


Abstract
Long noncoding RNAs (lncRNAs) are key regulators of diverse cellular processes. Recent advances in high-throughput sequencing have allowed for an unprecedented discovery of novel lncRNAs. To identify functional lncRNAs from thousands of candidates for further functional validation is still a challenging task. Here, we present a novel computational framework, lncFunNet (lncRNA Functional inference through integrated Network) that integrates ChIP-seq, CLIP-seq and RNA-seq data to predict, prioritize and annotate lncRNA functions. In mouse embryonic stem cells (mESCs), using lncFunNet we not only recovered most of the functional lncRNAs known to maintain mESC pluripotency but also predicted a plethora of novel functional lncRNAs. Similarly, in mouse myoblast C2C12 cells, applying lncFunNet led to prediction of reservoirs of functional lncRNAs in both proliferating myoblasts (MBs) and differentiating myotubes (MTs). Further analyses demonstrated that these lncRNAs are frequently bound by key transcription factors, interact with miRNAs and constitute key nodes in biological network motifs. Further experimentations validated their dynamic expression profiles and functionality during myoblast differentiation. Collectively, our studies demonstrate the use of lncFunNet to annotate and identify functional lncRNAs in a given biological system.
INTRODUCTION
Long noncoding RNAs (lncRNAs) are non-protein coding transcripts >200 nucleotides (1,2). They have gained widespread attention in recent years as crucial components of gene regulatory networks and have been revealed to play key roles in many biological processes such as cell differentiation (3–5), imprinting control (6), immune responses, human diseases, tumorigenesis etc. (7–9). The advancement of high-throughput genomic technologies such as next generation sequencing (NGS) has resulted in an unprecedented ability to detect thousands of novel lncRNA transcripts from direct assembling of transcriptome sequencing data (10). Intensive efforts from many groups (11–13) have focused on functional exploration of these lncRNAs, however, the functional studies require direct perturbation experiments, such as loss-of-function and gain-of-function assays that are very time consuming. Therefore, only a few lncRNAs have been clearly characterized so far while the function of the vast majority is still an enigma (14). This situation is exemplified in the system of skeletal muscle cell differentiation during which proliferating myoblasts (MBs) exit cell cycle and are fused into multi-myonucleotided myotubes (MTs). At the transcriptional level, the process is orchestrated by a complex networks intertwining transcription factors, epigenetic regulators, miRNAs and lncRNAs. Using a murine myoblast cell line, C2C12, we have identified thousands of lncRNAs through de novo assembly of the transcriptome sequencing data that expressed in MBs or MTs (3,5). Despite the intensive efforts during the past few years, we and others were only able to characterize a few of them (3–5,12,15); we are thus in need of a high confidence computational approach which can systematically identify and prioritize potentially functional lncRNAs from large reservoirs of novel lncRNAs before we dive into time and labor consuming experimental testing for their functional studies.
However, this type of computational prediction of lncRNA function is still at its early stage. As the sequences and secondary structures of lncRNAs are generally not conserved, functional prediction of lncRNAs using the comparative genomic approach is limited (8,14). Recent studies revealed that lncRNAs mainly execute their functions through interacting with other types of molecules such as chromatin modifiers, transcription factors (TFs) (3–5), microRNAs (miRNAs) (11,12), protein complexes (4,13,16) and DNAs (17) in an integrated gene network. These observations open the possibilities that the functions of lncRNAs can be inferred through their interactions with other molecules within the interaction networks (i.e. lncRNA interactome). In fact, co-expressed gene network approaches have been commonly used for the functional prediction using gene expression data only (18) or combined with protein–protein interaction (PPI) data (19). This approach however is based on the interactions (mostly indirect interactions) inferred by gene expression correlations and PPI networks, thus, can only catch the tip of the iceberg of the entire interactome among a particular lncRNA and its partners. For example, emerging evidence demonstrates the intimate connection between lncRNAs and TFs. Similar to mRNAs, the transcription process of a lncRNA can be regulated by TFs through binding to its promoter (3,5); a lncRNA can also regulate the transcriptional activity of a TF through physically interacting with the TF (5,13). In addition, lncRNA–miRNA interaction has become an increasingly accepted phenomenon existing in cells (20,21); the most well-known mode of the interaction is the ceRNA model, i.e. lncRNAs acting as sponges for miRNAs competing for their binding to bona fide mRNA targets but increasing evidence also shows that miRNAs can bind and degrade lncRNAs post-transcriptionally. Luckily, the development of CLIP-seq (cross-linked immunoprecipitation followed by next generation sequencing) has allowed the mapping of lncRNA–miRNA interactions genome-wide; several databases are now available to obtain the CLIP-seq data for inferring miRNA–lncRNA interactome (22,23). Taking together, we reason that integration of lncRNAs into gene networks based on different types of lncRNA interactions will gain greater prediction power for reliable functional outcomes.
Here, we developed lncFunNet, a computational framework to predict, prioritize, and annotate functional lncRNAs by systematically exploring gene networks established on the lncRNA interactomes. It integrates a number of interactions (i.e. TF–lncRNA, miRNA–lncRNA, lncRNA–PCG (protein coding gene)) and provides a trained scoring scheme to calculate the functional information score (FIS) for each evaluated lncRNA, thus helps to elucidate and evaluate the functional importance of newly discovered lncRNAs in different biological systems. When applied in mouse embryonic stem cells (mESCs), the evaluation demonstrated a high accuracy of lncFunNet in identifying known functional lncRNAs. When further applying to skeletal muscle C2C12 cells lncRNAs with potential functions were identified in MBs or MTs and display distinct genomic features compared to non-functional lncRNAs. In addition, we showed that lncRNAs are key motif components in the integrative gene networks which in turn can be further used to infer the functional mechanisms. Lastly, wet-lab experiments were conducted to validate the functionality of the selected lncRNAs during C2C12 cell differentiation. Altogether lncFunNet provides a new tool for identification of functional lncRNAs in a given biological system.
MATERIALS AND METHODS
Identification of TF–gene interactome through chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-seq) data analysis
To establish the TF–gene interactions, we downloaded ChIP-seq data from Gene Expression Omnibus (GEO) database (Supplementary Tables S1 and S2) (24). The raw reads were processed with the protocols described in our previous publication (5). Briefly, the adapter and low quality sequences were trimmed from 3΄ to 5΄ ends. After trimming, reads shorter than 36 bp were discarded and the preprocessed reads were aligned to mouse reference genome (mm9) using SOAP2 (25). Model-based Analysis for ChIP-seq (MACS v 2.1.0) (26) was then used to detect TF binding peaks with either input DNA or IgG sample as the control. During the peak calling, the q-value cutoff was set to 0.05 for all ChIP-seq datasets to identify the positive binding peaks. We associated TF with its target gene by searching if there is at least one peak within the regulatory region (10 kb upstream and 5 kb downstream of the transcription start site (TSS)). The interactions between TF and its directed target genes (including lncRNAs) form the TF–gene interactome and were used for network construction.
Establishing miRNA–gene interactome through CLIP-Seq data analysis
To establish miRNA–gene interactions, the Argonaute 2 (Ago2) CLIP-seq data sets were downloaded from GEO (24) (Supplementary Tables S1 and S2). Raw reads were preprocessed with the same protocols used in processing ChIP-seq data to trim adaptors and low quality reads; duplicated reads and short reads (<17 bp) were then discarded. Next, the preprocessed reads were aligned to mm9 using bowtie2 (version 2.2.3) (27) following the procedure in (28) with adjusted parameters (–very-sensitive –rdg 5, 2 –score-min L, -0.6, -0.7); Ago2 binding peaks within the gene locus were then identified with Pirhana (-bin_size_reponse 200, -p_threshold 0.01) (29). To further establish the interactions between miRNA and lncRNA or miRNA and PCG, we used miRanda (default parameters) (30) to predict miRNA binding sites within the peaks. MiRNA–gene interactions were established if there is at least one binding site within the corresponding Ago2 binding peaks.
Constructing gene–gene interactions through co-expression gene analysis
To establish the interacting relationship among genes (mostly PCGs), we used a series of RNA-seq data across the cell lineage progression for both mESCs (31) and C2C12 cells (32) (Supplementary Figures S1 and S2). RNA-seq raw data was converted into FASTQ file using ‘fastq-dump’ from the SRA Toolkit (https://github.com/ncbi/sratoolkit), then filtered using FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), and aligned by commonly used software Tophat2 using default settings (33). Reads mapped to multiple locations within the genome were discarded. The expression level of each gene was quantified by fragment per kilobase exon model per million sequencing reads (FPKM) using Cufflinks (32). Pearson correlation coefficients (PCCs) of a gene pair was calculated by using the gene expression levels at different time points across the lineage progression for both mESCs and C2C12 (Supplementary Figures S1 and S2). The interaction between a pair of genes was established through the co-expression analysis if (i) their gene expression patterns are highly correlated across the time course (i.e. PCC > 0.95); (ii) both of them are expressed at any given time point (i.e. FPKM > 0.5).
Network integration
The integrated network was constructed through combining the above inferred three sub-networks of TF–gene, miRNA–gene and co-expression interactions using an in-house Perl script. We first merged the TF–gene and miRNA–gene interactions to obtain a core sub-network controlled by key TFs and functional miRNAs and then merged the co-expression interactions, during which steps we only included the edges in co-expression network with shared nodes in the aforementioned core sub-network established from TF–gene and miRNA–gene interactions.
Calculating functional information score (FIS) for identifying and prioritizing functional lncRNAs
To identify functional lncRNAs from thousands of known and de novo assembled lncRNA transcripts in a given biological system, we implemented a scoring system FIS to evaluate the functional importance of each lncRNA based on the assumption that, in the integrated network, the importance of a specific lncRNA is associated with the type and strength of the interactions between the lncRNA and its neighboring genes. Specifically, the FIS is calculated using the following equations:
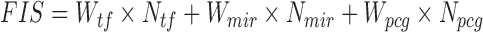 |
(1) |
where Wtf, Wmir and Wpcg are weights (contributions) of each type of network edge (i.e. lncRNA–TF, lncRNA–miRNA and lncRNA–PCG interaction edge) toward the FIS of a specific lncRNA. 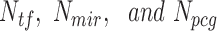 are the normalized numbers of network edges connecting lncRNA node with TF, miRNA and PCG nodes, respectively; they are calculated by the following equations:
are the normalized numbers of network edges connecting lncRNA node with TF, miRNA and PCG nodes, respectively; they are calculated by the following equations:
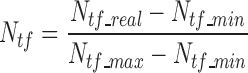 |
(2) |
where Ntf_real, is the total number of lncRNA–TF edges for a lncRNA under the evaluation; Ntf_min and Ntf_max are the minimal and maximal numbers of lncRNA–TF edges among all lncRNA nodes within the network.
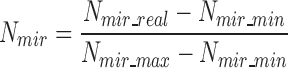 |
(3) |
where Nmir_real, is the total number of lncRNA–miRNA edges for a lncRNA under the evaluation; Nmir_min and Nmir_max are the minimal and maximal numbers of lncRNA–miRNA interaction edges among all lncRNA nodes within the network.
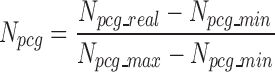 |
(4) |
where Npcg_real, is the total number of lncRNA–PCG edges for a lncRNA under the evaluation; Npcg_min and Npcg_max are the minimal and maximal numbers of lncRNA–PCG interaction edges among all lncRNA nodes within the network.
Inferring the network edge weight for FIS calculation
A machine learning approach using logistic regression model was employed to determine the weight for each kind of lncRNA–gene interaction edge:
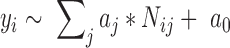 |
(5) |
Here, 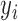 is the indicator of whether a lncRNA is functional
is the indicator of whether a lncRNA is functional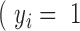 ) or nonfunctional (
) or nonfunctional ( 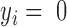 ).
). 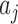 is the vector of regression coefficients, which represents the weight of different type of interaction edge,
is the vector of regression coefficients, which represents the weight of different type of interaction edge, 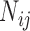 is the normalized number of network edges connecting lncRNA node with TF, miRNA or PCG node, which represents the interaction strength between lncRNA i and type j neighboring nodes (i.e.
is the normalized number of network edges connecting lncRNA node with TF, miRNA or PCG node, which represents the interaction strength between lncRNA i and type j neighboring nodes (i.e. 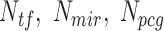 in Equation 1) and
in Equation 1) and 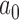 is the bias vector, which was defined as empty. The weight was inferred by using logistic model with a defined training dataset.
is the bias vector, which was defined as empty. The weight was inferred by using logistic model with a defined training dataset.
Generating randomized network for FIS cutoff calculation
Randomized networks for estimating FIS background distribution were generated by a Python library named NetworkX (https://networkx.github.io/). Briefly, 100 randomized networks with the same nodes used in the original network were generated by ‘powerlaw_cluster_graph’ function in NetworkX python package. Then, these randomized networks were used as background to calculate false discovery rate (FDR) in order to determine FIS cutoff for functional lncRNA prediction and prioritization.
Histone ChIP-seq analysis
H3K27ac (GSE37525) and H3K4me3 (GSE25308) ChIP-seq data were downloaded from Gene Expression Omnibus (GEO) database (Supplementary Tables S1 and S2) (24). The raw reads were processed with the protocols described in our previous publication (5). Briefly, the adapter and low quality sequences were trimmed from 3΄ to 5΄ ends. After trimming, reads shorter than 36 bp were discarded. Subsequently, the preprocessed reads were aligned to mouse reference genome (mm9) using a popular software SOAP2 (25). Following alignment, the aligned reads were converted to bed format using Samtools (34) and duplicates were removed by Picard (http://broadinstitute.github.io/picard). Read density over the defined TSS proximal regions of lncRNAs was calculated using homer (35) and plotted by R (36).
Cell cultures
Mouse C2C12 myoblasts (MB) were obtained from ATCC (American Type Culture Collection, Cat. No. CRL-1772) and cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U ml–1 penicillin and 100 μg of streptomycin at 37 °C in 5% CO2. For obtaining differentiating myotubes (MTs), cells were seeded in 60- or 100-mm plates and shifted to DMEM containing 2% horse serum (HS) when 90% confluent to induce differentiation.
Plasmids
For constructing the Snhg1 reporters, a 250 bp region encompassing the 50 bp predicted Yy1 binding site was cloned into the promoter region of a pGL3-Basic-vector between KpnI and HindIII. And a 240 bp region encompassing the 17 bp predicted miR-200b binding site was cloned into the 3΄end region of a pMIR-REPORT Luciferase vector between HindIII and SpeI. For constructing the 9530072K05Rik reporters, a 389 bp region encompassing the 259 bp MyoD binding site was cloned into the promoter region of a pGL3-Basic-vector between KpnI and HindIII. And a 377bp region encompassing the 22bp miR-29b binding site was cloned into the 3΄end region of a pMIR-REPORT Luciferase vector between HindIII and SpeI.
Transient transfection
All the mature miRNA oligos and siRNAs for lncRNAs were purchased from GenePharma. The sequences of siRNAs are listed in Supplementary Table S9). For the transfection of miRNA and siRNA oligos, C2C12 cells were seeded into six-well plates with Lipofectamine 2000 reagent as suggested by the manufacturer (Invitrogen). For luciferase reporter assays, C2C12 cells were transfected with various luciferase reporters in six-well plates. Cell extracts were prepared 48 h after transfection and luciferase activity was monitored as previously described (5,37,38) using Dual-Luciferase kit (Promega).
RNA extraction and qRT-PCR
Total RNAs from cells were extracted using TRIzol reagent (Life Technologies) according to the manufacturer's instructions. For the lncRNAs, cDNAs were prepared using M-MLV (Moloney murine leukemia virus) Reverse Transcriptase (Life Technologies) and Oligo(dT)20 primer. Expression of mRNA analysis was performed with SYBR Green Master Mix (Life Technologies) as described (39,40) on an ABI PRISM 7900HT Sequence Detection System (Life Technologies) using glyceraldehydes 3-phosphate dehydrogenase for normalization. Expression of mature miRNAs were determined using the miRNA-specific Taqman microRNA probe and assay kit (Applied Biosystem) in a 7900HT system (Applied Biosystem) using U6 as a normalization control.
RESULTS
LncFunNet: a computational framework to identify functional lncRNAs
To identify functional lncRNAs from a variety of biological systems, we implemented lncFunNet, a computational framework by systematically exploring gene network constructed by utilizing high-throughput sequencing data. It has three key consecutive modules: (i) network integration; (ii) lncRNA functionality prediction and (iii) lncRNA functional annotation (Figure 1).
Figure 1.

Schematic view of lncFunNet. The lncFunNet composes of three consecutive modules: network integration, lncRNA functionality prediction, and lncRNA functional annotation modules. (A) Inferring TF–lncRNA, TF–miRNA and TF–PCG interactions using ChIP-seq data. (B) Establishing miRNA mediated interactions among miRNAs, lncRNAs, TFs and PCGs. (C) Using gene expression correlation from RNA-seq to infer interactions among lncRNAs and other network components. (D) Constructing a gene regulatory network by integrating the above three sub-networks. (E) Optimizing the weights for the above three types of nodes by logistic regression and calculating a functional information score (FIS) for each lncRNA based on its network interactions (left panel) and selecting functional lncRNAs by calculating false discovery rate (FDR) obtained through comparing to the randomized networks (right panel). (F) Annotating lncRNA functions using GO terms or KEGG pathways associated with its interacting partners.
Network integration module
To construct the integrated network through network integration module, three types of sub-gene networks are included: (a) TF regulatory network; (b) miRNA regulatory network; (c) gene co-expression network. In these subnetworks, key TFs, lncRNAs, miRNAs and PCGs act as nodes; and the interactions among them form the edges (Figure 1). First, to construct the TF regulatory subnetworks formed by key TFs and their target genes (i.e. lncRNAs, miRNAs, PCGs), we selected a list of key TFs based on their biological importance and the availability of ChIP-seq data. The binding peaks of the TFs were defined in the gene regulatory regions (see Materials and Methods). The interactions (regulatory relationships) between a key TF and their targets (i.e. other TFs, lncRNAs, miRNAs and PCGs) can be established if there are TF binding peaks within the regulatory regions of the corresponding loci (Figure 1A). Second, to build the miRNA regulatory sub-networks, we first employed the data from CLIP-seq that allowed us to map the physical interactions between a miRNA and lncRNA or mRNA. We then scanned for specific miRNAs that may bind within those regions using miRanda (30) (Figure 1B, see Materials and Methods). Third, to integrate gene co-expression network into the framework, RNA-seq data was used to calculate PCC for each gene pair if both paired genes are expressed (FPKM > 0.5) (see Materials and Methods). Co-expression network was constructed by selecting the gene pairs with PCC higher than 0.95 (Figure 1C). Lastly, we integrated three sub-networks by merging the shared nodes and edges to form the integrated gene networks using an in-house Perl script (Figure 1D).
LncRNA functionality prediction module
The main aims of this module are: (a) to quantitatively evaluate the functional importance of the tested lncRNA with FIS, a score that measures the likelihood of a lncRNA to be functional in a certain biological system and (b) to decide the cutoff of FIS for ranking and prioritizing functional lncRNAs for further experimental validation. To this end, briefly, the FIS is calculated as the sum of the functional contributions from different lncRNA-Gene interactions (Equation 1, see Materials and Methods). For example, the contribution of the TF–lncRNA gene interaction (formed by the TF binding to the promoter of a lncRNA) is measured by multiplying the weight of TF–lncRNA interaction with the normalized total number of the interactions (Equation 2). The normalized edge number is a number from 0 to 1. The higher the number is, the more interactions are associated with the tested lncRNA. This number can be obtained by counting the edge numbers from the constructed network (see Methods, Equations 2, 3 and 4).
To infer the weight for each type of the network edge, we took advantage of the machine algorithm using logistic regression model (see Materials and Methods). After inferring the weight and the normalized edge number for all lncRNAs tested, we calculated FIS score for each one using the Equation (1). Next, to estimate the cutoff of FIS for differentiating functional and non-functional lncRNAs, we calculated the false discovery rate (FDR) for each cutoff level by generating ∼100 randomized networks (see Method); the FDR was calculated at each FIS cutoff and a value of <0.05 was used as minimal threshold (Figure 1E, see Materials and Methods).
LncRNA functional annotation module
To annotate lncRNA functions, we adopted the approaches used previously (19,41) that associate the function of a lncRNA with its directly connected genes with known functions. Briefly, for a given lncRNA, we obtained a list of directly connected neighboring genes (i.e. TFs, miRNAs and PCGs) within the gene regulatory network, and then retrieved the GO terms annotated to each neighboring node. The most enriched GO terms (adjusted P-value < 0.05) evaluated by hyper-geometric test (42) were assigned to the lncRNA as its annotated functions (Figure 1F). In addition to using the GO terms, we also used the KEGG pathway information for the functional annotation. Briefly, the neighboring genes of the annotated lncRNA were tested for any enriched KEGG pathways (obtained from KEGG database via http://www.kegg.jp/kegg/rest/ using REST-style API). The most enriched KEGG pathways (adjusted P-value < 0.05) evaluated by hyper-geometric test were assigned to the lncRNA as its annotated functions.
LncFunNet accurately identified known functional lncRNAs in mESCs
First, as a proof of concept study, we applied lncFunNet to mESCs for screening lncRNAs that are functionally important to maintain mESC pluripotent considering the availability of a wealth of lncRNA knowledge as well as many available high-through sequencing data for network construction. To this end, we collected 12 ChIP-seq data sets conducted on pluripotent (undifferentiated) mESCs that correspond to 12 key TFs maintaining pluripotency of mESCs (i.e., Pou5f, Nanog, Sox2, Tcf3, Brd4, Esrrb, Klf4, Nr5a2, Prdm14, Smad3, Stat3, Tfcp2l1) (Supplementary Table S1, Supplementary Figure S1) (43–47), one CLIP-seq dataset from pluripotent mESCs (48), and a series of RNA-seq datasets representing four time points of mESC differentiation toward cardiomyocytes (ES cells, mesoderm, cardiac precursor cells and cardiomyocytes) (Supplementary Table S1, Supplementary Figure S1) (31). A list of annotated lncRNA genes from RefSeq (49) and assembled lncRNA transcripts from the published literatures were obtained (1,10,50). To identify functional lncRNAs from the above list, we first filtered out those with an expression level lower than 0.01 FPKM in all stages, which resulted in a list of 2584 lncRNAs for further analyses. To construct the gene network, we first established the interactions between TFs and their target genes (lncRNA, miRNA and PCGs) by analyzing ChIP-seq, CLIP-seq and RNA-seq data. As a result, we created a network that consists of 12 TFs, 246 miRNAs, 2584 lncRNAs and 17 947 PCGs as nodes (Figure 2A, Supplementary Table S3). More than 2 million edges (interactions) were discovered including 8660 TF–lncRNA, 1672 miRNA–lncRNA and 451 372 PCG–lncRNA interactions. To screen functional lncRNAs from the above list using FIS approach, it is critical to obtain positive and negative training datasets for the machine learning model. This has benefited from the large number of lncRNAs experimentally tested in mESCs. In a study conducted by Guttman et al., a set of lncRNAs were subjected to systematic loss of function screening and 137 were identified to cause significant changes of the global gene expression upon knockdown, indicating a possible role in ES cells (13). However, when closely examining this set of 137 lncRNAs by iSeeRNA (51), we found some of them were now updated as coding genes or with length smaller than 200 bp, leaving only 81 available as the positive training dataset. It is even more difficult to obtain a negative training dataset due to the lack of definitive experimental evidence. To overcome the problem, we reasoned that a lncRNA is not expressed in ESC could not be possibly functional in the state. Accordingly, we selected 215 lncRNAs showing no expression (FPKM < 0.01) and also negative for GRO-seq signal in ESC stage, but expressed in differentiated stage (i.e. mesoderm, cardiac precursor cells or cardiomyocytes; FPKM > 0.01) (Supplementary Figure S3A). We then used logistic regression model to learn the weight for different type of Gene-lncRNA interaction by 5-fold cross validation inside the training dataset (see Materials and Methods). As a result, we obtained the optimized weights as 0.57, 0.21 and 0.22 for TF–lncRNA, PCG–lncRNA and miRNA–lncRNA interactions, respectively, which yielded the maximal AUC performance from the regression model (Supplementary Figure S3B). To calculate the FIS cutoff, we used the randomized scale-free network as background (see Materials and Methods) and obtained a cutoff as 0.247 (FDR < 0.05) to determine the functionality of a lncRNA (Figure 2B). As a result, 0.247% (707/2, 584) lncRNAs within the network were predicted as functional. Further analysis showed that the expression levels of these lncRNAs are higher those non-functional lncRNAs in pluripotent stage, which is consistent with the expected roles of these lncRNAs in maintaining pluripotency and suggested the accuracy of our prediction (Figure 2C). To further validate our findings, we compared the functional lncRNAs predicted by our approach with lncRNAs that have been experimentally validated (13). The first set of lncRNAs included 18 lncRNAs (Supplementary Table S4) whose functions are to maintain mESC pluripotency as validated using Nanog luciferase reporter as a read out in a loss-of-function screening (13). Among 18 validated lncRNAs, 14 were computationally predicted as functional in maintaining mESC pluripotency by our approach (77.8%), suggesting the high accuracy of the lncFunNet approach (Figure 2D, top panel). The second validation data set contains 81 lncRNAs affecting mESC global gene expression assessed by loss-of-function assays using lentiviral-based short hairpin RNAs (shRNAs) (13). Our results showed that lncFunNet successfully predicted 60.5% (49/81) of these lncRNAs (Figure 2D bottom panel). Collectively, these results demonstrate that our integrated computational framework can indeed predict the functionality of lncRNAs with high accuracy. In addition, among 707 predicted functional lncRNAs, 94.0% (658/707) are previously uncharacterized. Using lncFunNet to annotate their functions with the statistically significant GO terms, we found that the majority of these lncRNAs are related to stem cell maintenance (30.7%), others were annotated as endodermal cell fate specification, or somatic stem cell maintenance, suggesting that most of the predicted functional lncRNAs are indeed associated with mESC maintenance and fate specification (Figure 2E, Supplementary Table S5). In addition, we also applied KEGG pathway database, consistent with the GO analysis, we found that many of the lncRNAs are assigned to signaling pathways regulating pluripotency of stem cells (29.5%) (Supplementary Figure S4).
Figure 2.

Identification of functional lncRNAs from mESCs. (A) Bird's view of gene regulatory network in mESCs. The network consists of 12 TFs, 246 miRNAs, 2584 lncRNAs and 17 947 PCGs. (B) Plots of FIS distribution for observed (red) and randomized (black) networks. The cutoff of FIS, to identify functional lncRNAs, is 0.247 with FDR < 0.05. (C) Boxplots showing gene expression levels of 707 predicted functional lncRNAs across different stages of mESC lineage progression. ESC: embryonic stem cell; MES: mesoderm; CP: cardiac precursor; CM: cardiomyocyte. (D) Venn diagrams showing the overlapping between 707 predicted functional lncRNAs and (i) 18 experimentally validated functional lncRNAs that are critical to maintain mESC pluripotency (top panel); (ii) 81 lncRNAs whose knockdown caused significant global gene expression change in mESCs (bottom panel). Within 707 predicted functional lncRNAs, 658 (94.0%) have not been annotated before. (E) Bar chart representing percentages of lncRNAs assigned to the indicated GO terms. (F) ROC curves or (G) AUPRC curves for LncFunNet (blue) compared to the model using co-expression only network (green).
To test whether LncFunNet gives rise to a higher accuracy in predicting the functionality of lncRNAs compared with the commonly used co-expression network approach (18,19), we built a co-expression network by using the RNA-seq datasets from the mESC differentiation toward cardiomyocytes. A co-expression edge was defined if the PCC for the co-expressed gene pairs is >0.95 or <–0.95. The normalized number of interactions between lncRNA and other genes was used to evaluate the functionality of each lncRNA. For comparison purpose, we performed receiver operator characteristic curves (ROCs) and the area under the precision and recall curves (AUPRCs) analysis by a python library named scikit-learn (52), respectively. We found that the accuracy using LncFunNet is much higher than the model using co-expression only network in terms of both sensitivity and specificity (Figure 2F and G).
LncFunNet uncovers functional lncRNAs in skeletal muscle cells
To test whether lncFunNet with the trained weights can be adopted to other biological systems, next, we applied lncFunNet to skeletal muscle cells (C2C12) that go through myogenic differentiation process in which, upon serum withdraw, proliferating myoblast (MB) exit cell cycle and is fuse to form multinucleated myotube (MT) (3–5). To this end, we started from a lncRNA gene list that was collected from RefSeq (49) and de novo assembled lncRNA transcripts from our previous study (5). After passing the expression filter (FPKM > 0.01), we obtained 2164 and 2538 lncRNAs expressed in MB and MT, respectively. To build the integrated gene networks, a collection of 11 and 14 TF ChIP-seq binding profiles were obtained from ENCODE (53) and other published studies (Supplementary Table S2) to infer TF–gene interactome in MB and MT, respectively. To identify lncRNA–miRNA interactions, CLIP-seq data in MB and MT was downloaded to define the interaction site (28); specific lncRNA–miRNA interactions were then predicted using miRNA target prediction program miRanda (30) within the Ago2 binding sites (see Materials and Methods); Lastly, RNA-seq data from the muscle cells corresponding to four time points along the differentiation course, i.e. –24 h (MB), 60, 120 and 168 h MT, were also collected (10) (Supplementary Figure S2 and Supplementary Table S2). Using these data, two integrated gene networks were constructed separately in MB and MT. The MB gene network contains 11 TFs, 2164 lncRNAs, 173 miRNAs and 11 204 PCG nodes, as well as 1 399 951 edges (interactions) (Figure 3A, Supplementary Table S3). Similarly, in MT gene network, there are 14 TFs, 2538 lncRNAs, 173 miRNAs and 11 966 PCG nodes and 2 096 061 edges (Figure 3B, Supplementary Table S3). To further identify functional lncRNAs, we calculated FIS for each lncRNA node in the gene networks and used FIS of 0.242 (MB, Figure 3C) and 0.259 (MT, Figure 3D) as cutoffs to predict the functional lncRNAs (FDR < 0.05). As a result, we obtained 752 and 860 functional lncRNAs in MB and MT, respectively (Figure 3E). Next, comparing the two sets, we classified the lncRNAs into three categories: (i) MB-specific (317), (ii) MT-specific (435) and (iii) constitutive lncRNAs (425), which were shared in both MB and MT (Figure 3E). To further annotate the functionality of the identified functional lncRNAs using lncFunNet, we found that the majority of the predicted MB-specific lncRNAs are associated with regulation of transcription (Figure 3F), while in MT, they are related to transcription from RNA polymerase II promoter and muscle contraction (Figure 3G), suggesting stage specific functions of lncRNAs during myogenesis (Supplementary Tables S6 and S7).
Figure 3.

Identification of functional lncRNAs in MB and MT. (A, B) Bird's view of gene regulatory networks in MB (A) and MT (B). In MB, the network consists of 11 TFs, 163 miRNAs, 2164 lncRNAs and 11 240 PCGs; while in MT, the network consists of 14 TFs, 173 miRNAs, 2538 lncRNAs and 11 966 PCGs. (C, D) FIS distributions in MB (C) and MT (D). Using FDR < 0.05, the cutoff of FISs for MB and MT are 0.242 and 0.259, respectively. (E) Venn diagram showing the overlapping of predicted functional lncRNAs in MB and MT. 752 lncRNAs was predicted as functional in MB. Within them, 42.2% (317/752) are MB specific, and 57.8% (435/752) are found shared in both MB and MT. In MT, a total of 860 lncRNAs were predicted functional, among which 49.4% (425/860) is MT specific. (F) Functional annotation of lncRNAs in MB by GO analysis. (G) Functional annotation of lncRNAs in MT by GO analysis.
To further explore the genomic characteristics of the predicted functional lncRNAs in C2C12 cells, we first investigated the expression patterns of MB and MT-specific, and the constitutive sets of lncRNAs across four time points of myogenic differentiation (–24, 60, 120 and 168 h) (Supplementary Figure S2). As expected, MB-specific lncRNAs are highly expressed in MBs but down-regulated gradually during the differentiation (Figure 4A); MT-specific lncRNAs are expressed significantly lower in MBs than in MTs (Figure 4A); and expectedly, the expression levels of the constitutive lncRNAs during myogenesis are comparatively stable (Figure 4A). In addition, as expected, in both MB and MT, the functional lncRNAs exhibited higher expression levels than non-functional ones (Figure 4B). It is believed that multiple TF binding in the promoter of lncRNA or miRNA binding within lncRNA transcript are important features for functional lncRNAs in variety of biological systems (12,13,21,54,55). Indeed, when calculating the total number of TFs and miRNAs binding to each lncRNA, we found functional lncRNAs are bound by a much higher number of TFs compared to non-functional lncRNAs. For example, in both MBs and MTs, >90% of function lncRNAs are bound by at least four TFs, while 50% non-functional lncRNAs are bound by less than two TFs (Figure 4C). Similarly, 6% of functional lncRNAs in MBs and 8% in MTs are bound by more than five miRNAs. In contrast, only 3% of non-functional lncRNAs has more than five miRNAs potentially regulating them (Figure 4D). We next examined the characteristics of histone marks associated with active transcription such as H3K4me3 (histone 3 lysine 4 trimethylation) and H3K27ac (histone 3 lysine 27 acetylation) in the promoter region of the lncRNAs by calculating the tag densities using 20 bp bins within the flanking regions (±5 kb) of putative TSSs defined by a least one H3K4me3 peaks at 5΄ end. In line with findings from previous studies (56), we found that the enrichment of these histone marks for functional lncRNAs are much higher compared with non-functional ones (57). Furthermore, the levels of H3K4me3 and H3K27ac sharply decreased within 100 bp upstream of the TSS, nucleosomes are depleted near the TSS to facilitate binding of transcriptional machineries including RNA polymerase II and associated TFs (58) (Figure 4E–H). Taken together, our results demonstrated significant differences between functional and non-functional lncRNAs in terms of associated genomic features.
Figure 4.

Integrative analysis of functional lncRNAs in MB and MT. (A) Box plots showing the global gene expression patterns for MB and MT specific lncRNAs, and those shared in both MB and MT across four time points from proliferation (–24 h) to differentiation (60, 120 and 168 h). MB specific lncRNAs (green) are expressed higher in MB (–24 h); MT specific lncRNAs (red) are expressed higher in MT. The shared lncRNAs (blue) are constitutively expressed high in both stages. (B) Gene expression patterns for functional and non-functional lncRNAs in MB (left) and MT (right). In both stages, functional lncRNAs are expressed higher than non-functional lncRNAs. (C) The total number of interacting TFs for functional and non-functional lncRNAs. The data indicates that functional lncRNAs are more likely to be bound by TFs comparing to non-functional lncRNAs. (D) Comparison of the total number of interacting miRNAs for functional and non-functional lncRNAs. (E) Enrichment of H3K4me3 mark around TSSs of lncRNAs in MB. (F) Enrichment of H3K27ac mark around TSSs of lncRNAs in MB. (G) Enrichment of H3K4me3 histone mark around TSSs of lncRNAs in MT. (H) Enrichment of H3K27ac histone mark around TSSs of lncRNAs in MT.
Functional lncRNAs in muscle cells are key motif components of integrative gene networks
Previous studies suggest that network motifs, a set of recurring and statistically significant sub-graphs or patterns normally compositing of three or four nodes, are responsible for carrying out specific information-processing functions (59–61). Here, we explored several types of network motifs in myogenesis. Specifically, we focused on 3- and 4-node network motifs that are typical TF mediated feed-forward loops (FFLs) with lncRNA involved. As a result, we have categorized those FFL network motifs into the five different types based on their node compositions and regulatory relationships among the nodes (Figure 5A (I–V)). Briefly, these are (I) TF–miRNA–lncRNA motif in which TF regulates lncRNA and miRNA loci, and miRNA regulates lncRNA; (II) TF–TF–lncRNA motif in which both TFs regulate lncRNA and one TF can also regulate the other; (III) TF–lncRNA–lncRNA motif in which one TF regulates two lncRNA genes; (IV) TF–PCG–lncRNA motif in which one TF regulates one PCG and one lncRNA gene; (V) In addition to 3-node motifs, we also identified 4-node, i.e. TF–TF–lncRNA–lncRNA motif (also called bi-fan motif) in which two TFs coordinately regulate two lncRNAs (Figure 5A (V)). We also noticed that the majority of these bi-fan network motifs tend to contain at least one functional lncRNA node, suggesting that lncRNA is a crucial component of functional networks; vice versa, the functionality of a lncRNA can be inferred from the network motifs it involves in (Figure 5B). Further analysis revealed that most of lncRNAs are involved in more than one types of network motif. Among 2164 expressed lncRNAs in MBs, 57 (2.6%) formed type I network motifs, 2045 (94.5%) formed type II network motifs, 712 (32.9%) formed type III motifs, 714 (33.0%) formed type IV motifs and 1931 (89.2%) formed type V network motifs (Figure 5C). Similarly, in MTs, among 2538 lncRNAs, 97 (3.8%), 2474 (97.5%), 790 (31.1%), 792 (31.2%) and 2436 (96.0%) formed types I to V network motifs, respectively (Figure 5C). In addition, we also found that many functional lncRNAs are shared by 3-node and 4-node network motifs in both MB and MT (Supplementary Figure S5A and B). Taken together, our results suggest that functional lncRNAs predicated by lncFunNet are key players in the biological network motifs in C2C12 cells. Interestingly, within the gene networks, some well-studied network motifs can be found. Linc-md1 is an outstanding example which was found to act as sponge for miR-133a (12). As shown in Figure 5D, in MBs, Linc-md1 interacts with a few TFs/miRNAs and the interaction is much stronger in MTs where it connects to many miRNAs including miR-133a, miR-19b, miR-152 and miR-324 and TFs such as MyoD, Myog and Rest (Figure 5D and E). Such lncRNA mediated motifs were commonly seen in the networks as illustrated in two other examples lncRNAs C130080G10Rik and CUFF.35670 (Supplementary Figure S5C and D). Altogether the above analyses demonstrate that the integrated network approach is not only useful for identification of functional lncRNAs, but also for further inferring their functional mechanisms.
Figure 5.

Analysis of network motifs in lncRNA involved gene regulatory networks. (A) Five types of lncRNA involved network motifs. Types I to IV are three-node motifs that form the feed-forward loop (FFL) circuitries. Type V motif is a four-node bi-fan motif. The network motif nodes are TF (orange), lncRNA (purple), miRNA (green) and PCG (blue). (B) In each of the above motif types the percentage of motifs with functional lncRNA as a node is higher than those with non-functional lncRNA as a node. (C) Venn diagrams show the overlapping of functional lncRNAs in type I to type IV motifs in MB (left) and MT (right). The majority of lncRNAs involve in type II network motif in which two TFs regulate one lncRNA node. In addition, many lncRNAs can be found in more than one type of network motifs. (D) Gene regulatory sub-networks involving Linc-md1 in MB (left) and MT (right). Linc-md1 displays stronger interaction with a higher number of TFs and miRNAs in MT than in MB, thus a large size of the node. (E) Illustration of type I network motif (left) involves Linc-md1. TFs (orange) regulate both Linc-md1 (purple) and miRNAs (green); MiRNA negatively regulates Linc-md1. Right panel shows the tracks of a list of TFs binding to the Linc-md1 promoter from ChIP-seq data and the binding of miRNAs to Linc-md1 in the identified CLIP-seq peak in MB (top) and MT (bottom).
Experimental validation of functional lncRNAs in MB and MT
The ultimate approach for testing the accuracy of our results is to experimentally validate the functionality of the identified lncRNAs and the associated interactions. To this end, we selected 10 predicted functional lncRNAs in both MB and MT with two criteria: (i) comparatively higher FIS (Supplementary Figure S6) and (ii) the ability to form interactions with miRNAs because miRNA binding appears to confer a higher FIS (Supplementary Figure S6). First, we confirmed their expression profiles. All 10 of the selected lncRNAs (Supplementary Table S8) were readily detected by quantitative qRT-PCR during muscle cell differentiation. Some were highly induced during the course of differentiation whereas others were repressed. The differential expression dynamics of these lncRNAs indicate their diverse roles in myogenesis, for example, the lncRNAs with increasing expression during differentiation may play a promoting role in the process (Figure 6A). Indeed, when five of such lncRNAs were knocked down by a siRNA oligo (Figure 6B), C2C12 differentiation was inhibited as assessed by a Myogenin luciferase reporter assay (Figure 6C). To further validate the involvement of functional lncRNAs in three-node network motifs and their interactions, we selected Snhg1 (small nucleolar RNA host gene 1) for further experimentation. It has been reported to promote cell proliferation in non-small cell lung cancer and hepatocellular carcinoma tumorigenesis (62,63) but with unexplored function in skeletal myogenesis. By our prediction, Snhg1 interacts with YY1 and miR-200b through direct binding with them to form a type I three-node FFL network motif. An YY1 binding site was identified upstream of Snhg1 TSS (6.5 kb upstream of TSS) by analyzing YY1 ChIP-seq; an Ago2 binding peak was found and four binding sites for miR-200b were predicted (Figure 7A). Indeed, by ChIP-PCR, an enrichment of YY1 on Snhg1 promoter was detected (Figure 7B). Furthermore, overexpression of YY1 in C2C12 increased its expression (Figure 7C) while knockdown of YY1 repressed it (Figure 7D), suggesting YY1 binding to Snhg1 causes the increase of its transcription. This was further confirmed by cloning a 250 bp fragment of Snhg1 promoter encompassing the YY1 binding site into a luciferase reporter; results from the reporter assay showed that YY1 positively regulates Snhg1 expression (Figure 7E and F). To dissect miR-200b-Snhg1 regulation, as expected, overexpression of miR-200b inhibited Snhg1 expression (Figure 7G). To further demonstrate the possibility of post-transcriptional degradation by miR-200b through direct binding, we selected the predicted binding site that resides within the Snhg1 exon 10. A 240 bp fragment encompassing the site was fused with luciferase gene; when transfected into C2C12 cells with miR-200b mimic oligos, a significant decrease of the reporter activity was observed. (Figure 7H). These experimental evidences demonstrate that Snhg1, a pro-myogenic lncRNA, is regulated by YY1 and miR-200b.
Figure 6.

Experimental validation of the predicted functional lncRNAs in C2C12 myogenic differentiation. (A) Gene expression levels of 10 predicted functional lncRNAs were measured by qRT-PCR at various time points during C2C12 differentiation. All PCR data were normalized to GAPDH mRNA and represent the average of three independent experiments ± S.D. (B and C) siRNA oligos targeting each of the selected lncRNAs were transfected into C2C12 cells together with a Myogenin luciferase reporter, 48 h after differentiation, Luciferase activities were determined and normalized to Renilla protein. Relative Luciferase Unit (RLU) is shown with respect to wild type and negative control oligos (siNC) transfection where luciferase activities were set to a value of 1. The results suggested promoting functions of the lncRNAs in myogenic differentiation. All luciferase data represent the average of three independent experiments ± S.D. The P-value was determined by Student's t-test: *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 7.

Experimental validation of two lncRNA mediated network motifs in C2C12 cells. (A) A TF–miRNA–lncRNA network motif involving lncRNA, Snhg1 (left). A Snapshot of genomic features (Pol 2, YY1 and MyoD1 ChIP-seq as well as CLIP-seq in separate tracks) on Snhg1 transcript (red track on top) in MB. (B) Validation of the binding of YY1 on the Snhg1 promoter by ChIP-PCR. Enrichment fold was calculated as percentage of enrichment of total input. (C) Overexpression of YY1 in C2C12 increased Snhg1 expression by RT-qPCR. (D) Knockdown of YY1 by shYY1 plasmid repressed Snhg1 expression by RT-qPCR. (E) C2C12 cells were transfected with a Luciferase reporter encompassing the binding site of YY1 from Snhg1 promoter and together with an YY1 expression plasmid and Renilla luciferase reporter plasmid. Luciferase activities were determined at 48 h post-transfection and normalized to Renilla protein. Relative luciferase unit (RLU) is shown with respect to wild type and vector control transfection where luciferase activities were set to a value of 1. The result showed that overexpression of YY1 increased the reporter activity. (F) Knockdown of YY1 inhibited the above reporter activity. (G) miR-200b mimic or negative control oligos were transfected into C2C12 cells and its overexpression inhibited Snhg1 expression. (H) A luciferase reporter encompassing the miR-200b binding site on Snhg1 (the last exon of 3΄ end) were transfected into C2C12 cells together with the miR-200b mimic oligos and Renilla luciferase reporter plasmid. Luciferase activities were determined at 48 h post-transfection and normalized to Renilla protein. The result showed that overexpression of miR-200b decreased the reporter activity. (I) A TF–miRNA–lncRNA network motif mediated by MyoD1 (left). A Snapshot of genomic features (Pol II, MyoD and MyoG ChIP-seq as well as CLIP-seq in separate tracks) around lncRNA 9530072K05Rik transcript (red track on top) in MB. (J) Validation of MyoD binding on lncRNA 9530072K05Rik's promoter by ChIP-PCR. (K) Overexpression of MyoD in C2C12 cells repressed 9530072K05Rik expression. (L) Knocking down of MyoD increased 9530072K05Rik expression. (M, N) C2C12 cells were transfected with a Luciferase reporter encompassing the binding site of MyoD from 9530072K05Rik promoter and together with a MyoD expression plasmid and Renilla luciferase reporter plasmid. Luciferase activities were determined at 48h post-transfection and normalized to Renilla protein. The result showed that overexpression of MyoD decreased the reporter activity. (O) Overexpression of miR-29 by transfecting mimic oligos inhibited 9530072K05Rik expression as compared to negative control oligos. All PCR data were normalized to GAPDH mRNA or U6 (for miRNA expression) and represent the average of three independent experiments ± S.D. All luciferase data were normalized to Renillia protein and represent the average of three independent experiments ± S.D. The P-value was determined by Student's t-test: *P < 0.05, **P < 0.01, ***P < 0.001.
To strengthen the findings, we further tested another lncRNA, 9530072K05Rik which was predicted to form type I network motif by interacting with MyoD and miR-29b through direct binding (Figure 7I) but its function in muscle cells remains unknown. Interestingly, MyoD binding on its promoter (350 bp upstream of TSS) was found to suppress its expression (Figure 6J–L); miR-29b was also found to decrease its level through binding to the 3΄ end of 9530072K05Rik (Figure 7O and P). Altogether these results strengthened the reliability of our network approach in inferring the lncRNA functional mechanisms. It also demonstrated the previously underappreciated complexity of interactions that lncRNAs are involved in through binding with TFs and miRNAs during the process of myogenic differentiation.
DISCUSSION
With the tremendous increasing of the transcriptome sequencing data from different cell/tissue types, thousands of lncRNAs have been identified through de novo assembling the data. To develop computational framework that can be used to systematically identify potentially functional lncRNAs and lead towards the understanding of their functions provides a huge impetus in the field, but remains a challenging task (8,14). Increasing studies demonstrate the multi-potency of lncRNAs as interacting hubs with other macromolecules including proteins, DNAs and RNAs (3–5,12,13,16,17,45); inferring lncRNA functions through its interacting partners has thus been made possible by the advancement in detecting these interactions through a variety of high-throughput sequencing methodologies such as ChIP-seq, RNA-seq and CLIP-seq (21,22). In this study, we described a robust and accurate computational framework lncFunNet to predict and prioritize functional lncRNAs and further annotate their functions in a specific biological system based on the integrated gene networks constructed by harnessing the molecular interaction data. By applying lncFunNet to mESCs, we have accurately recovered more than 60% functional lncRNAs that were previously identified by different experimental assays. In addition, we also identified ∼700 novel lncRNAs that have not been annotated previously, which opened new avenues for future research investigation. When applying lncFunNet to C2C12 muscle cells, we also identified 1177 novel lncRNAs with distinct genomic features. For the selected 10 novel lncRNAs with high FISs and high FPKM value, we successfully validated their expression by RT-PCR and functions in myogenesis using loss of function assays, demonstrating the reliability of lncFunNet; this effort thus has filled the gap of knowledge in the lncRNA involved regulation of myogenesis.
The unique features of lncFunNet are multiple folds. First, it is the first attempt to systematically integrate multiple genomic data to predict and prioritize functional lncRNAs. The commonly used co-expression network approach uses only the correlations among lncRNAs and their co-expressed genes to annotate lncRNA functions, thus is limited by the fact that (i) some lncRNAs do not have co-expressed genes; (ii) co-expression does not always infer co-function. LncFunNet, on the other hand, expands the network construction by including TF–lncRNA and miRNA–lncRNA interactions derived from ChIP-seq and CLIP-seq data. Indeed, ROC and AURPC analyses showed that LncFunNet yielded a much higher accuracy than co-expression analysis alone. In mESCs, lncFunNet yielded a very low (6%) false positive rate, while the false negative rate is high (35.8%). The integration of a greater number of high-throughput sequencing datasets in particularly the TF ChIP-seq data in the network construction is expected to increase the likelihood of predicting the true functional lncRNAs and decrease the false negative rate. Second, advanced machine learning algorithm using logistic regression model was implemented in lncFunNet to allow us to calculate FIS and thus stands as the first framework to provide a systemic scoring system for quantitative evaluation of the functionality of lncRNAs. Indeed, the optimized weight, when applying in mESCs has significantly increased the prediction accuracy of lncFunNet as compared to the model relying only on the co-expression network.
Thirdly, not only our approach can answer whether an lncRNA is functional or not, but also can provide insights into how an lncRNA functions through identifying the interacting partners i.e. the binding TFs and miRNAs in network motifs. As a result, in C2C12, Snhg1, an lncRNA with previously unknown function was identified to promote myogenic differentiation. Moreover, we verified that Snhg1 is bound by YY1 on its promoter and positively regulated in myoblast cells. It is also bound by miR-200b and down-regulated. Interestingly, transfection of miR-200b could maintain C2C12 cells in an undifferentiated stage. The functional antagonization further suggested that miR-200b probably binds and down-regulates Snhg1 expression, supporting the lncRNA–miRNA interaction in a non-ceRNA manner. Similarly, we also validated another predicted network motif, 9530072k05Rik-miR-29b-MyoD. Interestingly, in contrast to the known transcriptional activating effect, MyoD binding to 9530072k05Rik, appeared to down-regulate its expression, which needs to be further investigated. MiR-29b, a previously known myogenic regulator repressed its level, again supporting that miRNA binding lncRNA can down-regulate their expression transcriptionally or post-transcriptionally. These two examples illustrated the predicting power of our approach and opened the door for further mechanistic investigations for many novel lncRNAs.
Several studies have established comprehensive databases to collect the miRNA-lncRNA interactions in various species and tissue/cell types, thus provide useful resources for lncRNA functional prediction (22,23). In our study, combining Ago2 CLIP-seq data and in silico predictions is proven to be an effective way of defining miRNA-lncRNA binding and significantly reduced the false positive rate compared to using the in silico prediction alone. Subsequently, we identified 1.78% (46/2584), 3.79% (82/2164) and 5.08% (127/2538) lncRNAs interacting with miRNAs in mESC, MB and MT, respectively. These observations are in line with the numbers of miRNA–lncRNA interactions reported by Li et al. and others in their databases for different cell types (22,23). With the improving sensitivity in detecting RNA–protein interaction, we believe a higher number of such interactions can be detected. Even though only ∼3–5% lncRNAs were predicted to interact with miRNAs in C2C12 cells, these lncRNAs showed much stronger functional potential comparing to those without interacting miRNAs, suggesting the important contribution of miRNA–lncRNA interactions to lncRNA functions (Supplementary Figure S7).
We should also point out that the integration of the comprehensive TF–lncRNA interactions from multiple ChIP-seq data in lncFunNet significantly increased the prediction accuracy, however, it also constraints the usage of lncFunNet in cells/tissues where such comprehensive TF ChIP-seq data is lacking. To solve the problem, emerging studies demonstrated that the genome-wide TF and gene interactions can be accurately predicted using the open chromatin regions identified through DNase-seq (64) or ATAC-seq (65), thus providing an alternative way to establish the TF–lncRNA interactome without ChIP-seq.
Finally, although in this study three types of genomic data including ChIP-seq, RNA-seq and CLIP-seq were employed, in the future the network approach can be expanded to include many other types of genomic interaction data such as RNA-DNA interactions obtained from Chromatin Isolation by RNA Purification (CHIRP) (66) or Capture Hybridization Analysis of RNA Targets (CHART) (67) data. More complex networks can be built to not only understand the functionality of lncRNAs but also to better study the complex molecular interactomes in a given cellular system.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Kevin Yuk-Lap Yip for the thoughtful discussion and the reviewers for their insightful comments on the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
General Research Funds (GRF) from the Research Grants Council (RGC) of the Hong Kong Special Administrative Region [Project Code: 473713, 14113514, 14102315 to H.S. and 476113, 14133016, 14116014, 14100415 to H.W.]; RGC Collaborative Research Fund (CRF) [Project Code: C6015-14G to H.S. and H.W.]; a grant from the Ministry of Science and Technology of China (Project Code: 2014CB964700 to H.W.). Funding for open access charge: General Research Funds (GRF) from the Research Grants Council (RGC) of the Hong Kong Special Administrative Region.
Conflict of interest statement. None declared.
REFERENCES
- 1. Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P. et al. . Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ponting C.P., Oliver P.L., Reik W.. Evolution and functions of long noncoding RNAs. Cell. 2009; 136:629–641. [DOI] [PubMed] [Google Scholar]
- 3. Lu L., Sun K., Chen X., Zhao Y., Wang L., Zhou L., Sun H., Wang H.. Genome-wide survey by ChIP-seq reveals YY1 regulation of lincRNAs in skeletal myogenesis. EMBO J. 2013; 32:2575–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang L., Zhao Y., Bao X., Zhu X., Kwok Y.K., Sun K., Chen X., Huang Y., Jauch R., Esteban M.A. et al. . LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015; 25:335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou L., Sun K., Zhao Y., Zhang S., Wang X., Li Y., Lu L., Chen X., Chen F., Bao X. et al. . Linc-YY1 promotes myogenic differentiation and muscle regeneration through an interaction with the transcription factor YY1. Nat. Commun. 2015; 6:10026. [DOI] [PubMed] [Google Scholar]
- 6. Lee J.T., Bartolomei M.S.. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013; 152:1308–1323. [DOI] [PubMed] [Google Scholar]
- 7. Wapinski O., Chang H.Y.. Long noncoding RNAs and human disease. Trends Cell Biol. 2011; 21:354–361. [DOI] [PubMed] [Google Scholar]
- 8. Kung J.T.Y., Colognori D., Lee J.T.. Long noncoding RNAs: past, present, and future. Genetics. 2013; 193:651–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carpenter S., Aiello D., Atianand M.K., Ricci E.P., Gandhi P., Hall L.L., Byron M., Monks B., Henry-Bezy M., Lawrence J.B. et al. . A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013; 341:789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guttman M., Garber M., Levin J.Z., Donaghey J., Robinson J., Adiconis X., Fan L., Koziol M.J., Gnirke A., Nusbaum C. et al. . Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010; 28:503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tripathi V., Ellis J.D., Shen Z., Song D.Y., Pan Q., Watt A.T., Freier S.M., Bennett C.F., Sharma A., Bubulya P.A. et al. . The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell. 2010; 39:925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cesana M., Cacchiarelli D., Legnini I., Santini T., Sthandier O., Chinappi M., Tramontano A., Bozzoni I.. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011; 147:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guttman M., Donaghey J., Carey B.W., Garber M., Grenier J.K., Munson G., Young G., Lucas A.B., Ach R., Bruhn L. et al. . lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011; 477:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mattick J.S., Rinn J.L.. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015; 22:5–7. [DOI] [PubMed] [Google Scholar]
- 15. Anderson D.M., Anderson K.M., Chang C.-L., Makarewich C.A., Nelson B.R., McAnally J.R., Kasaragod P., Shelton J.M., Liou J., Bassel-Duby R. et al. . A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015; 160:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsai M.-C., Manor O., Wan Y., Mosammaparast N., Wang J.K., Lan F., Shi Y., Segal E., Chang H.Y.. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010; 329:689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grote P., Wittler L., Hendrix D., Koch F., Währisch S., Beisaw A., Macura K., Bläss G., Kellis M., Werber M. et al. . The tissue-specific lncRNA Fendrr Is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013; 24:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liao Q., Liu C., Yuan X., Kang S., Miao R., Xiao H., Zhao G., Luo H., Bu D., Zhao H. et al. . Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011; 39:3864–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo X., Gao L., Liao Q., Xiao H., Ma X., Yang X., Luo H., Zhao G., Bu D., Jiao F. et al. . Long non-coding RNAs function annotation: a global prediction method based on bi-colored networks. Nucleic Acids Res. 2012; doi:10.1093/nar/gks967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P.. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language?. Cell. 2011; 146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Du Z., Sun T., Hacisuleyman E., Fei T., Wang X., Brown M., Rinn J.L., Lee M.G.-S., Chen Y., Kantoff P.W. et al. . Integrative analyses reveal a long noncoding RNA-mediated sponge regulatory network in prostate cancer. Nat. Commun. 2016; 7:10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J.-H., Liu S., Zhou H., Qu L.-H., Yang J.-H.. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paraskevopoulou M.D., Vlachos I.S., Karagkouni D., Georgakilas G., Kanellos I., Vergoulis T., Zagganas K., Tsanakas P., Floros E., Dalamagas T. et al. . DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016; 44:D231–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barrett T., Troup D.B., Wilhite S.E., Ledoux P., Rudnev D., Evangelista C., Kim I.F., Soboleva A., Tomashevsky M., Edgar R.. NCBI GEO: mining tens of millions of expression profiles–database and tools update. Nucleic Acids Res. 2007; 35:D760–D765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li R., Yu C., Li Y., Lam T.-W., Yiu S.-M., Kristiansen K., Wang J.. SOAP2: an improved ultrafast tool for short read alignment. Bioinforma. Oxf. Engl. 2009; 25:1966–1967. [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. . Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang X., Zuo X., Yang B., Li Z., Xue Y., Zhou Y., Huang J., Zhao X., Zhou J., Yan Y. et al. . MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014; 158:607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uren P.J., Bahrami-Samani E., Burns S.C., Qiao M., Karginov F.V., Hodges E., Hannon G.J., Sanford J.R., Penalva L.O.F., Smith A.D.. Site identification in high-throughput RNA–protein interaction data. Bioinformatics. 2012; 28:3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Betel D., Wilson M., Gabow A., Marks D.S., Sander C.. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008; 36:D149–D153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wamstad J.A., Alexander J.M., Truty R.M., Shrikumar A., Li F., Eilertson K.E., Ding H., Wylie J.N., Pico A.R., Capra J.A. et al. . Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012; 151:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup . The sequence alignment/map format and SAMtools. Bioinforma. Oxf. Engl. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ihaka R., Gentleman R.. R: a language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996; 5:299–314. [Google Scholar]
- 37. Zhou L., Wang L., Lu L., Jiang P., Sun H., Wang H.. Inhibition of miR-29 by TGF-beta-Smad3 Signaling through Dual Mechanisms Promotes Transdifferentiation of Mouse Myoblasts into Myofibroblasts. PLoS ONE. 2012; 7:e33766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou L., Wang L., Lu L., Jiang P., Sun H., Wang H.. A novel target of MicroRNA-29, Ring1 and YY1-binding protein (Rybp), negatively regulates skeletal myogenesis. J. Biol. Chem. 2012; 287:25255–25265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang L., Zhou L., Jiang P., Lu L., Chen X., Lan H., Guttridge D.C., Sun H., Wang H.. Loss of miR-29 in myoblasts contributes to dystrophic muscle pathogenesis. Mol. Ther. 2012; 20:1222–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu L., Zhou L., Chen E.Z., Sun K., Jiang P., Wang L., Su X., Sun H., Wang H.. A novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PLoS ONE. 2012; 7:e27596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Necsulea A., Soumillon M., Warnefors M., Liechti A., Daish T., Zeller U., Baker J.C., Grützner F., Kaessmann H.. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014; 505:635–640. [DOI] [PubMed] [Google Scholar]
- 42. Martin D., Brun C., Remy E., Mouren P., Thieffry D., Jacq B.. GOToolBox: functional analysis of gene datasets based on Gene Ontology. Genome Biol. 2004; 5:R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marson A., Levine S.S., Cole M.F., Frampton G.M., Brambrink T., Johnstone S., Guenther M.G., Johnston W.K., Wernig M., Newman J. et al. . Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008; 134:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. et al. . Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008; 133:1106–1117. [DOI] [PubMed] [Google Scholar]
- 45. Heng J.-C.D., Feng B., Han J., Jiang J., Kraus P., Ng J.-H., Orlov Y.L., Huss M., Yang L., Lufkin T. et al. . The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell. 2010; 6:167–174. [DOI] [PubMed] [Google Scholar]
- 46. Ma Z., Swigut T., Valouev A., Rada-Iglesias A., Wysocka J.. Sequence-specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat. Struct. Mol. Biol. 2011; 18:120–127. [DOI] [PubMed] [Google Scholar]
- 47. Mullen A.C., Orlando D.A., Newman J.J., Lovén J., Kumar R.M., Bilodeau S., Reddy J., Guenther M.G., DeKoter R.P., Young R.A.. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011; 147:565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leung A.K.L., Young A.G., Bhutkar A., Zheng G.X., Bosson A.D., Nielsen C.B., Sharp P.A.. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat. Struct. Mol. Biol. 2011; 18:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pruitt K.D., Maglott D.R.. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 2001; 29:137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sigova A.A., Mullen A.C., Molinie B., Gupta S., Orlando D.A., Guenther M.G., Almada A.E., Lin C., Sharp P.A., Giallourakis C.C. et al. . Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sun K., Chen X., Jiang P., Song X., Wang H., Sun H.. iSeeRNA: identification of long intergenic non-coding RNA transcripts from transcriptome sequencing data. BMC Genomics. 2013; 14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. et al. . Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 53. Sloan C.A., Chan E.T., Davidson J.M., Malladi V.S., Strattan J.S., Hitz B.C., Gabdank I., Narayanan A.K., Ho M., Lee B.T. et al. . ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016; 44:D726–D732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tan J.Y., Sirey T., Honti F., Graham B., Piovesan A., Merkenschlager M., Webber C., Ponting C.P., Marques A.C.. Extensive microRNA-mediated crosstalk between lncRNAs and mRNAs in mouse embryonic stem cells. Genome Res. 2015; doi:10.1101/gr.181974.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leucci E., Patella F., Waage J., Holmstrøm K., Lindow M., Porse B., Kauppinen S., Lund A.H.. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci. Rep. 2013; 3:2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sati S., Ghosh S., Jain V., Scaria V., Sengupta S.. Genome-wide analysis reveals distinct patterns of epigenetic features in long non-coding RNA loci. Nucleic Acids Res. 2012; 40:10018–10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luo S., Lu J.Y., Liu L., Yin Y., Chen C., Han X., Wu B., Xu R., Liu W., Yan P. et al. . Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell. 2016; 18:637–652. [DOI] [PubMed] [Google Scholar]
- 58. Hassan A.H., Neely K.E., Vignali M., Reese J.C., Workman J.L.. Promoter targeting of chromatin-modifying complexes. Front. Biosci. J. Virtual Libr. 2001; 6:D1054–D1064. [DOI] [PubMed] [Google Scholar]
- 59. Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007; 8:450–461. [DOI] [PubMed] [Google Scholar]
- 60. Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U.. Network motifs: simple building blocks of complex networks. Science. 2002; 298:824–827. [DOI] [PubMed] [Google Scholar]
- 61. Tran N.H., Choi K.P., Zhang L.. Counting motifs in the human interactome. Nat. Commun. 2013; 4:2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang M., Wang W., Li T., Yu X., Zhu Y., Ding F., Li D., Yang T.. Long noncoding RNA SNHG1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomed. Pharmacother. 2016; 80:73–79. [DOI] [PubMed] [Google Scholar]
- 63. You J., Zhou Q., Fang N., Gu J., Zhang Y., Li X., Zu L.. Noncoding RNA small nucleolar RNA host gene 1 promote cell proliferation in nonsmall cell lung cancer. Indian J. Cancer. 2014; 51:99. [DOI] [PubMed] [Google Scholar]
- 64. Jankowski A., Tiuryn J., Prabhakar S.. Romulus: robust multi-state identification of transcription factor binding sites from DNase-seq data. Bioinforma. Oxf. Engl. 2016; doi:10.1093/bioinformatics/btw209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J.. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013; 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chu C., Quinn J., Chang H.Y.. Chromatin isolation by RNA purification (ChIRP). J. Vis. Exp. JoVE. 2012; doi:10.3791/3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Simon M.D. Capture hybridization analysis of RNA targets (CHART). Curr. Protoc. Mol. Biol. Ed. Frederick M Ausubel Al. 2013; doi:10.1002/0471142727.mb2125s101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.