological contexts, with no requirements for accessory proteins or auxiliary DNA sequences. This property is reminiscent of the tyrosine recombinases Cre and Flp (6), but is quite different from the bacteriophage λ integrase, which requires a 240 bp supercoiled attP site and the Escherichia coli integration host factor to carry out the attPxattB integration reaction (7).
Despite their simplicity, the serine integrases display a high level of directionality. The attL and attR sites generated following integration are not recombined to form attP and attB sites by the integrase protein alone; a phage-encoded recombination directionality factor (RDF) is required for the excision reaction to proceed (8–13). The question of how serine integrases are able to achieve this level of regulation with simple attachment sites has motivated genetic, biochemical and structural investigations of these systems (reviewed in (1,14)). The structure of a Listeria phage integrase carboxy-terminal domain (CTD) bound to an attP half-site has provided insight into how the enzyme recognizes attachment sites and how the integrase domains are organized on those sites (15). Together with the results of biochemical studies in several serine integrase systems, these structural data have led to a model in which interactions between DNA-bound integrase domains are responsible for promoting integration and preventing excision in the absence of the RDF.
The current model for understanding how directionality is regulated by the serine integrases is shown in its simplest form in Figure 1. The key structural element is an extended, antiparallel coiled-coil (CC) domain that is flexibly attached to each of the four integrase subunits involved in recombination. During the integration reaction, the CC domains from attP-bound integrases interact with the CC domains from attB-bound integrases to facilitate association and stabilization of a synaptic complex where recombination can be initiated. Following recombination, the distinct domain organization on attP-derived half-sites (P and P’) versus attB-derived half-sites (B and B’) facilitates intra-molecular interactions between CC domains on attL and attR. These interactions prevent recombination between attL and attR sites, making the integration reaction effectively irreversible. RDF proteins are thought to function by disrupting the CC interactions on attL and attR to facilitate excision, but little is currently known about RDF structures or the nature of the interactions between RDFs and the DNA-bound integrases.
Figure 1.

Serine integrase domain structure and model for control of recombination directionality. (A) Domain structure of LI integrase. NTD: N-terminal catalytic domain; αE: conserved helix mediating oligomerization, DNA-binding and subunit rotation during recombination; RD: recombinase domain, ZD: zinc ribbon domain; CC: coiled-coil motif. The CC motif is embedded in the ZD. See Supplementary Figure S1 for a comparison to other serine integrases. (B) Model for control of recombination directionality. In the integration reaction, Int binds as a dimer to attP and attB sites and associates the sites using Int–Int interactions involving the NTDs and the CC motifs. The attP half-sites are labeled P and P’ and the attB half-sites B and B’. Recombination involves double-strand cleavage of the sites, 180° relative rotation of the P’ and B’ half-sites about a horizontal axis through the center of the complex, and re-ligation of the DNA segments (see references (1,42) for reaction schemes showing these steps). The resulting attL and attR sites have a spatial arrangement of integrase domains that allows formation of intramolecular interactions between CC motifs. The reversal of the integration reaction is thereby prevented in the absence of a phage-encoded RDF protein (see text). This model requires flexibly linked CC motifs that can adopt distinct trajectories, as illustrated schematically here.
A central tenet of the model shown in Figure 1 is that the CC domains self-interact. The crystal structure of the CTD from Listeria innocua integrase (LI Int) bound to DNA revealed that the CC domain is flexibly linked to the zinc ribbon domain (ZD) of the integrase and does not interact with the DNA (15). The CC domains are poorly resolved in the four independent copies of the complex present in the crystal structure and there are no CC self-interactions in the crystal lattice that can be easily interpreted. Biochemical data supporting a CC self-interaction comes from the φC31 integrase system, where a maltose binding protein fusion to a region containing the predicted CC region of the integrase was an apparent dimer based on its gel-filtration retention volume (16). However, the purified CTDs of the A118, φC31 and Bxb1 integrases were reported to be monomeric based on similar criteria, arguing against a strong CC self-interaction (17–19).
Here, we use the LI Int model system (15,18) to probe the structure and function of the CC motif. We chose LI Int for this study because structural data are available for the CTD bound to att-site DNA and structure-based models of how the CC might function have been proposed (15). We show that the CC domain promotes dimerization in the context of full-length Int, the Int CTD and in isolation. Three independent crystal structures of the CC domain reveal the molecular nature of the CC dimer interface, providing both redundant and high resolution models to design experiments. Functional analyses of alanine substitutions at key residues in the CC dimer interface provide support for both the significance of the structural model and for the idea that the same interface is responsible for promoting integration and for inhibiting excision. A striking feature of the CC dimer is its lack of symmetry. The helical domains do not self-interact via a dyad of symmetry, but instead form a front-to-back interface. An updated model of a LI Int•attL complex that incorporates the high resolution CC dimer structure explains the need for an asymmetric dimer. Together, these findings help to explain why Int•attL and Int•attR complexes are inert to recombination and will be useful in designing experiments to test the mechanism of action of the phage RDFs.
MATERIALS AND METHODS
Strains, plasmids and reagents
Strains BW25113 (F-, Δ(araD-araB)567, lacZ4787(del)::rrnB-3, LAM-, rph-1, Δ(rhaD-rhaB)568, hsdR514) and CSH142 (F−, ara-600 Δ(gpt-lac)5 LAM- relA1 spoT1 thi-1) were obtained from the Coli Genetic Stock Center (Yale University). Buffer pH values were measured at 25°C. The Listeria innocua prophage-derived att site sequences used in recombination assays are the same as in (15). A table of plasmids used in this study is provided as Supplementary Table S1.
Purification of LI Int and LI Int CTD
Full-length integrase variants were sub-cloned into the NdeI and XhoI sites of pACYCBad1 (15) and expressed in strain BW25113 for 5 h at 20°C in 2xYT medium supplemented with 2 mM MgSO4 and 10 μM ZnSO4 after inducing with 1 mM l-arabinose. Bacterial cells from 2 to 3 l culture were lysed and the integrase purified as previously described (15), except the final sizing buffer was 20 mM tris(hydroxymethylamino) methane chloride (Tris•HCl), pH 7.4, 100 mM ammonium sulfate, 0.5 M NaCl and 10 mM 2-mercaptoethanol. Int CTD variants were cloned into the NdeI and XhoI sites of pET29b or pCDFDuet and expressed without affinity tags in strain BL21(DE3) as described above for full-length integrase, but with 100 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) induction. CTDs were purified using the Int protocol as modified above, with small adjustments to the SP-sepharose and hydroxyapatite gradients. Biophysical analyses of Int and Int CTD variants were performed in 20 mM Tris•HCl pH 7.0, 100 mM ammonium sulfate, 500 mM NaCl and 1 mM tris (2-carboxyethyl)phosphine (TCEP), unless otherwise stated.
Purification of LI Int CC domains
Int CC349-400, CC350-399 and CC345-405 were sub-cloned into pCDFDuet (Novagen) in-frame with an N-terminal His6-FLAG-Smt3 affinity tag and expressed in strain BL21(DE3) with 2xYT medium for 3 h at 37°C after inducing with 100 μM IPTG. Cells from 2 to 3 l culture were lysed using an Avestin homogenizer in 20 mM sodium/potassium phosphate, 300 mM NaCl and 10 mM imidazole, pH 7.0, with protease inhibitors. Cleared lysates were purified on Ni-NTA affinity resin (Qiagen), eluting with 20 mM sodium/potassium phosphate, 300 mM NaCl and 250 mM imidazole, pH 8. The His6-FLAG-Smt3-CC fusion was purified on an 8 ml MonoQ column (GE Healthcare), followed by overnight digestion at 4° with His6-Ulp1 protease (LifeSensors) and dialysis to 20 mM TrisCl, pH 7.4, 300 mM NaCl. The affinity tag and protease were removed by passage through a second Ni-NTA column. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry confirmed the approximate molecular mass of the CC domain. The protein was concentrated, aliquoted, frozen in liquid nitrogen in 20 mM Tris–Cl pH 7.4, 300 mM NaCl and 10% glycerol, and stored at −80°C. Biophysical analyses were performed in 20 mM Tris–Cl pH 7.4 and 300 mM NaCl.
Sedimentation equilibrium (SE)
Analytical ultracentrifugation experiments were performed with an XL-A analytical ultracentrifuge (Beckman-Coulter) and a TiAn60 rotor with six channel charcoal-filled epon centerpieces and quartz windows. SE data were collected at 4°C with detection at 280 nm for 2–5 sample concentrations. Analyses were carried out using global fits to data acquired at multiple speeds for each concentration with strict mass conservation using the program SEDPHAT (20). Error estimates for equilibrium constants were determined from a 1000-iteration Monte Carlo simulation. The partial specific volume (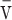 ), solvent density (ρ) and viscosity (η) were derived from chemical composition by SEDNTERP (21). For wild-type Int, the best fit to four concentrations and five rotor speeds (20 distributions) gave Kd = 32 ± 1 nM. However, Kd values down to 0.1 nM also gave acceptable fits, indicating that this value is not well-determined at the concentrations required for protein detection. Since the fits become poor at values higher than 32 nM, we regard this as an estimate of the upper limit for the dimerization Kd.
), solvent density (ρ) and viscosity (η) were derived from chemical composition by SEDNTERP (21). For wild-type Int, the best fit to four concentrations and five rotor speeds (20 distributions) gave Kd = 32 ± 1 nM. However, Kd values down to 0.1 nM also gave acceptable fits, indicating that this value is not well-determined at the concentrations required for protein detection. Since the fits become poor at values higher than 32 nM, we regard this as an estimate of the upper limit for the dimerization Kd.
Size-exclusion chromatography and multi-angle light scattering (SEC-MALS)
Absolute molecular weights were determined by multi-angle light scattering coupled with refractive interferometric detection (Wyatt Technology Corporation) and a Superdex 200 Increase 5/150 GL column (G.E.Healthcare) at 25°C, as previously described (22).
Structure determination
Crystals of CC-I (LI Int345-405) and CC-II (LI Int350-399) were obtained by hanging drop vapor-diffusion at 21°C at 20–25 mg/ml in 20 mM Tris, pH 7.4, 300 mM NaCl, and 0.8–1.3 M sodium citrate and cryo-protected in 1.3 M sodium citrate (23). Crystals of CC-I grown at 21°C were long rods that diffracted poorly (∼6–8 Å), but growth at 4°C yielded trigonal crystals that diffracted well. Construct CC-III (LI Int349-400) was crystallized at 21°C by hanging drop vapor diffusion at 20–25 mg/ml in 2.0 M ammonium sulfate, 100 mM HEPES pH 7.5 and 5% glycerol. The crystals were cryo-protected in a reservoir solution that additionally contained 20% sucrose, flash-frozen and stored in liquid nitrogen prior to diffraction experiments.
Diffraction data were collected at the Advanced Light Source beam line 5.0.3 or the Cornell High Energy Synchrotron Source beam line F1 at 100K and data were processed using either HKL3000 (24) or MOSFLM (25). Analysis of the diffraction data from the trigonal CC-I construct revealed severe twinning (law -h, -k, l; fraction 0.48). The structure was determined by molecular replacement using a CC motif from the LI Int CTD structure (PDB code: 4KIS; chain A), revealing four subunits in the asymmetric unit. Tetragonal crystal forms II and III are isomorphous and each is a nearly perfect merohedral twin (law h, -k, -l; fraction 0.48) with eight CC subunits per asymmetric unit. After refinement of CC-I, a single CC domain was used to phase CC-II by molecular replacement. Following refinement of CC-II, the CC-III structure was determined by the same molecular replacement procedure and could also be refined with the CC-II coordinates as a starting point. Molecular replacements were performed with Phaser (26), models were adjusted with COOT (27) and the structures refined with PHENIX (26). A summary of diffraction data and refinement results is given in Table 2. Coordinates for each of the three structures have been deposited in the Protein Data Bank with codes 5UAE, 5UDO and 5U96 for CC-I, CC-II and CC-III, respectively.
Table 2. Summary of X-ray data processing and refinement statistics.
| CC-I | CC-II | CC-III | |
|---|---|---|---|
| Residues | 345–405 | 350–399 | 349–400 |
| PDB code | 5UAE | 5UDO | 5U96 |
| Beamline | CHESS F1 | CHESS F1 | ALS 5.0.3 |
| HKL3000 | HKL3000 | MOSFLM | |
| Wavelength (Å) | 0.9760 | 0.9760 | 1.00 |
| Temperature (K) | 100 | 100 | 100 |
| Resolution (Å) (outer shell) | 35.42–2.75 | 48.94–2.36 | 35.7–1.95 |
| (2.848–2.749) | (2.44–2.36) | (2.02–1.95) | |
| Space Group | P31 | P41 | P41 |
| Unit cell (Å) | a = b = 75.4 c = 103.1 | a = b = 97.9 c = 52.7 | a = b = 97.2 c = 52.6 |
| Total reflections | 69 598 | 80 952 | 71 799 (7035) |
| Unique reflections | 17 032 (1722) | 20 864 (2030) | 36 021 (3588) |
| Multiplicity | 4.1 (3.7) | 4.1 | 7.3 (4.5) |
| Completeness (%) | 99.7 (99.0) | 99.90 (99.4) | 99.9 (100) |
| Mean I/sigma(I) | 15.3 (1.8) | 14.6 (2.4) | 21.4 (2.2) |
| Wilson B-factor (Å2) | 91.1 | 67.0 | 31.5 |
| R-merge (%) | 5.2 (62.3) | 6.5 (47.2) | 1.9 (35.3) |
| Reflections used for R-free | 1722 (10%) | 2029 (9.7%) | 1996 (5.5%) |
| R-work | 0.2133 (0.3808) | 0.2111 (0.4302) | 0.1991 (0.3634) |
| R-free | 0.2450 (0.3949) | 0.2283 (0.4137) | 0.2144 (0.3781) |
| Twin Law | -h, -k, l | h, -k, -l | h, -k, -l |
| Number of non-hydrogen atoms | 2286 | 4589 | 4850 |
| macromolecules | 1972 | 3078 | 3173 |
| ligands (citrate) | 65 | 0 | 0 |
| water | 249 | 1511 | 1677 |
| Protein residues | 238 | 372 | 385 |
| RMS(bonds) | 0.002 | 0.004 | 0.002 |
| RMS(angles) | 0.59 | 0.73 | 0.45 |
| Ramachandran favored (%) | 96.0 | 94.0 | 95.0 |
| Ramachandran allowed (%) | 4.0 | 5.2 | 5.0 |
| Ramachandran outliers (%) | 0.0 | 0.8 | 0.0 |
| Average B-factor (Å2) | 90.3 | 73.8 | 40.3 |
| Macromolecules (Å2) | 89.9 | 68.0 | 38.1 |
| Solvent (Å2) | 77.1 | 85.6 | 44.5 |
Statistics for the highest-resolution shell are shown in parentheses.
R merge = Σhkl Σi |Ii(hkl) − <I(hkl)>|/ΣhklΣiIi(hkl).
In vivo recombination
Intramolecular recombination in E. coli was tested using the F'-reporter and assay previously described (15,28), where deletion of a transcriptional terminator flanked by attachment sites leads to streptomycin resistance and a lac+ phenotype. For the current experiments, a pACYC-derived plasmid expressing LI Int under control of the E. coli arabinose promoter was transformed into strain CSH142 containing the reporter F’. Following heat shock, cells were incubated in SOB (2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, pH 7) containing 10 mM L-arabinose for 60 min at 37°C. Cells were plated on MacConkey lactose agar (Difco) containing 25 μg/ml chloramphenicol to assess overall recombination activity and were plated on both LB/chloramphenicol and LB containing 20 μg/ml streptomycin for quantitation. Activity was defined as the percentage of colonies that are streptomycin resistant following transformation of an integrase expression plasmid.
Intermolecular recombination in E. coli was measured by transformation of a suicide R6kγ plasmid containing a single attP site (pGV2345) into a strain containing an attB site in single copy on an F'-episome and an integrase expression plasmid as described (15), with the following modifications. Here, we used strain BW25113 and incubated transformed cells in SOB containing 100 μM L-arabinose for 60 min at 37°C before plating on LB containing 50 μg/ml ampicillin. To provide an internal control for transformation efficiency, we co-transformed pCDFSK, a compatible pCDFDuet (Novagen) derivative with the T7 expression cassettes replaced by a pBluescript cloning site (GV, unpublished). Activity was defined as the transformation efficiency of R6kγ-attP, normalized by the transformation efficiency of pCDFSK. Wild-type LI Int typically yielded 50–150 colonies/ng attP plasmid. All assays were performed three or more times.
In vitro excision and integration
Intramolecular recombination was performed using 4 kb plasmids pGV1895 (PxB) or pGV1894 (LxR) containing two att sites separated by 1 kb and arranged as direct repeats with respect to the central crossover dinucleotides. The 20-μl reactions contained 250 ng (5 nM) plasmid and 250 nM integrase in buffer SR3 (20 mM Tris–Cl, 150 mM KCl, 1 mM spermidine, 2 mM dithiothreitol, 5 mM MgCl2, 25 μg/ml bovine serum albumin, 5% glycerol, pH 8.0) and were incubated at 32° for 2.5 h. Intermolecular recombination was performed between 100 bp PCR-generated DNA fragments containing one att site (attP or attL) and 2.3 kb plasmids pGV2693 (attB) or pGV2695 (attR) containing a second att site. The 20-μl reactions contained 250 ng (8 nM) plasmid, 125 ng linear fragment (95 nM) and 250 nM integrase in buffer SR3 and were incubated at 32°C for 2.5 h. Reactions were analyzed on 0.9% agarose gels and post-stained with GelStar (Lonza).
Intermolecular PxB reaction time courses were performed between a 100 bp attP-containing DNA fragment and the 4 kb attB-containing plasmid pGV1741. Reaction volumes of 80 μl contained 5 μg plasmid (24 nM) and 400 ng attP segment (76 nM) in SR3 buffer, were initiated by addition of integrase to 250 nM, and incubated at 32°. Aliquots of 10 μl were quenched with 2 μl 1% sodium dodecyl sulphate, 0.02% bromophenol blue at various times and samples were analyzed on 0.8% agarose gels containing 0.5 μg/ml ethidium bromide. Agarose gels were quantitated by fluorescence using a Typhoon scanner (GE Healthcare) with 532 nm excitation and 610 nm (BP30) emission. Three replicate experiments were performed.
Modeling attL and attPxattB complexes
An attL complex model was constructed starting with the Int•attL model reported by Rutherford et al. (15). The attL site was smoothly unbent by distributing 15° in roll angle over the central 8-bp, removing most of the kink from the site. To reduce steric clashes created by the DNA bend, the NTD dimer (originally placed by superposition onto the γδ-resolvase•DNA complex structure (29)) was translated 3 Å along its dyad axis away from the DNA. The CC dimer was then superposed onto the complex, where the sum of the square distances between residues 347 and 400 of the CC motifs and the same residues in the ZD domains bound at P and B’ was minimized.
An attPxattB synaptic complex model was also constructed starting with the one previously reported (15). The four ZDs in the complex, which originated from chain A of structure 4KIS, were replaced by the ZDs from chain B of 4KIS. CC dimers were then superposed on the P-B and P’-B’ ZDs as described above, using residues 347 and 400 from the CC motifs and residues 338 and 400 of the ZDs, respectively. The CC dimers were further optimized using a local grid search to minimize the connection distances between residue 400 in the ZDs and CCs. Due to the asymmetric nature of the CC dimer, only one choice of superposition resulted in attL and attPxattB models with juxtaposed residue 400 and little steric clash. For both complexes, the CC dimer can be rotated by several degrees about an axis connecting residue 400 in the two subunits, since the αK connection is not well-defined. The range of rotation is more limited for the attL complex, due to steric clashes that would occur with the P half-site RD. All calculations were carried out using locally written python scripts.
RESULTS
Recombination terminology
For the experiments described below, integration refers to intermolecular recombination between DNA substrates containing attachment (att) sites and excision refers to intramolecular recombination between att sites whose central crossover dinucleotide sequences are oriented in the same direction. The sites being tested in a given experiment are given explicitly, as in ‘PxB integration’ or ‘LxR excision’, where P = attP, B = attB, etc.
The LI Int CC self-interacts
The hypothesis of a self-interacting CC motif in LI Int leads to testable predictions regarding the properties of the integrase protein and its isolated CTD. For example, the CC motifs could stabilize dimers formed by interactions between NTDs. Alternatively, they could bring dimers together to form tetramers or higher oligomers (30,31). To examine these possibilities, we compared the sedimentation equilibrium (SE) properties of Int to those of Int-ΔCC, an integase mutant where the CC motif has been deleted (15). As anticipated, both Int and Int-ΔCC form dimers at μM concentrations (Figure 2A and Table 1). We found that Int does not form stable species larger than dimers, but does form more stable dimers than Int-ΔCC. Loss of the CC domain results in a modest monomer-dimer Kd of 5.7 μM, which implies that Int-ΔCC is largely monomeric at sub-μM concentrations. Int forms dimers with at least 100-fold higher affinity (Kd ≤ 32 nM; see ‘Materials and Methods’ section).
Figure 2.

The integrase CC domain mediates oligomerization. (A) Sedimentation equilibrium (SE) analysis of LI Int. The integrase forms dimers with Kd ≤ 32 nM. When the CC domain is removed, LI Int still forms dimers, but with Kd = 5.7 μM (Table 1). Radial distributions for 1.6 μM Int are shown. (B) SE analysis of LI Int CTD. The isolated CTD forms weak dimers, with Kd = 19 μM. Radial distributions for 8.7 μM CTD are shown. (C) Linearized plot of SE data from a single rotor speed for 14 μM LI Int CTDΔCC. Without the CC domain, the CTD is monomeric. M and D indicate the calculated data lines expected for a CTD monomer and dimer, respectively. Scale = 2RT/[M (1-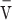 ρ)ω2]. (D) Linearized plot of SE data from a single rotor speed for the isolated CC domain at 350 μM, demonstrating that the domain is dimeric at this concentration. The low extinction coefficient of the domain precluded analysis at lower concentrations with UV absorbance optics. A summary of the oligomeric properties of the LI Int constructs studied, along with their residue ranges, is given in Table 1.
ρ)ω2]. (D) Linearized plot of SE data from a single rotor speed for the isolated CC domain at 350 μM, demonstrating that the domain is dimeric at this concentration. The low extinction coefficient of the domain precluded analysis at lower concentrations with UV absorbance optics. A summary of the oligomeric properties of the LI Int constructs studied, along with their residue ranges, is given in Table 1.
Table 1. Sedimentation equilibrium results.
| Protein | Concentrations analyzed (μM) | Rotor peeds (krpm) | Model fit | Massa (kDa) | K d (μM) | Global reduced χ2 |
|---|---|---|---|---|---|---|
| LI Int | 1.6, 3.2, 6.4, 9.5 | 8, 12, 16, 18, 20 | M-D | 53 | 0.032 ± 0.001 | 1.27 |
| IntΔCC (Δ342-416) | 5.2, 8.7, 12.2, 15.7 | 8, 12, 16, 18, 20 | M-D | 44 | 5.7 ± 1.0 | 1.93 |
| CTD (133-452) | 5.7, 6.9, 9.7 | 22, 28, 30 | M-D | 38 | 18.8 ± 0.9 | 1.63 |
| CTDΔCC (133-452; Δ342-416) | 14.3, 16 | 22, 28, 30 | SS | 29 (25.1 ± 0.1) | N.A. | 0.84 |
| CTDK262A (133-452) | 1.3, 3.6, 4.9 | 22, 28, 30 | M-D | 38 | 45.2 ± 0.9 | 1.21 |
| CTDY369A (133-452) | 4.4, 7.2, 10.6 | 22, 28, 30 | M-D | 38 | 289 ± 1 | 0.92 |
| CC-I (345-405) | 309, 348, 423 | 28, 30, 32 | SS | 7.2 (13.9 ± 0.3) | N.A. | 0.38 |
| His6Flag-Smt3-CC-I | 30, 60 | 18, 20, 22, 24 | M-D | 21 | 20.1 ± 2.8 | 0.52 |
M = monomer, D = dimer, SS = single species.
aMonomeric masses are given, with single-species fits to the mass given in parentheses, where applicable.
A self-interacting CC model also predicts that the isolated Int CTD should form dimers, whereas Int CTDΔCC should be monomeric. This is indeed the case; the isolated CTD dimerizes with Kd = 19 μM, whereas CTDΔCC is monomeric at the 14–16 μM concentrations studied (Figure 2B and C). We also analyzed the oligomeric properties of the isolated CC domain (residues 345–405). Despite the absence of the remainder of the CTD into which the CC is embedded, the isolated CC polypeptide is soluble and forms stable dimers at >300 μM concentration (Figure 2D). We were not able to analyze lower CC concentrations due to the low extinction coefficient of the CC polypeptide, but we were able to study the Smt3-CC fusion protein generated during expression and purification of the CC. This fusion has properties similar to the Int CTD, with a dimerization Kd of 20 μM (Table 1). Thus, the CC domain of LI Int stabilizes Int dimers, mediates CTD self-interaction and forms dimers in isolation.
Role of the CC in LI Int recombination
We previously showed that in E. coli, IntΔCC carries out intramolecular PxB and LxR recombination, but is inefficient at intermolecular PxB recombination (15). To further explore how the CC motif influences site selectivity, we compared the ability of Int versus IntΔCC to carry out PxB and LxR excision and PxB and LxR integration in vitro (Figure 3). For the excision reactions, Int converts both the supercoiled and open circle forms of a PxB plasmid to the expected deletion products (Figure 3A, lane 2). IntΔCC relaxes supercoils of this plasmid, but yields a smaller fraction of deletion products compared to Int (lane 3). This result suggests that synapsis and strand exchange of the supercoiled substrate by IntΔCC is efficient, but site alignment during synapsis is biased against formation of recombinant products. The open circle form accumulates, but is not efficiently converted to deletion products by IntΔCC. When the LxR plasmid substrate is used, Int generates no products (lane 5). IntΔCC relaxes supercoils and produces a small fraction of deletion products (lane 6), similar to that observed for the PxB substrate.
Figure 3.

Effects of CC deletion on LI Int recombination. (A) Intramolecular recombination between att sites separated by 1 kb and oriented in the same direction with respect to their central crossover dinucleotides. A total of 250 ng (5 nM) plasmid was incubated with buffer, 250 nM Int or 250 nM IntΔCC for 2.5 h. For the PxB reaction, Int converts both supercoiled (sc) and open circle (oc) substrates primarily to 1 and 3 kb free circles (supercoiled and open circle products labeled p). IntΔCC relaxes the supercoiled substrate to the open circle form, but produces only small amounts of deletion circles. For the LxR reaction, no products are observed with Int and IntΔCC produces similar products as observed in the PxB reaction. (B) Intermolecular recombination between a 100 bp linear DNA fragment containing one att site (not shown) and a 2.3 kb plasmid (sc, oc) containing a second att site. Recombination results in linearization of the plasmid (p). For the PxB integration reaction, Int efficiently linearizes the plasmid but IntΔCC generates only small amounts of integration product. For the LxR reaction, Int produces no product and IntΔCC results in small amounts of product similar to that observed for PxB integration. For A and B, reactions were analyzed on 0.9% agarose gels and post-stained with GelStar.
To test intermolecular recombination, we examined the ability of Int or IntΔCC to integrate a 100-bp linear fragment containing an attP or attL site into a supercoiled plasmid containing attB or attR, respectively (Figure 3B). In this case, the product of recombination is linearized plasmid. As expected, Int carries out the PxB integration reaction efficiently (lane 2), but only a small fraction of product is generated by IntΔCC (lane 3). For the LxR integration reaction, Int generates no products (lane 5), but IntΔCC yields a small amount of product (lane 6), similar to the PxB reaction.
Thus, the CC motif is required for efficient intermolecular PxB recombination and for efficient inhibition of intramolecular and intermolecular LxR recombination. In the absence of the CC motif, IntΔCC becomes promiscuous, displaying similar activities for PxB and LxR reactions. The CC motifs therefore confer identities and distinct properties to the four attachment sites involved in phage integration and excision.
Structure of a serine integrase coiled-coil dimer
In order to understand the basis for Int CC association, we attempted to crystallize isolated CC dimers. We obtained diffracting crystals using three different LI Int CC constructs that differed by the lengths of their polypeptides. We refer to these constructs and their corresponding crystal forms as CC-I, CC-II and CC-III (Table 2). All three crystal forms grow as merohedral twins. CC-I is the longest construct (Glu345-Lys405) and formed trigonal crystals with two CC dimers per asymmetric unit. We determined this structure by molecular replacement, where the search model was the best resolved CC motif from the LI Int CTD•DNA complex structure (PDB code: 4KIS). Two identical CC dimers were readily identified and the structure rapidly converged during refinement at 2.8 Å resolution.
CC-II (Ser350-Ala399) formed tetragonal crystals with four CC dimers per asymmetric unit which diffracted to 2.4 Å. This structure was determined by molecular replacement using a single, refined CC-I domain as a search model. The CC-III construct (Asp349-Asn400) differs by only two residues from CC-II and forms crystals that are isomorphous to CC-II. However, the CC-III crystals diffract to 1.95 Å, providing a higher resolution structural model than was obtained for CC-II. Four CC dimers were identified in CC-III (and CC-II, which is isomorphous), with dimerization interfaces similar to those seen in the trigonal form. A summary of diffraction data and refinement results for the three crystal forms is given in Table 2.
From the CC-I and CC-III crystal forms (CC-II is identical to CC-III), we obtained a total of six independent structures of the CC dimer and twelve independent CC domains. Four of the six dimers (two from CC-I and two from CC-III) are nearly superposable, with pairwise r.m.s.d. values ranging from 0.63 to 1.19 Å for all main chain atoms. Orthogonal views of the overall CC dimer structure are shown in Figure 4A and B and superpositions of the four most similar dimers are shown in Figure 4C. The two additional CC dimers from CC-III are similar to one another (main chain r.m.s.d = 1.38 Å), but adopt slightly different conformations from the other four (superimposed in Figure 4D). Despite the subtle structural differences in these CC subunits, the dimer interfaces are similar in all six independent structures. In the figures and discussion that follow, we use the A-B dimer (chains A and B) from CC-III as the representative structure.
Figure 4.

Structure of the LI Int CC dimer. (A and B) Orthogonal views of the first of four dimers (the A and B dimer; see text) from crystal structure CC-III. The antiparallel helical segments in each subunit are connected by a five-residue turn. The CC subunits interact in a front-to-back manner, with a dihedral angle (Ω) between αK helices of 108°. The CC subunits in the dimer are therefore related by a rotation of ∼108°, followed by a translation, as shown schematically by the two left hands. Residues comprising the core of the hydrophobic dimer interface are drawn as sticks and are labeled. (C) Superposition of the four most similar CC dimers observed in the CC-I, and CC-III crystal forms. The pairwise r.m.s.d. values for all main chain atoms range from 0.67 to 1.38 Å. The dimers superimposed are CC-I chains A and B, CC-III chains A and B, and CC-III chains C and D. (D) Addition of the remaining two CC dimers (CC-III chains E and F and chains G and H) to the superposition, illustrating structural differences, primarily for the top subunit, where the αK-αL turn adopts a different conformation.
The CC dimer interface is formed by both the CC region and the 5-residue turn that connects the K and L helices of the CC domain (Figure 4A and B). The interacting CC domains are not aligned in an anti-parallel configuration, but instead form an inter-helical dihedral angle (Ω) of 108° between their αK segments (32). The interface formed between CC motifs buries 1024 Å2 of solvent-accessible surface and appears highly specific, with a shape complementarity (SC) index of 0.73 (33). For comparison, typical protein-protein and oligomeric protein interfaces have SC values of 0.71–0.74.
A surprising feature of the CC dimer structure is that it is not symmetric. Rather than forming a 2-fold symmetric arrangement where identical surfaces from each subunit interact with one another, the domains interact in a ‘front-to-back’ or ‘head-to-tail’ manner. The CC domains avoid formation of extended polymers in the crystals and in solution because the turn that connects K and L helices in the A subunit differs in conformation from the turn in the B subunit (Figure 4). The αK-αL packing angles also differ slightly between the front and back subunits, with the average B subunit angle 9° more acute than that observed in subunit A. Thus, an induced fit occurs between CC domains in which the A and B subunits become specialized as ‘front’ and ‘back’. As discussed below, an Int-attL model provides a plausible explanation for the asymmetric nature of the CC dimer.
A related interface between CC motifs was also observed in the LI Int CTD•attP half-site crystal structure (15), where the CC from chain A forms a crystal packing interaction with αK of a symmetry-related copy of the CC from chain B. The poor electron density of the CC regions in that structure (PDB code: 4KIS) and the missing helical turn in the chain B CC preclude a meaningful comparison, but the inter-helical angles and side chain positions are qualitatively similar.
A conserved core interface mediates coiled-coil function
Close-up views of the CC dimer interface and electron density for the CC-III structure are shown in Figure 5A and B. Leu368, Tyr369 and Leu379 are at the center of a hydrophobic core that is augmented by Phe366, Ile370, Tyr374, Val376 and Met383. This core is flanked by polar interactions involving Lys362 and Arg364. Lys362 forms salt bridges with Asp387 from the same CC subunit and Glu378 from the partner subunit (Figure 5B). Arg364 and Tyr369 hydrogen bond to each other's backbone carbonyl oxygen atoms in the partner helices, while Tyr374 hydrogen bonds to Asp380 in the partner helix (Figure 5A). Leu368, Tyr369 and Leu379 are moderately conserved among serine integrases (Figure 5C), suggesting that CC domains may form a similar interface in integrases from diverse species.
Figure 5.

The LI Int CC dimer interface. (A and B) Views from opposite sides of the CC dimer, with weighted 2Fo-Fc electron density from CC-III contoured at 1.4 σ shown. The CC subunits are in magenta and brown, with residues substituted by alanine drawn in gray. Select hydrogen bonds are indicated in red. (C) Sequence alignment of six serine integrases (LI refers to the Listeria innocua prophage; the next five refer to the bacteriophages) and the serine transposase TnpX in the CC region. The longest construct crystallized, CC-I is indicated by the boxed region. Residues substituted by alanine are indicated above the alignment. The a and d positions of the helical repeat, which mediate interactions between helices in the CC, are indicated below the alignment. The alignment was made based on the CC dimer structure, functional importance of key interface residues and the expectation of heptad repeats in the helical regions. See (14) for alignments outside of the CC region.
To examine the functional significance of the CC dimer interface, we made alanine substitutions for several key residues (listed in Table 3). To confirm that the integrase variants are expressed and are catalytically active, we tested their ability to delete a transcriptional terminator flanked by attP and attB sites in E. coli, resulting in expression of the lacZYA gene products (28). This intramolecular PxB reaction does not require a functional CC domain in vivo (15). Each of the Int variants tested except the catalytic S10A mutant resulted in lac+ colonies on MacConkey agar, indicating a functional integrase (Table 3).
Table 3. Recombination activities of coiled-coil mutants.
| Integrase | PxB excision MacConkey | LxR excision MacConkey | LxR excision in vivoa | PxB integration in vivob | PxB integration in vitroc |
|---|---|---|---|---|---|
| Wild-type | red | white | 0 | 35 ± 11 | 0.32 ± 0.08 |
| S10A | white | white | 0 | 0 | N.D. |
| ΔCC | red | red | 62 ± 12 | 0.97 ± 0.34 | N.D. |
| K362A | red | red | 5.7 ± 1.9 | 0.55 ± 0.04 | 0.12 ± 0.04 |
| R364A | red | white | 0.21 ± 0.22 | 4.5 ± 1.4 | N.D. |
| F366A | red | red | 13 ± 9 | 2.8 ± 1.1 | N.D. |
| L368A | red | red | 0.027 ± 0.026 | 11 ± 3 | N.D. |
| Y369A | red | white | 0.34 ± 0.13 | 1.5 ± 1.0 | 0.00 ± 0.02 |
| Y374A | red | white | 0.10 ± 0.11 | 1.8 ± 0.6 | N.D. |
| L379A | red | pink | 0.088 ± 0.022 | 4.3 ± 1.6 | N.D. |
| L368A Y369A L379A | red | red | 1.5 ± 0.9 | 0.25 ± 0.25 | N.D. |
aExcision activity is the percent of transformants that are streptomycin resistant when the LxR reporter strain is transformed with an integrase-expressing plasmid.
bIntegration activity is the number of ampicillin-resistant colonies obtained when an attB reporter strain is transformed with an attP suicide plasmid, normalized for transformation of an internal control.
cThe fraction of attB plasmid that is integrated by linear attP with saturating integrase (250 nM) in 20 min is given.
We next tested the ability of the integrase mutants to carry out LxR excision. Since the CC domain is required for inhibition of LxR recombination (Figure 3), (15,16), our hypothesis was that disruptions to the CC dimer interface would compromise this function by weakening the intramolecular CC interactions present on attL and attR sites. Intramolecular recombination could then occur, resulting in excision of a terminator flanked by attL and attR sites and expression of lacZYA (shown schematically in Figure 6A). To broadly assess activity, we transformed our LxR tester strain with plasmids expressing Int, Int-ΔCC or alanine-substituted Int variants. As we reported previously, Int-ΔCC is efficient at LxR excision in vivo (15). This integrase mutant therefore results in red (lac+) colonies on MacConkey agar. Wild-type Int results in white colonies because LxR excision is strongly inhibited in the absence of the phage-encoded RDF. Examples of this assay are shown in Figure 6B. The K362A substitution and the triple L368A, Y369A, L379A substitution result in partial loss of CC inhibition, whereas Int R364A is still able to inhibit excision. The results for our panel of Int CC mutants are given in Table 3.
Figure 6.

Recombination activity of CC-substituted LI Int. (A) Schematic of the F’ used to report intramolecular recombination in Escherichia coli. The LxR excision reaction is shown, where removal of a strong terminator allows transcription of streptomycin resistance and lac genes. A similar reporter with attP and attB sites monitors intra-molecular PxB recombination. (B) Examples of LxR excision in E. coli, where an arabinose-inducible plasmid expressing Int is transformed into a reporter strain containing the F’ shown in A. Cells are plated on MacConkey agar, with red colonies resulting when recombination is efficient (e.g. K362A) and white colonies resulting when recombination is blocked or very slow (e.g. wild-type). The results for all alanine-substitutions are given in Table 3. (C) Quantitative comparison of in vivo LxR excision activities, where activity is the percentage of transformants that are streptomycin resistant. No colonies have been observed in many such experiments for wild-type Int. A range of excision activities, indicating partial loss of CC function, are observed for the Int variants studied. (D) Results of PxB integration experiments in E. coli, where an attP-containing R6kγ plasmid (which cannot replicate in the tester strain) is transformed into a strain containing a single copy of attB. Activity is the number of ampicillin-resistant transformants, normalized by an internal transformation control. All CC-substitutions lead to defective integrases, with low PxB integration integration activity correlated with high LxR excision activity. All experiments were performed three or more times and are summarized in Table 3. Error bars represent ±SD.
To more carefully evaluate the LxR inhibition activity of Int mutants, we carried out quantitative LxR excision assays, where transformed cells have 60 min to carry out recombination and express a streptomycin marker before being plated on LB/streptomycin and scored for activity. As shown in Figure 6C and Table 3, all of the Int CC substitutions result in measurable excision activity, but K362A, F366A and the triple substitution are particularly active. Thus, we observe a wide range of CC defects, with L368A the least defective and K362A and F366A the most defective at supressing LxR recombination.
We next examined the ability of Int CC mutants to carry out intermolecular PxB recombination in E. coli, a reaction where the Int CC domain is required. We used an assay that we previously reported (15), where integration activity is scored as the transformation efficiency of an attP-containing R6kγ plasmid into a F’-attB strain expressing an integrase variant (see ‘Materials and Methods’ section). As shown in Figure 6D and Table 3, each of the alanine-substituted Int variants is defective at PxB integration. The most defective mutants for integration (K362A and the triple substitution) also had high LxR excision activity, and the least defective mutant for integration (L368A) had the lowest LxR excision activity. The F366A substitution showed a similar correlation, but was more active in integration than might be expected based on the high rates of excision allowed. These results support a model where a similar interface between CC domains is responsible for both facilitating PxB integration and inhibiting LxR excision.
Coiled-coil mutants defective at in vitro recombination
To test whether the recombination defects observed in E. coli are also observed in vitro, we purified the Int K362A and Y369A mutants and tested their ability to integrate a small linear attP site into supercoiled plasmid containing an attB site (Figure 7). As anticipated, both mutants are defective at PxB integration, relative to wild-type Int. However, Int K362A displays higher activity in vitro than might be expected from the in vivo experiments (Table 3). Since we do not control for expression levels in vivo, we cannot rule out the trivial explanation that the Int K362A concentration is lower in E. coli. It is also possible that our in vitro buffer conditions (e.g. 5 mM Mg2+) compensate for the loss of Lys362, elevating integration activity above that found in E. coli. Interestingly, the φC31 integrase Y475H mutant has properties similar to LI integrase Y369A (aligned in Figure 5C). The Y475H variant was first identified in an E. coli screen for disrupted CC function, resulting in increased LxR excision. However, purified Y475H integrase was found to be poorly active in vitro (16).
Figure 7.

in vitro PxB integration activity of Int variants. (A) SDS-PAGE of purified Int, Int Y369A and Int K362A (calculated MW = 53 kDa). (B) Integration of a 100 bp attP-containing DNA segment into a 4-kb supercoiled plasmid containing an attB site (sc). Recombination results in linearized plasmid (p). One of the three independent experiments is shown for each Int. (C) Plot of the reaction time courses shown in B, where errrors are ±SD.
Coiled-coil mutants defective at oligomerization
Alanine substitutions in the CC motif that result in diminished recombination function would also be expected to show defects in self-association if the interface shown in Figure 5 is functionally relevant. We therefore constructed and purified Int CTDs containing the K362A and Y369A substitutions and determined their oligomeric properties using size-exclusion chromatography with in-line multi-angle light scattering detection (SEC-MALS) and SE ultracentrifugation. As shown in Figure 8, both K362A and Y369A CTDs show increased SEC elution volumes and reduced weight-average molecular weights compared to the wild-type CTD. Global fits to SE radial scans indicate increased monomer-dimer Kd values of 45 and 289 μM for K362A and Y369A, respectively, compared to the wild-type CTD Kd of 19 μM (Table 1), consistent with the reduced mass values observed by SEC-MALS. The relative self-association properties of these mutants mirror the relative in vitro PxB integration activities of the corresponding full-length integrases (Figure 7).
Figure 8.

Oligomeric properties of LI Int CTDs. (A) SDS-PAGE of purified Int CTD, CTD Y369A and CTD K362A (calculated MW = 38 kDa). (B) Size-exclusion chromatography with multi-angle light scattering detection (SEC-MALS) analyses. The SEC elution profiles are shifted to longer retention times for the alanine-substituted CTDs relative to wild-type CTD and the weight-averaged molecular mass (Mw) across the profiles indicate primarily monomeric species. The wild-type CTD has an increased Mw. Eluted peak concentrations ranged from 0.04 to 0.1 mg/ml, as determined by refractive index. SE analyses support these findings, with dimer Kd values of 19, 290 and 45 μM for wild-type, Y36149A and K362A CTDs, respectively (Table 1).
The defective nature of Int constructs containing the K362A substitution indicates that this residue contributes to stabilizing CC self-association. The CC dimer interface shown in Figure 5B provides a structural basis for this stabilization. As noted above, two of the six independent dimer structures determined in this study have slightly different conformations of the CC subunits, but maintain similar hydrophobic packing between subunits in the dimers. In these two alternative dimers, however, Lys362 plays no obvious structural role in self-association. These results suggest that the four dimer structures represented by Figure 4A–C and Figure 5A and B (the ‘A-B dimer’) are more functionally relevant than are the two shown superimposed in Figure 4D.
An improved Int-attL model
We previously proposed a model for LI Int bound to attL in which distinct CTD positioning on the left (P) versus right (B’) half-sites could facilitate an intra-molecular interaction between CC domains (15). However, the CC motifs were poorly resolved in the experimental structure used to build that model and there was no readily interpretable structural information available to inform how the CC domains might interact. Using the experimental CC dimer structures described here, we have updated the Int-attL model as shown in Figure 9. In addition to positioning the CC dimer, we have removed the sharp bend located at the center of attL DNA that was present in the original model. The unbent site is more consistent with the lack of evidence for bending in serine integrase-att site complexes and provides the maximum separation between CTDs for testing whether the CC dimer structure can span between ZDs bound at the P and B’ half-sites.
Figure 9.

Model of the LI Int•attL complex. The attL DNA from the model reported by Rutherford et al. (15) was straightened by removal of the kink located at the center. The CC dimer structure was then docked onto the complex, maximizing alignment and overlap with the connecting points in the P and B’ ZD domains. Two orthogonal views are shown. The αL helices match well with their connecting helical segments in the ZD domains (labeled αL(ZD)), but the αK helices require 8-residue linkers that adopt different conformations or are disordered in the LI Int CTD•DNA complex structure (indicated by dashed curves). The bottom view illustrates the spatial match between the CC dimer N- and C-termini and the ZD connection points. A front-to-back CC dimer matches the ZD arrangement, but a symmetric dimer would not. A potential interaction surface between the magenta CC subunit and the P half-site RD domain is also evident in this view.
As shown in Figure 9, the CC dimer easily spans the distance between P and B’ half-site CTDs. The αL helix in the CC can connect to the ordered region of αL in the P half-site ZD, since their termini are nearly juxtaposed. However, a bend or kink in αL would be required near the junction between helical segments, since they are not co-linear. Indeed, the four independent CC motif structures available from the LI Int CTD•attP crystal structure provided evidence for considerable flexibility in these motifs, with multiple examples of helical segments disrupted by kinks or turns. We did not attempt to model the eight-residue segment linking Ala338 in the ZD to Glu347 at the start of αK in the CC (shown as a dashed curve in Figure 9).
In the B’-half-site, the αL helical segments from the CC dimer and the ZD are nearly co-linear, suggesting that a continuous αL may form. The Ala338-Glu347 linker has a slightly longer span to cover (19 Å in the B’ half-site versus 17 Å in the P half-site), but could be readily accommodated in both cases by a mixed α/turn conformation. This region of serine integrase sequences is not strongly conserved, with variation in both the length and composition of the connecting residues (Figure 5C).
The Int-attL model provides a structural explanation for the front-to-back arrangement of subunits in the CC dimer. The αL helices of the dimer can be connected to the ordered segments of αL (residues 401–417) in the ZDs, while positioning the αK helices for simple connection to Ala338 via a short peptide linker. If one of the CC motifs in the dimer structure were rotated so that its helices exchange positions, a symmetric dimer could be generated with similar overall geometry. This symmetric dimer, however, would require structural rearrangements and crossing of linkers in order to accommodate the connections, since αK would now be juxtaposed onto αL in one of the ZDs. Thus, the helices in the front-to-back CC dimer match the spatial arrangement of ZDs, whereas the helices in a dyad-symmetric CC dimer with similar geometry would not align well with the connecting structural elements.
A second insight provided by the updated Int•attL model is that the CC motif could potentially contact the recombinase domain (RD) in the P half-site, but not the B’ half-site. The juxtaposition of the CC motif and the P half-site RD is evident in Figure 9. Since there is some rotational and translational flexibility in positioning of the CC dimer, it is not clear which regions would be most likely to interact with the RD.
An updated attPxattB synaptic complex model
To test whether the CC dimer can also bridge the P-B and P’-B’ half-sites in the synaptic complex formed during the phage integration reaction, we constructed a model of the PxB complex based on a γδ-resolvase reaction intermediate structure (34). Three views of the model are shown in Supplementary Figure S2. The CC dimer readily spans the distance between half-site ZDs, with Asn400 residues at the ends of the αL helices in position to connect to Glu401 of the corresponding ZDs. There are no steric clashes and a short gap allows for eight missing residues to connect the αK helices to their corresponding ZDs. An important difference between the PxB synaptic complex model and the attL model is the dramatic difference in CC trajectories that are used, as illustrated schematically in Figure 1. This may be accomplished by utililization of bent αL helices in the P and P’ half-sites for attL and attR, but in all four half-sites for the attP-attB synaptic complex. One consequence of this difference in trajectories is that the CC motifs are not positioned to contact other Int domains in the attP-attB complex as they appear to be in attL and attR.
DISCUSSION
CCs are ubiquitous in biology, playing important roles in diverse processes such as solute transport, vesicle tethering, transmembrane signaling, membrane fusion, chromosome condensation and cohesion, DNA sequence recognition, and movement along microtubule and actin fibers (for recent reviews, see (35,36). The functional roles of CC motifs within their host proteins is also diverse on a molecular level and include structural rigidity, oligomerization, channel formation, allosteric communication and measuring molecular distances. The serine integrases use their CC motifs by combining these themes; they act as molecular rulers to identify att site context and they provide inter-subunit oligomerization. The serine integrase CC domains also serve as allosteric regulators, promoting formation of a cleavage-competent synaptic assembly in an RDF and att site-dependent manner.
Despite the widespread use of heptad repeats in the proteomes of all organisms, the interaction between terminal ends of antiparallel CC motifs shown in Figure 4 for the LI integrase appears to be rarely used. Based on the alignment in Figure 5C, we expect the structures of CC domains from other serine integrases to be similar. The LI Int CC dimer structure allowed us to update this alignment, with some important changes relative to our previous structural predictions (14,15). The size of insertions into the αK-αL turn shown in Figure 5C is smaller than we previously proposed, particularly for the φC31 integrase. At the same time, there is reasonable consensus among the A and D positions of the helical repeat patterns for αK and αL. We suggest that the serine integrases have only small variations in the length of the helical turn and a moderately well-conserved hydrophobic CC dimer interface. There is, however, variability in both length and composition of the sequence at the ends of the helical segments, consistent with the need for flexible positioning of the CC domain.
We found that the LI integrase CC domain contributes toward stabilization of integrase dimers in solution. The wild-type integrase self-associates tightly, with a dimer Kd ≤ 32 nM. Without the CC domain, LI Int dimerization is more than 100-fold weaker, with Kd = 5.7 μM. These results explain the gel filtration behavior of serine integrase truncations that lack the ZD or both the ZD and the recombinase domain; in both cases, apparent monomers and dimers have been observed by gel filtration (17,37). These results are also consistent with previous suggestions that the CC domain may be involved in inter-subunit interactions within Int dimers and that these interactions must be disrupted upon att site binding (14,16,17).
One of the puzzling observations from early studies in the Bxb1 and φC31 integrase systems was that the isolated integrase CTDs bound to att sites with affinities similar to those of the full length integrase dimers (17,38). Thus, the avidity expected from binding of the second CTD in an integrase dimer to an att site is somehow lost. The weakened dimerization of LI Int ΔCC provides an explanation for this phenomenon for attB and attP, since binding of an integrase dimer to each comes with the cost of disrupting intra-dimer interactions that involve the CC domains. Presumably, this cost is paid by binding of the second CTD. Using the same argument, we can rationalize why full-length Bxb1 integrase dimer binding to truncated att half-sites is so poor (17); in this case, there is no second CTD binding site to compensate for the cost of dimer reorganization.
The above arguments do not explain why Int binds to attL and attR with similar affinities as attP and attB in the φC31 and A118 systems (18,38), since CC interactions are presumably restored upon binding to attL and attR. If Int-half-site interactions and DNA conformations are the same in attL and attR as they are in attP and attB, then an increased affinity should be observed for attL and attR binding, as it is for Bxb1 Int (17). We suggest that for φC31 and A118 Int (and perhaps Bxb1 Int), the unsynapsed attL and attR complexes must differ in some way (in addition to formation of intramolecular CC interactions) from the hybrid sites expected from simple swapping of attP and attB half-sites. If the energetic cost of these conformational changes is compensated by the intramolecular CC interactions, then similar binding affinities for attL and attR could be explained by a redistribution of binding energy within the complexes. We do not know what these changes in attL and attR are likely to be, since there are currently no experimental structures of any serine integrase dimer bound to a full attachment site to provide insights.
We found that the LI Int CTD and the Smt3-CC fusion dimerize weakly, with Kd = 19 and 20 μM, respectively. The similarity of these values suggests that CC dimerization is the primary contributor to CTD dimerization, with a CC dimer Kd of ∼20 μM. Although we were not able to determine the dimerization constant for the isolated CC, the finding that it is entirely dimeric at concentrations 15× Kd is consistent with our estimate. Despite the weak nature of this bi-molecular association, in an intra-molecular context such as that found in integrase-dimers, this is a considerable affinity. Indeed, a stronger interaction between CCs would be expected to decrease binding affinity to attP and attB sites. A stronger interaction might also make phage excision a difficult reaction to promote at modest concentrations of an RDF, which must use inter-molecular interactions to compete. If we assume that other serine integrase CTDs have similar dimerization affinities, then our findings would explain reports in several LSR systems that the isolated CTDs appear monomeric by gel filtration (17–19). Indeed, the LI CTD also runs as an apparent monomer on size-exclusion columns unless very high concentrations are injected (i.e. Figure 8B).
The Int•attL model shown in Figure 9 provides a possible explanation for the surprising phenotypes of φC31 Int mutations in the CC region (16). Several glutamate residues (E449, E452, E456 and E463) are predicted to lie on the same solvent-exposed surface of αK. When substituted by lysine, each results in an increase in LxR recombination, while maintaining PxB recombination. The CC dimer structure and Int•attL model are consistent with these results, in that the mutations should not directly affect the ability of the CC motifs to interact, but could affect the positioning of the CCs in a way that disfavors intramolecular dimer formation on attL and attR, while permitting dimer formation on attP and attB. The idea that CC-RD interactions could be present within attL and attR complexes but absent in the attP–attB synaptic complex supports this interpretation. A detailed understanding of what interface is being perturbed by these substitutions will require an Int•attL or Int•attR structure in order to determine the exact positioning of the CC dimer and the conformations of the ZD-CC hinge regions that are involved in establishing CC trajectories.
The Int-attL model also raises the important question of how integrase-bound attL and attR sites are inhibited from undergoing recombination in the absence of an RDF. The simplest mechanism is that the intramolecular CC interaction prevents attL and attR from forming synaptic complexes, regardless of whether the CCs participate in stabilizing those complexes. This would be consistent with the idea that attL and attR complexes differ structurally from attP and attB complexes. This mechanism would also be consistent with the lack of experimental support for LxR synapse formation in systems where PxB synapsis with Int and LxR synapsis with the CC mutants discussed above can be readily detected on native polyacrylamide gels (16,17,38). A synapsis inhibition mechanism is not supported by a recent kinetic model described for φC31 Int recombination by Pokhilko et al. (39), where a synaptic LxR complex is predicted to be the most stable species in the P + B → L + R pathway. We note, however, that the model's prediction of distinct reaction intermediates that may differ by the nature of their CC interaction partners is consistent with the structures and models described here.
If attL and attR complexes become competent for synapsis upon release of the intramolecular CC interaction, then the role of the CC domains in this context is to maintain a kinetic barrier that prevents recombination. Given the very high on-rate expected for the intramolecular CC interaction, the concentration of ‘activated’ attL and attR complexes that have dissociated CC domains and are therefore competent for synapsis would be very low. Thus, the likelihood of a productive collision beween activated attL and attR would also be low, resulting in little or no LxR recombination observed. Future studies will focus on testing this mechanism quantitatively, as done for other systems (39–41).
Our Int•attL model also suggests two simple mechanisms by which RDFs could break inhibitory CC interactions in the attL and attR sites. The simplest is that the RDF could bind to the CC domains and compete directly with the intramolecular interaction. Alternatively, the RDF could bind to the zinc ribbon or recombinase domain and sterically block the CC–CC interaction. Finally, some combination of these mechanisms could be used, where attP and attB-derived half-sites may differ markedly in the nature of the RDF interactions. The RDF may also participate directly in synapsis of attL and attR sites. The RDF for the Listeria phage A118 has recently been identified, and indeed, it binds to the CC domain of Int, with evidence for contacts with other domains as well (13). The RDFs for phages Bxb1, φC31, φBT1, φRV1 and TP901 have been studied (8–12), but their specific integrase binding regions are not known. Understanding the structural and mechanistic details of how RDFs influence the recombination pathway is an important priority in the quest to learn how the serine integrases achieve such remarkable RDF-regulated directionality.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM108751 to G.V.]; National Science Foundation (to CHESS) [DMR-1332208]; National Institutes of Health (to MacCHESS) [GM103485]; National Institutes of Health (to Berkeley Center for Structural Biology) (in part); National Institute of General Medical Sciences; Howard Hughes Medical Institute; Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy (to Advanced Light Source) [DE-AC02-05CH11231]. Funding for open access charge: Office of Extramural Research, National Institutes of Health [GM108751].
Conflict of interest statement. None declared.
REFERENCES
- 1. Smith M.C.M. Phage-encoded serine integrases and other large serine recombinases. Microbiol. Spectr. 2015; 3, doi:10.1128/microbiolspec.MDNA3-0059-2014. [DOI] [PubMed] [Google Scholar]
- 2. Groth A.C., Calos M.P.. Phage integrases: biology and applications. J. Mol. Biol. 2004; 335:667–678. [DOI] [PubMed] [Google Scholar]
- 3. Brown W.R.A., Lee N.C.O., Xu Z., Smith M.C.M.. Serine recombinases as tools for genome engineering. Methods. 2011; 53:372–379. [DOI] [PubMed] [Google Scholar]
- 4. Chavez C.L., Calos M.P.. Therapeutic applications of the ΦC31 integrase system. Curr. Gene Ther. 2011; 11:375–381. [DOI] [PubMed] [Google Scholar]
- 5. Fogg P.C.M., Colloms S., Rosser S., Stark M., Smith M.C.M.. New applications for phage integrases. J. Mol. Biol. 2014; 426:2703–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jayaram M., Ma C.-H., Kachroo A.H., Rowley P.A., Guga P., Fan H.-F., Voziyanov Y.. An overview of tyrosine site-specific recombination: from an Flp perspective. Microbiol. Spectr. 2015; 3, doi:10.1128/microbiolspec.MDNA3-0021-2014. [DOI] [PubMed] [Google Scholar]
- 7. Landy A. The λ integrase site-specific recombination pathway. Microbiol. Spectr. 2015; 3, doi:10.1128/microbiolspec.MDNA3-0051-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Breüner A., Brøndsted L., Hammer K.. Novel organization of genes involved in prophage excision identified in the temperate lactococcal bacteriophage TP901-1. J. Bacteriol. 1999; 181:7291–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bibb L.A., Hatfull G.F.. Integration and excision of the Mycobacterium tuberculosis prophage-like element, phiRv1. Mol. Microbiol. 2002; 45:1515–1526. [DOI] [PubMed] [Google Scholar]
- 10. Ghosh P., Wasil L.R., Hatfull G.F.. Control of phage Bxb1 excision by a novel recombination directionality factor. PLoS Biol. 2006; 4:e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khaleel T., Younger E., McEwan A.R., Varghese A.S., Smith M.C.M.. A phage protein that binds phiC31 integrase to switch its directionality. Mol. Microbiol. 2011; 80:1450–1463. [DOI] [PubMed] [Google Scholar]
- 12. Zhang L., Zhu B., Dai R., Zhao G., Ding X.. Control of directionality in Streptomyces phage φBT1 integrase-mediated site-specific recombination. PLoS One. 2013; 8:e80434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mandali S., Gupta K., Dawson A.R., Van Duyne G.D., Johnson R.C.. Control of recombination directionality by the Listeria phage A118 protein Gp44 and the coiled-coil motif of its serine integrase. J. Bacteriol. 2017; doi:10.1128/JB.00019-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Duyne G.D., Rutherford K.. Large serine recombinase domain structure and attachment site binding. Crit. Rev. Biochem. Mol. Biol. 2013; 48:476–491. [DOI] [PubMed] [Google Scholar]
- 15. Rutherford K., Yuan P., Perry K., Sharp R., Van Duyne G.. Attachment site recognition and regulation of directionality by the serine integrases. Nucleic Acids Res. 2013; 41:8341–8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rowley P.A., Smith M.C.A., Younger E., Smith M.C.M.. A motif in the C-terminal domain of phiC31 integrase controls the directionality of recombination. Nucleic Acids Res. 2008; 36:3879–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghosh P., Pannunzio N.R., Hatfull G.F.. Synapsis in phage Bxb1 integration: selection mechanism for the correct pair of recombination sites. J. Mol. Biol. 2005; 349:331–348. [DOI] [PubMed] [Google Scholar]
- 18. Mandali S., Dhar G., Avliyakulov N.K., Haykinson M.J., Johnson R.C.. The site-specific integration reaction of Listeria phage A118 integrase, a serine recombinase. Mob. DNA. 2013; 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McEwan A.R., Rowley P.A., Smith M.C.M.. DNA binding and synapsis by the large C-terminal domain of phiC31 integrase. Nucleic Acids Res. 2009; 37:4764–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R.A., Rouault T.A., Schuck P.. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 2004; 326:234–256. [DOI] [PubMed] [Google Scholar]
- 21. Laue T.M., Shah B., Ridgeway T.M., Pelletier S.L., Harding S.E., Rowe A.J., Horton J.C.. Harding SE, Rowe AJ, Horton JC. Computer-aided interpretation of analytical sedimentation data for proteins. Analytical Ultracentrifugation in Biochemistry and Polymer Science. 1992; Cambridge: Royal Society of Chemistry; 90–125. [Google Scholar]
- 22. Gupta K., Diamond T., Hwang Y., Bushman F., Van Duyne G.D.. Structural properties of HIV integrase. Lens epithelium-derived growth factor oligomers. J. Biol. Chem. 2010; 285:20303–20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bujacz G., Wrzesniewska B., Bujacz A.. Cryoprotection properties of salts of organic acids: a case study for a tetragonal crystal of HEW lysozyme. Acta Cryst. D. 2010; 66:789–796. [DOI] [PubMed] [Google Scholar]
- 24. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 25. Battye T.G.G., Kontogiannis L., Johnson O., Powell H.R., Leslie A.G.W.. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Cryst. D. 2011; 67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Cryst. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Struct. Biol. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 28. Gibb B., Gupta K., Ghosh K., Sharp R., Chen J., Van Duyne G.D.. Requirements for catalysis in the Cre recombinase active site. Nucleic Acids Res. 2010; 38:5817–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang W., Steitz T.. Crystal structure of the site-specific recombinase gamma delta resolvase complexed with a 34 bp cleavage site. Cell. 1995; 82:193–207. [DOI] [PubMed] [Google Scholar]
- 30. Yuan P., Gupta K., Van Duyne G.D.. Tetrameric structure of a serine integrase catalytic domain. Structure. 2008; 16:1275–1286. [DOI] [PubMed] [Google Scholar]
- 31. Rowley P.A., Smith M.C.M.. Role of the N-terminal domain of phiC31 integrase in attB-attP synapsis. J. Bacteriol. 2008; 190:6918–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chothia C., Levitt M., Richardson D.. Helix to helix packing in proteins. J. Mol. Biol. 1981; 145:215–250. [DOI] [PubMed] [Google Scholar]
- 33. Lawrence M., Colman P.. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 1993; 234:946–950. [DOI] [PubMed] [Google Scholar]
- 34. Li W., Kamtekar S., Xiong Y., Sarkis G.J., Grindley N.D.F., Steitz T.A.. Structure of a synaptic gammadelta resolvase tetramer covalently linked to two cleaved DNAs. Science. 2005; 309:1210–1215. [DOI] [PubMed] [Google Scholar]
- 35. Lupas A.N., Bassler J.. Coiled Coils—a model system for the 21st century. Trends Biochem. Sci. 2017; 42:130–140. [DOI] [PubMed] [Google Scholar]
- 36. Truebestein L., Leonard T.A.. Coiled-coils: the long and short of it. Bioessays. 2016; 38:903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adams V., Lucet I.S., Lyras D., Rood J.I.. DNA binding properties of TnpX indicate that different synapses are formed in the excision and integration of the Tn4451 family. Mol. Microbiol. 2004; 53:1195–1207. [DOI] [PubMed] [Google Scholar]
- 38. Thorpe H.M., Wilson S.E., Smith M.C.. Control of directionality in the site-specific recombination system of the Streptomyces phage phiC31. Mol. Microbiol. 2000; 38:232–241. [DOI] [PubMed] [Google Scholar]
- 39. Pokhilko A., Zhao J., Ebenhöh O., Smith M.C.M., Stark W.M., Colloms S.D.. The mechanism of ϕC31 integrase directionality: experimental analysis and computational modelling. Nucleic Acids Res. 2016; 44:7360–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bonnet J., Subsoontorn P., Endy D.. Rewritable digital data storage in live cells via engineered control of recombination directionality. PNAS. 2012; 109:8884–8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bowyer J., Zhao J., Subsoontorn P., Wong W., Rosser S., Bates D.. Mechanistic modeling of a rewritable recombinase addressable data module. IEEE Trans. Biomed. Circuits Syst. 2016; 10:1161–1170. [DOI] [PubMed] [Google Scholar]
- 42. Stark W.M. The serine recombinases. Microbiol Spectr. 2014; 2, doi:10.1128/microbiolspec.MDNA3-0046-2014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.