


Abstract
Chd1 (Chromodomain Helicase DNA Binding Protein 1) is a conserved ATP-dependent chromatin remodeler that maintains the nucleosomal structure of chromatin, but the determinants of its specificity and its impact on gene expression are not well defined. To identify the determinants of Chd1 binding specificity in the yeast genome, we investigated Chd1 occupancy in mutants of several candidate factors. We found that several components of the PAF1 transcription elongation complex contribute to Chd1 recruitment to highly transcribed genes and identified Spt4 as a factor that appears to negatively modulate Chd1 binding to chromatin. We discovered that CHD1 loss alters H3K4me3 and H3K36me3 patterns throughout the yeast genome. Interestingly, the aberrant histone H3 methylation patterns were predominantly observed within 1 kb from the transcription start site, where both histone H3 methylation marks co-occur. A reciprocal change between the two marks was obvious in the absence of Chd1, suggesting a role for CHD1 in establishing or maintaining the boundaries of these largely mutually exclusive histone marks. Strikingly, intron-containing genes were most susceptible to CHD1 loss and exhibited a high degree of histone H3 methylation changes. Intron retention was significantly lower in the absence of CHD1, suggesting that CHD1 function as a chromatin remodeler could indirectly affect RNA splicing.
INTRODUCTION
Chromodomain Helicase DNA Binding Protein 1 (CHD1) is a chromatin remodeler important for maintaining nucleosome structure over transcription units in yeast and CHD1 mutants exhibit a high degree of aberrant nucleosomal structures (1,2). As part of its fundamental role in maintaining well-positioned nucleosomes, CHD1 prevents histone replacement in the wake of RNA Polymerase II (RNAPII) and thereby represses cryptic transcriptional initiation over coding regions (3). Genome-wide studies investigating Chd1 localization along the genome have revealed that it binds at highly transcribed genes, which are marked with histone H3 tri-methylated at lysine 36 (H3K36me3) due to the histone methyltransferase activity of Set2 (2–4). Moreover, human Chd1 specifically recognizes tri-methylated H3 lysine 4 (H3K4me3) residues produced by Set1 (5) and thus Chd1 is considered an effector of this active histone modification (6,7).
In yeast, Chd1 interacts with Set1 and Set2 (8,9) but does not have the ability to directly bind to H3K4me3 (5). Despite its interaction with Set2, Chd1 binding in the yeast genome is not affected by loss of SET2 (2). Studies on a number of individual genes in yeast have implicated transcriptional elongation factors, including Rtf1 (a component of the PAF1 complex), Spt4–Spt5 (DSIF complex) and Spt16–Pob3 (FACT complex), in recruiting Chd1 to highly transcribed genes (10,11). Consistent with the earlier studies, we recently found that the genome-wide binding profile of Chd1 shows high concordance with the locations of RNA polymerase II phosphorylated at serine 5 of its C-terminal domain (RNAPII Ser-5P), which is an early elongation mark (2). However, the effects of these elongation factors on genome-wide Chd1 localization have not been tested systematically. Given the current evidence, the relationship between CHD1 and the two histone modifications (H3K4me3 and H3K36me3), as well as the determinants of Chd1 recruitment to its targets throughout the genome remain unclear.
In this study, we attempted to elucidate the basis for genome-wide Chd1 occupancy by examining ten candidate factors, including all components of the PAF1 complex (PAF1, CTR9, LEO1, CDC73 and RTF1), two histone methyltransferases (SET1 and SET2), one component of the DSIF complex (SPT4), a critical member of the Rpd3S histone deacetylase complex (RCO1) and the histone H2A.Z variant (HTZ1) (3,9,10,12). By investigating how genome-wide Chd1 occupancy is affected in deletion mutants of these factors, we discovered that the PAF1 complex (PAF1C), a RNAPII-associated factor involved in transcription elongation, is important for Chd1 recruitment to actively transcribed genes. Moreover, SPT4, a component of the conserved DSIF complex that regulates transcription elongation, plays a counteracting role in modulating Chd1 recruitment.
To test whether Chd1 could affect the deposition of H3K4me3 and H3K36me3 at the genes it is recruited to, we generated profiles for these two histone marks in the chd1Δ strain. We discovered that the loss of CHD1 causes global changes in H3K4me3 and H3K36me3 patterns throughout the yeast genome. Interestingly, the aberrant methylation patterns were predominantly observed within 1 kb of transcription start sites where both methylation marks co-localize. Additionally, we detected reciprocal changes in occupancy patterns between these two marks, suggesting a possible role for CHD1 in establishing or maintaining the boundaries of these histone modifications.
Intron-containing genes were over-represented among the genes that we identified as differentially methylated genes in the chd1Δ strain. This led us to analyze the effects of CHD1 on intron retention within the transcripts produced at these genes. Consequently, we discovered that intron retention was significantly lower in the absence of CHD1. This suggests that CHD1 affects RNA splicing, most likely by modulating the rate of RNAPII elongation.
MATERIALS AND METHODS
Yeast strains and cultures
To measure genome-wide Chd1 occupancy by Chromatin immunoprecipitation sequencing (ChIP-seq), we used a yeast strain expressing Chd1 with a 13 Myc epitope-tag, which was generated earlier (2). To create the ten deletion strains in this background, we replaced the ORF of interest with the cassette of a selectable marker, His3MX6, by homologous recombination according to the protocol described previously (13). The yeast strains generated were confirmed by polymerase chain reaction (PCR)-based genotyping. The Myc tagged-strains were also tested by immuno-blotting to validate that Chd1 was correctly tagged.
The chd1Δ strain used for H3K4me3 and H3K36me3 ChIP-seq was obtained from the yeast deletion collection (Open Biosystems, now GE Dharmacon, Lafayette, CO, USA) (14). For the wild-type (WT), we used the S288C-derivative laboratory strain, BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). All yeast cells were cultured in YPD (yeast extract, peptone and dextrose) media at 30°C with shaking at 250 rpm and collected at an O.D.600 (optical density)of 0.8 and immediately frozen in liquid nitrogen for use in subsequent experiments. For the heat-shock experiment (Supplementary Figure S1), yeast cells were collected at an O.D of 0.6–0.8 and split into two aliquots. The old YPD media was removed and one half was re-suspended in 30°C YPD (normal sample) and the other half was re-suspended in pre-warmed 39°C YPD (heat-shock sample). The normal and heat shock samples were incubated at 30 and 39°C, respectively, for 15 min and harvested and frozen in liquid nitrogen.
Chromatin immunoprecipitation sequencing (ChIP-seq)
We followed the ChIP-seq protocol described in (2) with 150 μl of anti-Myc conjugated agarose beads (Sigma Aldrich, St Louis, MO, USA, cat.# E6654), 10 μg of H3K4me3 antibody (EMD Millipore, Darmstadt, Germany, cat.# 07–473), 10 μg of H3K36me3 antibody (Abcam, Cambridge, MA, USA, cat.# ab9050), 10 μg of RNAPII S5p antibody (Abcam, Cambridge, MA, USA, cat.# ab5131) and 10 μg of RNAPII S2p antibody (Abcam, Cambridge, MA, USA, cat.# ab5095) to pull down Chd1, H3K4me3, H3K36me3, RNAPII Ser-5P and Ser-2P, respectively. For Set2 ChIP-seq, we used the TAP-tagged Set2 strain from the TAP-tag library (15) and 100 μl of anti-TAP conjugated sepharose beads (GE Healthcare, Pittsburgh, PA, USA, cat.# 17–0969) for immunoprecipitation. For the mock ChIP, we carried out immunoprecipitation with anti-Myc conjugated agarose beads in cells expressing no Myc-tagged protein.
Gene expression profiling
For the microarray experiments carried out in paf1Δ, spt4Δ and WT strains, we used NimbleGen Saccharomyces cerevisiae Gene Expression Array System (Roche NimbleGen, Madison, WI, USA) according to the manufacturer's instructions. For the RNA sequencing experiments performed in chd1Δ and WT, we isolated total RNA from the yeast cells through the hot acid phenol extraction method and incubated the total RNA with magnetic beads conjugated to oligo-dT (BIOO Scientific, Austin, TX, USA, cat.# 512980) to enrich poly-adenylated RNA. Next, a sequencing library with the poly-adenylated RNA was prepared using NEBNext Multiplex Small RNA Library Prep Set for Illumina (New England Biolabs, Ipswich, MA, USA, cat.# E7300L), then sequenced using the Illumina HiSeq 2500 System.
Data analysis
ChIP-seq and RNA-seq reads were mapped onto the sacCer3 reference genome using the aligner BWA (16) and TopHat (17), respectively. Subsequently, we visualized the data on a local mirror of the UCSC genome browser by loading the WIG files generated from the BAM files. The top 100 Chd1-occupied genes and the most actively transcribed genes were identified based on the occupancy of Chd1 and RNAPII Ser-5P, respectively, in the WT strain under normal growth conditions. The most actively transcribed genes were taken to be the high outlier genes showing high levels of RNAPII Ser-5P occupancy (>Q3 + 1.5 * inter-quartile range of RNAPII Ser-5P occupancy) and totaled 536 genes. The transcript coordinates, such as transcription start site (TSS) and transcription termination site (TTS), were consolidated by combining two previous published resources (18,19). To bin H3K4me3 and H3K36me3 according to their nucleosomal coordinates, we used the distances from the TSS in the intervals: 55, 110, 275, 440, 605, 770 and 935 to bin the +1 to + 6 nucleosomes respectively, derived from nucleosome positions in previous studies (2).
All statistical analyzes were performed using R and Bioconductor. Specifically, we used edgeR (20) and limma (21) packages for the differential H3K4me3/H3K36me3 analysis and differential gene expression analysis. All graphs from these analyzes were generated using ggplot2. Hierarchical clustering was performed using Cluster3 (22) and the output was visualized using Java TreeView (23).
To quantify intron retention as a measure of splicing efficiency, we counted the intronic and coding exonic sequencing reads separately for all intron-containing genes. To compare intron retention between chd1Δ and WT, we calculated the odds ratio (OR) of intron retention for individual introns as follows:
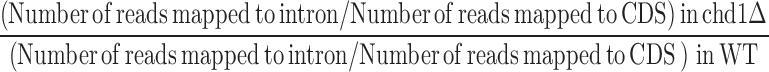 |
Essentially, this measurement informs us whether intron retention is higher or lower in RNA transcripts in the CHD1 mutant compared to WT depending on whether the OR is greater or less than 1, respectively, in conjunction with a lenient P-value threshold of <0.1 from Fisher's exact test.
RESULTS
The PAF1C elongation complex is the key determinant of Chd1 recruitment to chromatin
Although several factors have been reported to physically interact with Chd1 and recruit it to chromatin, their role in Chd1 binding has not been systematically examined on a genomic scale. To identify the key factor that gives rise to the specificity of Chd1 binding on chromatin, we prioritized ten candidate factors that have been reported to physically or functionally associate with CHD1, including all components of the PAF1C complex (PAF1, CTR9, LEO1, CDC73 and RTF1), two histone methyltransferases (SET1 and SET2), one component of the DSIF complex (SPT4), a critical member of the Rpd3S histone deacetylase complex (RCO1) and the H2A.Z histone variant (HTZ1) (3,9,10,12).
We created deletion mutants for each of these candidate genes and used ChIP-seq to investigate if overall Chd1 occupancy was affected in the deletions when compared with WT. All deletions of PAF1C components, with the exception of rtf1Δ, showed significantly lower Chd1 occupancy. Interestingly, spt4Δ showed higher levels of Chd1 binding when we considered either the top 100 genes highly bound by Chd1 or the most actively transcribed genes in WT cells (536 genes, see ‘Materials and Methods’ section) (Figure 1).
Figure 1.

Chd1 occupancy in the wild-type (WT) strain and deletion mutants of candidate recruitment factors. (A) Top: the heat map shows Chd1 occupancy in the indicated deletion mutants at the 100 genes where Chd1 occupancy was highest in WT cells. Each row indicates a gene. Chd1 occupancy was measured by counting Chd1 Chromatin immunoprecipitation sequencing (ChIP-seq) reads from the transcription start site (TSS) to transcription termination site (TTS) and normalizing by transcript length and sequencing depth. The level of occupancy of Chd1 is depicted in a green-to-red color scheme (green—low; red—high occupancy of Chd1) after being standardized into a z-score per row. Bottom: boxplots summarize the distribution of Chd1 occupancy values shown above. (B) Metagene average occupancy profiles of Chd1 in candidate recruitment factor deletions. The y-axis in the plots shows normalized Chd1 ChIP-seq read density (reads per million, RPM) obtained by averaging the read counts for the most actively transcribed genes, identified based on the occupancy of RNAPII Ser-5P in the WT strain under normal growth conditions (536 genes, see ‘Materials and Methods’ section). The x-axis shows the distance from 1 kb upstream to 2 kb downstream of the TSS. Each plot includes a positive control (Chd1 occupancy measured in the WT strain, blue) and a negative control (a mock ChIP, green) to compare Chd1 occupancy in the selected deletion mutants (red). The dark line is the mean and the shaded envelope indicates the 95% confidence interval for the mean Chd1 ChIP-seq read density.PAF1, CTR1 and SPT4 mutants show a highly significant difference in Chd1 occupancy. The arrows indicate the genomic regions showing the strongest and most significant differences in occupancy. RTF1, SET1 and SET2 mutants show a marginal difference in Chd1 occupancy.
We observed the greatest difference in Chd1 binding in paf1Δ, ctr9Δ and spt4Δ (Figure 1A). In order to examine how Chd1 binding was altered in these strains in more detail, we generated average Chd1 binding profiles from 1 kb upstream of the TSS to 2 kb downstream of the TSS for the most actively transcribed genes (Figure 1B).
The PAF1 and CTR9 mutants showed a considerable depletion of Chd1 binding over the gene body and a corresponding significant accumulation of Chd1 at the promoter, just upstream of the TSS (Figure 1B, see arrows). A possible explanation for this reciprocal pattern of Chd1 occupancy at promoters and gene bodies in the elongation factor mutants may be that Chd1 is loaded on to promoters but is transported to its sites of action at the coding regions of highly transcribed genes via interactions with elongation factors such as Paf1. In contrast to the PAF1C mutants, spt4Δ showed significantly more Chd1 binding near the TSS (Figure 1B), indicating that Spt4 is involved in negatively modulating Chd1 recruitment to the 5΄ ends of genes.
Rtf1, a member of the PAF1C complex, has been proposed to be a recruitment factor for Chd1 in yeast because of its direct interaction with Chd1 (10). However, rtf1Δ did not show any large changes in Chd1 occupancy and thus appeared to be distinct from the other members of PAF1C (Figure 1). We note that rtf1Δ showed a small increase in Chd1 occupancy (Figure 1B), but its magnitude was substantially smaller than the effects seen with Spt4. Moreover, loss of SET1 or SET2, two histone H3 methyltransferases known for their close functional relationship with CHD1, did not appreciably affect Chd1 occupancy (Figure 1B).
One possibility is that loss of the elongation components such as Paf1 and Spt4 causes a change in RNAPII occupancy and indirectly causes a change in Chd1 recruitment to chromatin. To distinguish between potential indirect and direct effects on Chd1 recruitment, we first examined changes in RNAPII Ser-5P occupancy in relation to changes in gene expression. RNAPII Ser-5P occupancy was highly correlated with changes in gene expression, showing that gene expression changes can provide a measure of RNAPII Ser-5P occupancy (Supplementary Figure S1). We then examined changes in Chd1 recruitment in paf1Δ and spt4Δ at two distinct sets of genes, those whose expression changed, corresponding to a decrease or increase in RNAPII Ser-5P occupancy and those actively transcribed genes whose expression did not change in the deletion. Chd1 occupancy was reduced at genes that were downregulated in paf1Δ, suggestive of an indirect effect, but there was a much larger set of 417 actively transcribed genes whose expression did not change and therefore had no change in RNAPII occupancy, and yet showed a clear reduction in Chd1 occupancy (Supplementary Figure S2). Similarly, spt4Δ also showed a small indirect effect on Chd1 recruitment at 93 genes whose expression increased in the deletion but a much larger direct effect at 528 actively transcribed genes with no change in gene expression or RNAPII occupancy, that nevertheless showed a significant increase in Chd1 recruitment (Supplementary Figure S2).
Chd1 controls the overlapping boundaries of the domains of H3K4me3 and H3K36me3
Although Set1 and Set2 did not affect Chd1 recruitment to chromatin, given the strong reported association between Chd1, Set1 and Set2, we were interested in how the two histone marks deposited by these histone methyltransferases (H3K4me3 and H3K36me3, respectively) were affected in the absence of CHD1. We utilized ChIP-seq to generate high-resolution chromatin profiles for H3K4me3 and H3K36me3 in chd1Δ and WT, and focused at the 5΄ end of genes where our recent data indicated strong Chd1 recruitment and nucleosome remodeling effects (2). A previous study by Radman-Livaja et al examined this question using lower resolution ChIP-chip, and focused on methylation changes at the 3΄ ends of genes in conjunction with CHD1 effects on histone replacement at long genes, although they did observe changes in H3K36me3 similar to what we observe near the 5΄ ends as well (24). We utilized ChIP-seq to generate higher-resolution chromatin profiles for H3K4me3 and H3K36me3 in chd1Δ and WT, and focused at the 5΄ end of genes where our recent data indicated strong Chd1 recruitment and nucleosome remodeling effects (2).
Our data revealed that CHD1 loss causes consistent changes in H3K4me3 and H3K36me3 profiles at most genes across the yeast genome, with the changes being localized relative to the 5΄ end of genes (Figure 2; Supplementary Figures S3 and 4). At most genes in WT cells, H3K4me3 is normally localized to the 5΄ end and H3K36me3 is distributed over the gene body. Although H3K36me3 is largely excluded from the 5΄ end, there is a narrow zone within 1 kb from the TSS where both marks overlap, which thus demarcates the H3K4me3 and H3K36me3 domains over genes. We predominantly observed changes in the methylation pattern in chd1Δ within 1 kb downstream of the TSS where the two marks overlap. In chd1Δ, the downstream boundary of the H3K4me3 domain and the upstream boundary of the H3K36me3 domain were both shifted upstream, in the direction of the TSS (Figure 2A). Although this shift was small, it was highly significant because first, we observed the same behavior in two independent ChIP-seq datasets and second, it was very consistent across all genes with good ChIP-seq signal, as indicated by the clear separation of the 95% confidence intervals around the ChIP-seq signal density profiles (Figure 2B). Moreover, it is the ratio of H3K4me3 to H3K36me3 that was affected, confirming that the effect of CHD1 loss is not simply due to a change in overall histone H3 or nucleosome occupancy in this region (Supplementary Figure S5).
Figure 2.

Chromodomain Helicase DNA Binding Protein 1 (CHD1) affects domains of H3K4me3 and H3K36me3. (A) Genome browser views of H3K4me3 and H3K36me3 across the SEC23 and EFT1 genes in WT and chd1Δ strains. (B) Average H3K4me3 and H3K36me3 profiles for all yeast genes for two replicate experiments from 500 bp upstream to 1500 bp downstream of the TSS. The dark line is the mean and the shaded envelope indicates the 95% confidence interval of the mean H3K4me3/H3K36me3 ChIP-seq read signal (RPM) for all genes. H3K4me3 and H3K36me3 signals in WT are colored sky blue and pink, respectively. H3K4me3 and H3K36me3 signals in chd1Δ are colored dark blue and purple, respectively. The inset for replicate 2 highlights the region between 500 and 700 bp downstream of the TSS to show the shift of H3K4me3 and H3K36me3 domain boundaries toward the TSS.
To specifically identify the nucleosomes showing differential depletion or enrichment of the histone methylation marks at genes, we used our earlier nucleosome positioning data generated using MNase-seq (2) to bin our methylation data with respect to nucleosome position. According to the nucleosome periodicity defined previously (2), we determined that the +1 nucleosome starts from 55 bp upstream of the TSS and each nucleosome spans a 165 bp region. To cover the ∼1 kb region from the TSS of all transcripts, where both H3K4me3 and H3K36me3 marks were prominent, we created genomic coordinates encompassing the +1 to +6 nucleosomes for all genes (see ‘Materials and Methods’ section). We then analyzed our histone mark ChIP-seq data to identify genes showing differential H3K4me3 and H3K36me3 in chd1Δ relative to WT at one or more nucleosome positions. We identified 2501 genes with at least a 2-fold-change in H3 methylation (fold-change > 2 and false discovery rate (FDR)-adjusted P < 0.01). This set represents the union of the differentially methylated H3K4me3 and H3K36me3 genes and is almost half of all the genes in the yeast genome (Supplementary Figure S6). Thus, our data shows that Chd1 affects histone methylation patterns at approximately half of all genes in the genome. Interestingly, the set of 818 genes showing differential methylation at both H3K4 and H3K36 tended to be longer and more highly transcribed genes, compared to those that showed alteration in only one modification (Supplementary Figure S6).
To elucidate the nature of the methylation signal changes in the absence of CHD1, we performed hierarchical clustering with the differentially tri-methylated genes for H3K4me3 and H3K36me3 (Figure 3). This analysis revealed a striking reciprocal change between the two methylation marks; if H3K4me3 was depleted at downstream nucleosomes, there was concurrently a higher level of H3K36me3 signal at upstream nucleosomes and vice versa (Figure 3). Thus, this analysis indicated that CHD1 is involved in the establishing the domains of these largely mutually exclusive histone modifications. In order to explore the mechanistic basis of this action, we considered nucleosome turnover, since this is a process known to be affected by Chd1. We obtained nucleosome turnover data from a previous study that measured nucleosome exchange rates over genes in WT and chd1Δ strains (3). The chd1Δ strain showed lower exchange rates over gene bodies for most genes, but at the subset of genes that showed marked reciprocal shifts in H3K4me3 and H3K36me3 in our data, nucleosome exchange rates were also markedly lower, over the corresponding gene regions (Supplementary Figure S7). This suggests that nucleosome exchange mediated by Chd1 could contribute to the establishment of normal H3K4me3 and H3K36me3 boundaries near the 5΄ ends of genes.
Figure 3.

A reciprocal change between H3K4me3 and H3K36me3 upon loss of CHD1. (A) Genes showing differential H3K4me3 in chd1Δ. Hierarchical clustering was performed on the H3K4me3 data (left) and the corresponding H3K36me3 data was plotted on the right to reveal the relationship between these two methylation marks. Each column in the heat maps indicates nucleosome positions from +1 to +6. Fold-change of H3 tri-methylation in chd1Δ over WT were transformed into a log2-scale and depicted in a blue-to-red color scheme (blue—less H3 tri-methylation in chd1Δ; red—more H3 tri-methylation in chd1Δ, when compared to WT). (B) Genes showing differential H3K36me3 in chd1Δ. Hierarchical clustering was performed on the H3K36me3 data (left) and the corresponding H3K4me3 data (right) was plotted on the right.
Determinants of histone H3 methylation sensitivity to CHD1
To better understand the factors that make H3K4me3 and H3K36me3 at certain genes dependent on Chd1, we examined several characteristics of these genes. To do this, we examined the methylation change sensitivity to CHD1 loss as a function of transcription level (measured as steady-state RNA levels) or transcript length (Figure 4A and B, respectively).
Figure 4.

Histone H3K4me3 and H3K36me3 sensitivity to CHD1 loss is correlated with transcription level but not transcript length. (A) The sensitivity of H3K4me3 and H3K36me3 changes to CHD1 loss, quantified using the statistical significance (−log10 FDR adjusted p-value) of methylation ratio in chd1Δ and WT, is plotted as a function of gene expression level at each of the +1, +2, +3, +4, +5 and +6 nucleosomes. The gene expression values are normalized RNA-seq data (reads per million, RPM) in WT and transformed into a log2-scale. Each dot represents one gene and its colors show fold changes of chd1Δ relative to WT in either H3K4me3 or H3K36me3 (blue—less H3 tri-methylation in chd1Δ; red—more H3 tri-methylation in chd1Δ, when compared to WT) and its sizes reflect the significance level (small—not significant, N.S; large—significant, S). (B) The sensitivity of H3K4me3 and H3K36me3 changes to CHD1 loss plotted as a function of transcript length at each of the same six nucleosomes.
There was a positive correlation between the dependence of H3K4me3 on CHD1 function and transcription level, specifically at the +4, +5 and +6 nucleosomes (Figure 4A). The overall trend between the alteration in H3K36me3 upon CHD1 loss and gene expression was also positive (Figure 4A), indicating that actively transcribed genes tend to show a high degree of methylation changes in the absence of CHD1. Moreover, increased transcription of the genes was associated with depletion of H3K4me3 and H3K36me3 in chd1Δ at downstream nucleosomes and increased H3K36me3 in chd1Δ at the +1 nucleosome (Figure 4A). However, there was no correlation between transcript length and histone methylation changes upon CHD1 deletion (Figure 4B). Rather, we observed a strong bias for H3K36me3 depletion at short genes in the absence of CHD1. Taken together, this data suggested that a specific group of genes with short transcript lengths and high transcription levels are strongly dependent on Chd1 for normal levels of H3K4me3 and H3K36me3.
Loss of Chd1 causes aberrant histone methylation patterns and affects intron retention in ribosomal protein genes
In yeast, the ribosomal protein (RP) genes are characterized by high levels of transcription and relatively short transcript lengths. We therefore examined H3K4me3 and H3K36me3 patterns specifically over the 137 RP genes in the chd1Δ strain. Strikingly, all RP genes, with the exception of five genes that did not meet the significance threshold (fold-change > 2 and FDR-adjusted P < 0.01) exhibited a strong and significant reduction in H3K4me3 and H3K36me3 deposition and a redistribution of this mark upon the loss of CHD1 (Figure 5).
Figure 5.

CHD1 strongly affects H3K4me3 and H3K36me3 at ribosomal protein (RP) genes. (A) Heat maps showing differential H3K4me3 (left) and H3K36me3 (right) in chd1Δ relative to WT for RP genes measured as fold changes. The fold changes are on a log2-scale and depicted in a blue-to-red color scheme (blue—less H3 tri-methylation; red—more H3 tri-methylation in chd1Δ). Each column in the heat maps represents nucleosome positions and the rows are sorted by transcript length. Gray areas reflect short genes that do not extend as far as the end of the displayed region. (B) Average H3K4me3 (left) and H3K36me3 (right) profiles for RP genes from 500 bp upstream to 1000 bp downstream of the TSS in chd1Δ and WT. The width of lines corresponds to the 95% confidence interval for the mean H3K4me3 and H3K36me3 ChIP-seq read density. (C) Genome browser views of H3K4me3 and H3K36me3 across the RPS14A, RPL4B and RPL20A genes in WT and chd1Δ strains.
RP genes in yeast are also distinguished by the fact that they are much more likely to contain an intron; 101 out of 137 RP genes (∼75%) have one or more intron(s), comprising about half of all intron containing genes in yeast. Most introns in RP genes occur toward the 5΄ end of the gene and we observed that in chd1Δ, the signal from H3K4me3 and H3K36me3 was significantly higher over the introns and lower in the exons (Figure 5 and Supplementary Figure S8). We have previously found that RNAPII Ser-5P accumulates at intron–exon junctions within highly transcribed genes in the absence of CHD1 (2). We therefore hypothesized that CHD1, as a consequence of its effect on nucleosome positions and histone modifications and thereby RNAPII elongation, could indirectly affect co-transcriptional RNA splicing at highly transcribed genes.
In support of this hypothesis, we found that Chd1 is recruited to introns within highly transcribed genes (Supplementary Figure S9). To directly test the hypothesis that Chd1 affects mRNA splicing, we performed RNA-seq of chd1Δ and WT, and quantified intron retention as a measure of splicing efficiency (see ‘Materials and Methods’ section). Table 1 shows the outcome of the intron retention analysis for all intron-containing genes considered. We found that 35 introns were significantly affected in splicing in the chd1Δ mutant. Interestingly, there was a strong skew in how intron retention was affected; 28 out of 35 introns (80%) showed lower intron retention and thus an improvement in splicing upon loss of CHD1 (Table 1).
Table 1. Introns significantly different with regard to intron retention between chd1Δ and WT, when all introns in the yeast genome were considered.
| This study, number of introns | Lee et al., (26) number of introns | |
|---|---|---|
| OR < 1, lower intron retention in chd1Δ | 28 | 98 |
| OR > 1, higher intron retention in chd1Δ | 7 | 6 |
OR is the odds ratio (see ‘Materials and Methods’ section).
As a control, we applied our analysis to RNA-seq data generated from a splicing factor mutant (temperature-sensitive prp40; prp40-1) (25) and found the opposite trend, namely that more than 95% of introns were impaired in splicing and showed higher intron retention in the mutant (data not shown), suggesting that our quantitation of intron retention was reliable. To assess whether the effect of CHD1 loss on splicing might be indirectly caused by altered expression of splicing-related genes, we analyzed the differentially expressed genes (DEGs) in the chd1Δ RNA-seq data. At the normally used threshold (FDR P < 0.05 and fold-change > 2), there were no DEGs involved in RNA splicing. At a more relaxed threshold (FDR P < 0.05 and fold-change > 1.4), there were only two genes relevant to RNA splicing that were slightly affected; however, one of these, PRP31 was activated (1.7-fold) and the other, DED1 was repressed (1.5-fold) at the level of transcription. Given that overall splicing efficiency increased in chd1Δ and that PRP31 is just one of a large number of spliceosomal components and that at least one other splicing factor was reduced in expression (which would reduce splicing efficiency), it is unlikely that CHD1 loss leads to an increase in splicing efficiency solely due to an effect on the transcription of splicing factors. Because our power to detect changes in intron retention was limited by the depth of our RNA-seq dataset, we repeated this analysis with more deeply sequenced RNA-seq data published in a previous study (26). A total of 94% of introns showed lower intron retention in the chd1Δ mutant, confirming the skew towards more efficient splicing in the mutant that we observed with our independent data. When we considered only the introns within RP genes, where Chd1 is most strongly enriched, the increased efficiency in splicing in the chd1Δ mutant (i.e., lower intron retention) was even more apparent (Table 2). Thus, CHD1, most likely by virtue of its effect on RNAPII elongation and H3K4me3/H3K36me3 domains, can indirectly affect the efficiency of pre-mRNA splicing.
Table 2. Introns significantly different with regard to intron retention between chd1Δ and WT, when only the introns at RP genes were considered.
| This study, number of introns | Lee et al., (26) number of introns | |
|---|---|---|
| OR < 1, lower intron retention in chd1Δ | 21 | 88 |
| OR > 1, higher intron retention in chd1Δ | 2 | 1 |
DISCUSSION
Here, we examined genome-wide Chd1 occupancy in ten candidate recruitment factor mutants, including five components of the PAF1C complex (PAF1, CTR9, LEO1, CDC73 and RTF1), two histone methyltransferases (SET1 and SET2), one component of the DSIF complex (SPT4), an essential member of the Rpd3S histone deacetylase complex (RCO1) and a histone H2A variant (HTZ1). Although all these factors have been reported to interact with Chd1 and proposed to recruit it to chromatin, our study examines their role on a genomic scale for the first time and clarifies the difference between positive and negative factors. We found that all components of the PAF1C, except for RTF1, are positively required for the normal level and distribution of Chd1 over highly transcribed genes (Figure 1A). SPT4 functions in negatively modulating Chd1 recruitment near the 5΄ ends of the genes (Figure 1B). Unexpectedly, none of the factors tested in this study completely abolished the specificity of Chd1 recruitment to actively transcribed genes in the yeast genome (Figure 1). This suggests that either there is some redundancy among the components of the PAF1C complex in recruiting Chd1, or that additional components not tested here, such as essential components of the transcription machinery, could be involved in recruiting Chd1 to its target loci.
Chd1 is engaged over the entire unit of transcription, at highly transcribed genes, from the transcription start site (TSS) to the transcription termination site (TTS). Given this behavior, the factor(s) maintaining Chd1 binding to chromatin is likely to have the same binding pattern as Chd1 and be necessary for active transcription. The most plausible factor would be RNAPII itself or a factor that moves with RNAPII, such as the PAF1C or DSIF complex (Spt4–Spt5), which was investigated in this study. We have previously found that the binding pattern of RNAPII Ser-5P closely matches Chd1 at active genes (2). Beyond the two elongation complexes, we observed that the most similar genome-wide binding profiles to Chd1 are those of Spt2, a DNA binding protein with HMG-like domains and Mbf1, a RNAPII coactivator, respectively (data not shown). According to the published data on SPT2 (27), its functional roles in transcription elongation are consistent with the known roles of CHD1. Investigating the factors with the most agreement to Chd1 binding profiles will likely shed some light on additional Chd1 co-factors that are important for its proper recruitment.
Our study also clarifies the relationship between CHD1 and two histone H3 modifications, H3K4me3 and H3K36me3, through high resolution sequencing and a binning-based strategy to detect differential methylation in the absence of CHD1. Loss of CHD1 led to significantly aberrant methylation patterns in approximately half of the yeast genome (Figure 3 and Supplementary Figure S6). These methylation changes were predominantly observed within 1 kb of the TSS of the genes, where both methyl marks partially overlap (Figure 2, Supplementary Figures S3 and 4). The changes in H3K4me3 and H3K36me3 were reciprocal between the promoter-proximal nucleosomes (+1, +2 and +3) and distal nucleosomes (+4, +5 and +6) within the 1 kb window (Figure 3). The disruption of these methylation marks due to loss of CHD1 was consistent and widespread across the genome (Figure 2 and Supplementary Figure S4). These results suggest a possible role for CHD1 in maintaining distinct domains for H3K4me3 and H3K36me3. A previous study by Radman-Livaja et al. showed a slight increase in H3K4me3 and a dramatic decrease in H3K36me3 at the 3΄ end of genes in chd1Δ (24). While our analysis was focused at the 5΄ end, we also observed a reduced level of H3K36me3 at the 3΄ end of genes in the absence of CHD1. However, we did not see a significant increase of H3K4me3 at the 3΄ end of genes in the CHD1 deletion. Although the impact of CHD1 loss on steady-state transcript levels was marginal (Supplementary Figure S10), it is noteworthy that an indirect consequence of its activity is a small reduction in splicing efficiency (discussed below).
Misregulation of histone modifications is known to lead a variety of human diseases, underscoring the significance of understanding how they are regulated on a genome-wide scale (28,29). Accordingly, many studies have been conducted to define the normal organization of histone modifications with regard to position and quantity. For example, Set1 deposits a gradient of H3K4 tri-, di- and mono-methylation, from the promoter towards the 3΄ ends of genes (28,30,31). Set2 can mono-, di- and tri-methylate H3K36 on bodies of genes (29). The histone H3K4 and H3K36 methylation tend to be inversely related to each other in terms of the location on genes, since as the level of H3K4 methylation diminishes, H3K36 methylation increases along genes (32).
It has been proposed that two distinct zones of tri- and di-methylated H3K4 loci are created via the recruitment of different histone modifying enzymes (31) and the presence of overlapping non-coding transcripts (33), defining disparate chromatin structures as a mechanism for fine-tuning transcriptional induction or repression. Furthermore, a separate study revealed that Set1-mediated methylation at H3K4 determines which chromatin-remodeling factor is recruited to cause changes in chromatin structure for condition-specific expression (34). In this study, we found that CHD1 loss disrupted H3K4me3 and H3K36me3 domains (Figure 4). Considering the importance for establishing distinct chromatin domain with proper epigenetic marks, CHD1 could potentially affect diverse chromatin events, as seen in the aforementioned studies.
H3K4me3 and H3K36me3 changes in the absence of CHD1 have been examined using ChIP-chip experiments in previous studies (3,24). One common observation was that H3K36me3 signal appeared to shift upstream in chd1Δ. However, these previous studies underestimated changes in H3K4me3 due to their low statistical significance. In contrast, we found a highly significant effect at a large number of genes that showed differential H3K4me3 and H3K36me3 at individual nucleosomes in the absence of CHD1. Our ability to detect this novel reciprocal shift in their occupancy patterns hinged on our use of ChIP-seq, which has a higher resolution than ChIP-chip experiments. When we considered the changes between chd1Δ and WT by summarizing the ChIP-seq reads only at the whole-gene level, the number of genes that showed differential H3K4 and H3K36 methylation were reduced by 85%. Representative false-negative examples (RPL15A and ADH1) are shown in Supplementary Figure S4B. This underscores the importance of analyzing histone modification data by employing a methodology to detect local signal changes such as ChIP-seq, coupled with appropriate binning or sliding window strategies (35,36) to detect changes at nucleosome resolution.
At last, we found a significant functional association of CHD1 with intron retention for the intron-containing genes that showed the most dramatic changes in H3K4me3 and H3K36me3 upon CHD1 loss (Table 1 and 2; Figure 5). How might Chd1 affect RNA splicing? One possibility could be through CHD1 function in releasing stalled RNAPII at introns. Recent studies have shown that RNAPII pausing is known to affect co-transcriptional splicing (37–39). Another potential mechanism might be through interactions between histone methylation and the splicing machinery, as demonstrated in a recent study where loss of SET2 recruits inadequate snRNPs to yeast chromatin (40). One caveat to our analysis is that we used polyA+ RNA to enriched for spliced transcripts, which was also the case in the external dataset analyzing splicing defects (25). Thus, non-polyadenylated transcripts generated due to splicing errors would be missed in these analyzes. Finally, it is possible that Chd1 directly affects splicing by interactions with splicing factors. Indeed, Chd1 has previously been reported to interact with splicing factors and affect the splicing of a reporter construct (41). Even if the effect of Chd1 on splicing is indirect, if it similarly affects splicing efficiency in mammalian cells, it might have important clinical consequences in light of the fact that CHD1 is known to be mutated in prostate and other cancers (42–45). It is possible that some of the phenotypic impact of mutations in CHD1 and other chromatin factors in cancer are mediated via an effect on splicing. Similar investigations examining the effect of chromatin remodeler mutations on histone modifications and splicing are likely to shed light on this question.
ACCESSION NUMBER
The genomic datasets from this study have been deposited in NCBI's Gene Expression Omnibus (GEO) database under accession number GSE90998.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Anna Battenhouse for mapping and pre-processing sequencing data and consolidating coordinates of yeast transcripts, Haridha Shivram for discussions and the Next Generation Sequencing Core Facility at the University of Texas MD Anderson Cancer Center Science Park, which was supported by CPRIT Core Facility Support Grant RP120348, for sequencing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funded in part by National Institutes of Health [CA095548 and CA198648] and Cancer Prevention and Research Institute of Texas [RP120194] to V.R.I. and KIST Intramural Research grant [2E26910] to D.P.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gkikopoulos T., Schofield P., Singh V., Pinskaya M., Mellor J., Smolle M., Workman J.L., Barton G.J., Owen-Hughes T.. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011; 333:1758–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Park D., Shivram H., Iyer V.R.. Chd1 co-localizes with early transcription elongation factors independently of H3K36 methylation and releases stalled RNA polymerase II at introns. Epigenet. Chromatin. 2014; 7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smolle M., Venkatesh S., Gogol M.M., Li H., Zhang Y., Florens L., Washburn M.P., Workman J.L.. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat. Struct. Mol. Biol. 2012; 19:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skene P.J., Hernandez A.E., Groudine M., Henikoff S.. The nucleosomal barrier to promoter escape by RNA polymerase II is overcome by the chromatin remodeler Chd1. Elife. 2014; 3:e02042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sims R.J. 3rd, Chen C.F., Santos-Rosa H., Kouzarides T., Patel S.S., Reinberg D.. Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J. Biol. Chem. 2005; 280:41789–41792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin J.J., Lehmann L.W., Bonora G., Sridharan R., Vashisht A.A., Tran N., Plath K., Wohlschlegel J.A., Carey M.. Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev. 2011; 25:2198–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sims R.J. 3rd, Millhouse S., Chen C.F., Lewis B.A., Erdjument-Bromage H., Tempst P., Manley J.L., Reinberg D.. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell. 2007; 28:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang L., Schroeder S., Fong N., Bentley D.L.. Altered nucleosome occupancy and histone H3K4 methylation in response to ‘transcriptional stress’. EMBO J. 2005; 24:2379–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krogan N.J., Kim M., Tong A., Golshani A., Cagney G., Canadien V., Richards D.P., Beattie B.K., Emili A., Boone C. et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 2003; 23:4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simic R., Lindstrom D.L., Tran H.G., Roinick K.L., Costa P.J., Johnson A.D., Hartzog G.A., Arndt K.M.. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J. 2003; 22:1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biswas D., Dutta-Biswas R., Stillman D.J.. Chd1 and yFACT act in opposition in regulating transcription. Mol. Cell. Biol. 2007; 27:6279–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quan T.K., Hartzog G.A.. Histone H3K4 and K36 methylation, Chd1 and Rpd3S oppose the functions of Saccharomyces cerevisiae Spt4-Spt5 in transcription. Genetics. 2010; 184:321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gietz R.D., Schiestl R.H.. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007; 2:31–34. [DOI] [PubMed] [Google Scholar]
- 14. Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H. et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999; 285:901–906. [DOI] [PubMed] [Google Scholar]
- 15. Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O'Shea E.K., Weissman J.S.. Global analysis of protein expression in yeast. Nature. 2003; 425:737–741. [DOI] [PubMed] [Google Scholar]
- 16. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Munster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M.. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009; 457:1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park D., Morris A.R., Battenhouse A., Iyer V.R.. Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res. 2014; 42:3736–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eisen M.B., Spellman P.T., Brown P.O., Botstein D.. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saldanha A.J. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004; 20:3246–3248. [DOI] [PubMed] [Google Scholar]
- 24. Radman-Livaja M., Quan T.K., Valenzuela L., Armstrong J.A., van Welsem T., Kim T., Lee L.J., Buratowski S., van Leeuwen F., Rando O.J. et al. A key role for Chd1 in histone H3 dynamics at the 3΄ ends of long genes in yeast. PLoS Genet. 2012; 8:e1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Volanakis A., Passoni M., Hector R.D., Shah S., Kilchert C., Granneman S., Vasiljeva L.. Spliceosome-mediated decay (SMD) regulates expression of nonintronic genes in budding yeast. Genes Dev. 2013; 27:2025–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee J.S., Garrett A.S., Yen K., Takahashi Y.H., Hu D., Jackson J., Seidel C., Pugh B.F., Shilatifard A.. Codependency of H2B monoubiquitination and nucleosome reassembly on Chd1. Genes Dev. 2012; 26:914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nourani A., Robert F., Winston F.. Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006; 26:1496–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012; 81:65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wagner E.J., Carpenter P.B.. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012; 13:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiao T., Shibata Y., Rao B., Laribee R.N., O’Rourke R., Buck M.J., Greenblatt J.F., Krogan N.J., Lieb J.D., Strahl B.D.. The RNA polymerase II kinase Ctk1 regulates positioning of a 5. histone methylation boundary along genes. Mol. Cell. Biol. 2007; 27:721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim T., Buratowski S.. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5΄ transcribed regions. Cell. 2009; 137:259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Narlikar G.J., Sundaramoorthy R., Owen-Hughes T.. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013; 154:490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim T., Xu Z., Clauder-Munster S., Steinmetz L.M., Buratowski S.. Set3 HDAC mediates effects of overlapping noncoding transcription on gene induction kinetics. Cell. 2012; 150:1158–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nadal-Ribelles M., Mas G., Millan-Zambrano G., Sole C., Ammerer G., Chavez S., Posas F., de Nadal E.. H3K4 monomethylation dictates nucleosome dynamics and chromatin remodeling at stress-responsive genes. Nucleic Acids Res. 2015; 43:4937–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schweikert G., Cseke B., Clouaire T., Bird A., Sanguinetti G.. MMDiff: quantitative testing for shape changes in ChIP-Seq data sets. BMC Genomics. 2013; 14:826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lun A.T., Smyth G.K.. csaw: a bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2016; 44:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alexander R.D., Innocente S.A., Barrass J.D., Beggs J.D.. Splicing-dependent RNA polymerase pausing in yeast. Mol. Cell. 2010; 40:582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saldi T., Cortazar M.A., Sheridan R.M., Bentley D.L.. Coupling of RNA polymerase II transcription elongation with pre-mRNA splicing. J. Mol. Biol. 2016; 428:2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shukla S., Oberdoerffer S.. Co-transcriptional regulation of alternative pre-mRNA splicing. Biochim. Biophys. Acta. 2012; 1819:673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sorenson M.R., Jha D.K., Ucles S.A., Flood D.M., Strahl B.D., Stevens S.W., Kress T.L.. Histone H3K36 methylation regulates pre-mRNA splicing in Saccharomyces cerevisiae. RNA Biol. 2016; 13:412–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tai H.H., Geisterfer M., Bell J.C., Moniwa M., Davie J.R., Boucher L., McBurney M.W.. CHD1 associates with NCoR and histone deacetylase as well as with RNA splicing proteins. Biochem. Biophys. Res. Commun. 2003; 308:170–176. [DOI] [PubMed] [Google Scholar]
- 42. Burkhardt L., Fuchs S., Krohn A., Masser S., Mader M., Kluth M., Bachmann F., Huland H., Steuber T., Graefen M. et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013; 73:2795–2805. [DOI] [PubMed] [Google Scholar]
- 43. Huang S., Gulzar Z.G., Salari K., Lapointe J., Brooks J.D., Pollack J.R.. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene. 2012; 31:4164–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu W., Lindberg J., Sui G., Luo J., Egevad L., Li T., Xie C., Wan M., Kim S.T., Wang Z. et al. Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene. 2012; 31:3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rodrigues L.U., Rider L., Nieto C., Romero L., Karimpour-Fard A., Loda M., Lucia M.S., Wu M., Shi L., Cimic A. et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res. 2015; 75:1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.