


Abstract
Most bacteria utilize the highly conserved parABS partitioning system in plasmid and chromosome segregation. This system depends on a DNA-binding protein ParB, which binds specifically to the centromere DNA sequence parS and to adjacent non-specific DNA over multiple kilobases in a phenomenon called spreading. Previous single-molecule experiments in combination with genetic, biochemical and computational studies have argued that ParB spreading requires cooperative interactions between ParB dimers including DNA bridging and possible nearest-neighbor interactions. A recent structure of a ParB homolog co-crystallized with parS revealed that ParB dimers tetramerize to form a higher order nucleoprotein complex. Using this structure as a guide, we systematically ablated a series of proposed intermolecular interactions in the Bacillus subtilis ParB (BsSpo0J) and characterized their effect on spreading using both in vivo and in vitro assays. In particular, we measured DNA compaction mediated by BsSpo0J using a recently developed single-molecule method to simultaneously visualize protein binding on single DNA molecules and changes in DNA conformation without protein labeling. Our results indicate that residues acting as hubs for multiple interactions frequently led to the most severe spreading defects when mutated, and that a network of both cis and trans interactions between ParB dimers is necessary for spreading.
INTRODUCTION
The partitioning (par) system in bacteria is essential for faithful chromosome segregation (1) and inheritance of low-copy-number plasmids during cell division (2). Chromosomally encoded Type I par systems (3) consist of three elements: an ATPase called ParA, a DNA-binding protein called ParB and a cis-acting centromere-like DNA sequence called parS located in close proximity to the origin of replication. This parABS system is widely conserved between bacterial species (4–11) and participates in chromosome organization and segregation by recruiting the structural maintenance of chromosomes (SMC) condensin complex to the origin (9,12,13) and repositioning the newly replicated origins to the cell poles (5,14–17).
ParB binds specifically to parS and non-specifically to flanking DNA over tens of kilobases in a phenomenon termed spreading. Despite structural and functional variations (18), spreading is a common feature shared by both chromosomal (4,19–21) and plasmid-encoded ParBs (22–25). Early ParB spreading models proposed that ParB forms a one-dimensional (1D) nucleoprotein filament along DNA (21,22,24). These models were largely based on roadblock experiments in which the insertion of other specific DNA-binding sites adjacent to parS attenuated ParB spreading. However, subsequent studies showed that the cellular concentration of ParB is insufficient to support a continuous 1D filament model given the amount of spreading observed in vivo (22,26,27). Recent studies using a combination of microscopy and biochemistry (26–28) as well as computational modeling (27,29) have argued that ParB spreading is rather a three-dimensional nucleoprotein complex assembled at parS, which requires cooperative interactions between ParB dimers. Consistent with the notion that ParB dimers can bridge DNA, single-molecule experiments (26,28) showed that they could compact DNA by trapping DNA loops within the same DNA molecule.
Structural studies of ParB homologs have provided further insights into the molecular mechanisms of ParB spreading. ParB family proteins are composed of three functionally similar domains connected by flexible linkers (18): an N-terminal domain involved in protein–protein interactions, a central DNA-binding domain containing a helix-turn-helix motif and a C-terminal domain for ParB dimerization. A recent structure of C-terminally truncated Helicobacter pylori ParB, also called Spo0J (HpSpo0J), co-crystallized with 24-bp parS double-stranded DNA (dsDNA) provided a direct visualization of the partition complex assembly for a chromosomal ParB (30). In the HpSpo0J–parS crystal structure, four HpSpo0J monomers (chains A, B, C and D) bound to four parS-containing DNAs oligomerize, forming an asymmetrical tetrameric complex (Figure 1A). Each HpSpo0J monomer is part of a dimer and binds to half of a parS site via its central DNA-binding domain. The overall structure of the HpSpo0J–parS complex can be well superposed to the structure of a C-terminally truncated Thermus thermophilus ParB (TtSpo0J) (31), which was crystallized in the absence of DNA. The structural alignment revealed a conformational change at the N-terminal domain that allows HpSpo0J dimers to interact with each other both in cis (for example, chains A and C in Figure 1A) and in trans (for example, chains A and B in Figure 1A) through this N-terminal domain (30). This suggested that binding to parS might induce a conformational change in ParB from a closed to an open conformation (30). Such an organization is reminiscent of previously proposed model in which spreading of ParB requires both DNA bridging in trans and nearest-neighbor interaction in cis (26,29).
Figure 1.

Crystal structure of HpSpo0J reveals a network of cis and trans interactions for ParB spreading (see colors online). (A) Crystal structure of the C-terminally truncated HpSpo0J–parS complex (30) (PDB code: 4UMK). (B) Cartoon representation of the crystal structure (figure is not drawn to scale). Each chain (chain A in blue, chain B in orange, chain C in red and chain D in green) is a C-terminally truncated HpSpo0J monomer that is part of a dimer bound to half of a 24-bp parS DNA duplex. Only two DNA molecules were available from the original PDB file. Note that tetramerization of the HpSpo0J monomers is asymmetric due to chain C failing to interact with chain D in the crystal structure. (C) Multiple interactions in trans between chain A (blue) and chain B (orange) coordinated by the two R89 residues on each chain. (D) Multiple interactions in cis between chain B (orange) and chain D (green) coordinated by the single R89 residue on chain D. (E) Multiple interactions in cis between chain A (blue) and chain C (red) coordinated by the single R89 residue on chain C. Yellow dashed lines indicate hydrogen bonds, and magenta dashed lines indicate hydrophobic interactions. Figures were prepared in PyMOL. (F) A 2D network map generated from the crystal structure (see ‘Materials and Methods’ section) indicating cis (blue) and trans (green) interactions within the HpSpo0J–parS tetrameric complex. Interactions between residues within the same HpSpo0J monomer are shown in gray. Highly conserved residues that act as hubs for multiple interactions are circled in magenta. Residue number corresponds to that in HpSpo0J.
Interestingly, the crystal structure of the HpSpo0J–parS complex identified an arginine-rich patch, which is located in the highly conserved ParB Box II region (32) and coordinates multiple cis and trans protein–protein interactions (30) (Figure 1C–E). We had previously shown that these same arginine residues are essential for DNA bridging and ParB spreading in Bacillus subtilis ParB (BsSpo0J) (26). Motivated by this observation, we computationally created a 2D network map (Figure 1F) based on the crystal structure for all participating residues at the intermolecular interaction surfaces (see ‘Materials and Methods’ section). From this map, we identified a number of additional residues that coordinate multiple cis and/or trans interactions, and we termed these residues interaction hubs. Guided by our interaction map and a sequence alignment between ParB homologs (Supplementary Figure S1), we selected hubs and their interacting partners with a high degree of conservation to perform comprehensive mutagenesis studies in BsSpo0J.
Here we report how these BsSpo0J mutants affect ParB spreading using a combination of in vivo fluorescence imaging, in vitro single-molecule microscopy and DNA binding assays. We found that mutating residues implicated in multiple interactions between ParB dimers abolished spreading without impacting the ability of ParB to bind parS or non-specific DNA. Many of these proteins are novel non-spreading ParB point mutants. Importantly, they implicate both cis and trans interaction surfaces as being essential for ParB spreading, and provide a direct physiological validation of the crystal structure of the HpSpo0J–parS complex.
MATERIALS AND METHODS
Generation of a 2D network map
A connection matrix was first generated in MATLAB for all residues participating in protein–protein interactions as revealed by the crystal structure of the HpSpo0J–parS complex (refer to Supplementary Table S2 in (30)). Specifically, all of the published interactions (53 in total) were documented in an interaction table that recorded the names and types of interactions (cis, trans or intramolecular interactions within the same ParB monomer) for each pair of interacting residues. A unique identification number was assigned for each participating residue based on its alphabetical name (for example, Ala86 = 1, Arg115 = 2 and so on). The connection matrix is a square matrix with size equal to the number of all participating residues (44 residues in total), and each entry in the matrix is the index of the row number in the interaction table, which can be used to identify the type of interaction between the residues at that entry. An interaction map (Figure 1F) was generated in MATLAB using the built-in function biograph. The graphical object was annotated in MATLAB to assign different colors representing different types of interactions. Interactions with the same kind of residue were omitted (for example, the trans interaction between the two Arg89 residues on chains A and B in Figure 1A, respectively). Interactions between the same pair of residues that involved different atoms or are located on different monomers were counted only once. Therefore, each interaction between residues shown on the map (Figure 1F) might occur simultaneously between multiple interfaces (for example, the cis interaction between Arg115 and Lys95 can be found both in the interfaces between chains A and C, as well as between chains B and D in Figure 1A). Hubs of multiple interactions and their severity in ParB spreading after being mutated (Figure 1F and Supplementary Figure S21) were annotated in Adobe Illustrator.
Fluorescence microscopy of bacterial cells
All B. subtilis strains were derived from the prototrophic strain PY79 (33). Tables of strains, plasmids, oligonucleotide primers and a description of plasmid construction can be found in the Supplementary Data. Fluorescence microscopy was performed on a Nikon Ti motorized inverted microscope equipped with a Plan Apo 100×/1.4 NA phase contrast oil objective and a Lumen Pro fluorescence illumination system (Prior Scientific). Cells were grown in defined rich (CH) medium (34) at 37°C to an optical density of 0.2–0.3, and were immobilized using 2% agarose pads containing growth medium. mGFPmut3-BsSpo0J and HBsu-mCherry fluorescence were collected using a FITC filter set (Nikon 96320) and a TRITC filter set (Nikon 96321), respectively. Images were acquired with a Hamamatsu ORCA-R2 cooled CCD camera controlled with MetaMorph software (Molecular Devices) using identical acquisition parameters. Image brightness and contrast were adjusted identically for compared image sets using ImageJ software. Final figures were prepared in Adobe Illustrator.
Protein purification
Wild-type and mutant B. subtilis Spo0J (BsSpo0J) proteins were expressed with an N-terminal His6-SUMO tag in BL21 (DE3) pLysS cells and purified by a two-step tandem affinity method (26) with modified buffer conditions as described here. Briefly, cell pellets were lysed by sonication in lysis buffer (20 mM Tris at pH 8.0, 1 M NaCl, 50 mM imidazole and 5 mM 2-mercaptoethanol (BME)) supplemented with 1 mM PMSF, and supernatants were clarified by ultracentrifugation. The clarified supernatant was bound to Ni-NTA resin (Qiagen) and washed extensively with lysis buffer and sequentially with salt-reduction buffer (lysis buffer with only 350 mM NaCl). His6-SUMO fusion proteins were manually eluted with elution buffer (20 mM Tris at pH 8.0, 350 mM NaCl, 250 mM imidazole and 5 mM BME) in a series of 1 ml aliquots. Peak fractions were collected and dialyzed overnight at 4°C against dialysis/storage buffer (20 mM Tris at pH 8.0, 350 mM NaCl, 10% glycerol, 10 mM imidazole and 5 mM BME) in the presence of His6-Ulp1 protease (26). The cleaved His6-SUMO tag and His6-Ulp1 were then removed from the proteins by incubating with Ni-NTA resin again on the second day. The flow-through, containing untagged BsSpo0J proteins, was collected and concentrated by centrifugation in Amicon Ultra-0.5 10 kDa cutoff spin filters (EMD Millipore) before being stored at −80°C.
Single-molecule PIFE experiments
DNA construction and flow cell design were performed as described (35). Briefly, 20-kb DNAs were generated from genomic DNA of B. subtilis strain PY79 using a two-round polymerase chain reaction (PCR) method (35) that randomly incorporated Cy3-dUTP (Enzo Life Sciences) at a low molar concentration. Primer sequences are listed in the Supplementary Data. The resulting PCR product was modified with biotin at one end, and there were 20 Cy3 dyes on average (35) incorporated at random positions along the 20-kb dsDNA. Cy3-labeled DNAs were then immobilized on a streptavidin-coated coverslip surface of a multi-channel flow cell connected to an automated syringe pump (Harvard Apparatus). Unbound DNAs were washed away with binding buffer (20 mM Tris at pH 7.5, 100 μg ml−1 bovine serum albumin and 100 mM NaCl unless indicated otherwise). Proteins that were diluted to the indicated concentrations with the same binding buffer were introduced into the flow cell at a flow rate of 100 μl min−1. Each channel of the flow cell was used only once after introducing the proteins.
Protein association was imaged on a homebuilt through-objective total internal reflection fluorescence microscope (26) equipped with a Plan Apo 60×/1.45 NA oil objective, a 1.6× magnification boost and a Hamamatsu ImagEM camera (EM-CCD 9100–13) controlled with HCImage software (Hamamatsu Photonics). Cy3 fluorescence was excited with a 532 nm laser (Coherent Compass 215M-75) and collected with a long-pass dichroic mirror (Chroma Z532RDC) and a HQ600/75M emission filter (Chroma). Images were acquired at 10 Hz with continuous exposure. A low incident laser power was used to minimize photobleaching.
Image analyses were performed as previously described (35). Briefly, 20–30 regions of interest containing single DNA molecules were first marked manually in ImageJ software. Time-lapse trajectories of integrated fluorescence intensity after subtracting background fluorescence and length of the Cy3-labeled 20-kb dsDNAs were generated using custom scripts written in MATLAB. Fold increase in integrated fluorescence intensity over time (Figure 3B and C; Supplementary Figures S8–12, 20B and C) was calculated by dividing each trajectory by the value averaged for the first 5–10 s before protein binding. Time zero was defined as the starting point of protein association. Lag time (Figure 3B, Supplementary Figure S8A and Table 1) was defined as the time between protein binding and the initiation of DNA compaction. Rate of DNA compaction (Table 1) was estimated by linear fitting the trajectory of DNA length between maximum and minimum values (Supplementary Figure S8C).
Figure 3.

Single-molecule DNA compaction by BsSpo0J (see colors online). (A) Schematic of the single-molecule protein-induced fluorescence enhancement (PIFE) assay (35). A 20-kb dsDNA sparsely labeled with Cy3 dyes (green) is tethered to the surface of a functionalized glass coverslip and extended by buffer flow. Step 1: binding of unlabeled proteins to DNA enhances the fluorescence of nearby Cy3 dyes (red) due to PIFE. Step 2: changes in DNA conformation mediated by proteins can be simultaneously detected as a change in length of flow-stretched DNAs. PEG, polyethylene glycol; SA, streptavidin. Figures are not drawn to scale. (B) Demonstration of the single-molecule PIFE assay with wild-type BsSpo0J (100 nM). Trajectories of individual DNAs are shown in gray and the average over all trajectories (n = 38) is shown in red. The fold increase in integrated fluorescence intensity over time was calculated by dividing each trajectory by the value averaged for the first 5–10 s before protein binding. DNA length was normalized to the maximum values in individual trajectories. Time zero was defined as the starting point of protein association. tlag is lag time between protein binding and the initiation of DNA compaction. Insert: kymograph of a single DNA. Scale bar = 6 s. (C) Fold increase in the integrated fluorescence intensity and DNA length trajectories for wild-type BsSpo0J (black; the same curve is reproduced in each panel) and mutants (red) at a protein concentration of 100 nM. Each trajectory was averaged over 20–30 Cy3-labeled DNAs. See Supplementary Figures S9 and 12 for trajectories of all characterized mutants.
Table 1. Quantification of the kinetics of DNA compaction for wild-type BsSpo0J and mutants.
| Lag time (s) | Rate of DNA compaction (μm s−1) | ||||
|---|---|---|---|---|---|
| Protein | Mean | SD | Mean | SD | n |
| WT | 5.7 | 1.3 | 0.33 | 0.11 | 160 |
| E52R | 5.8 | 0.8 | 0.89 | 0.25 | 45 |
| E78R | 4.2 | 0.9 | 1.43 | 0.60 | 36 |
| F81A | 59.7 | 23.6 | 0.05 | 0.04 | 49 |
| G77S | 6.3 | 1.3 | 1.07 | 0.36 | 67 |
| G77S + R79A | 6.8 | 1.2 | 1.38 | 0.56 | 44 |
| I74A | 2.8 | 0.7 | 2.51 | 0.98 | 41 |
| M104A | 24.9 | 8.0 | 0.19 | 0.07 | 45 |
| N112S | 2.5 | 0.7 | 0.79 | 0.29 | 44 |
| P62A | 3.6 | 1.0 | 1.14 | 0.30 | 33 |
| Q140A | 11.1 | 6.2 | 0.05 | 0.02 | 66 |
| Q140R | 3.2 | 4.1 | 0.05 | 0.02 | 47 |
| Q61A | 36.8 | 13.5 | 0.12 | 0.06 | 53 |
| Q61R | 3.9 | 0.6 | 1.17 | 0.28 | 39 |
| R105A | 19.6 | 9.0 | 0.09 | 0.03 | 59 |
| V75E | 4.9 | 1.7 | 1.94 | 0.72 | 70 |
| Y72A | 3.9 | 1.1 | 2.02 | 0.65 | 38 |
Lag time was defined as the time between protein binding and the initiation of DNA compaction (Figure 3B and Supplementary Figure S8A). Rate of DNA compaction was estimated by linear fitting of the DNA length trajectory between maximum and minimum values (Supplementary Figure S8C). Distributions of lag time and rate of DNA compaction were fit to a Gaussian distribution (Supplementary Figure S8B and D). The total number of trajectories, n, was obtained from analysis for at least two replicates of experiments. SD = standard deviation.
Electrophoretic mobility shift assay
The 24- and 39-bp DNA substrates containing either a parS or scrambled parS site were generated by annealing pairs of complementary oligonucleotides (Supplementary Table S4) in 1× TE + 100 mM NaCl. The annealed dsDNAs were then labeled with γ32P-ATP by phosphorylation with T4 polynucleotide kinase (NEB), and the free nucleotide was removed using illustra MicroSpin G-50 spin desalting columns (GE Healthcare Life Sciences). Proteins were first diluted to 5× the indicated concentrations in binding buffer (20 mM Tris at pH 7.5 and 300 mM NaCl), and they were then added to the indicated labeled DNA substrate (2 nM final concentration) in a 10 μl reaction volume. Cold 39-bp scrambled competitor oligo duplexes (20 nM final concentration) were added to reactions with labeled 39-bp parS DNA substrate (2 nM final concentration) (see Figure 4A, C and E; Supplementary Figure S14). Each reaction was assembled on ice and then incubated at room temperature for 10 min. Complexes were resolved on a 5% 0.5× Tris/Borate/Ethylenediaminetetraacetic acid (TBE)-polyacrylamide gel electrophoresis gel, which was pre-run at 200 V at room temperature for at least 30 min. 5 μl of loading dye (20 mM Tris at pH 7.5, 300 mM NaCl, 46% glycerol and trace amounts of bromophenol blue) was added to each reaction, and 5 μl of each was loaded on the gel to run at 200 V for ∼25 min at room temperature. Gels were dried under vacuum and exposed to a storage phosphorscreen, which was subsequently imaged on a Personal Molecular Imager (BioRad).
Figure 4.

In vitro characterization of the specific binding of BsSpo0J to parS DNA duplexes. EMSA of wild-type BsSpo0J and mutants binding to either a radioactively labeled 39-bp parS DNA substrate supplemented with cold 39-bp scrambled parS competitor DNA as shown in (A, C and E), or a labeled 24-bp parS substrate without competitor DNA as shown in (B, D and F) (see ‘Materials and Methods’ section). Protein concentrations were 0.2, 0.4, 0.8 μM in (A and B) and 0.2, 0.4, 0.8 and 1.0 μM in (C–F). Asterisk and arrow indicate position of the wells and free DNA respectively in each gel. See Supplementary Figures S13 and 14 for results of all characterized mutants.
Differential scanning fluorimetry
Proteins were diluted in storage buffer (20 mM Tris at pH 8.0, 350 mM NaCl, 10% glycerol, 10 mM imidazole and 5 mM BME) to 100 μg ml−1 in a total reaction volume of 20 μl on ice. SYPRO Orange from the Protein Thermal Shift Dye Kit (Life Technologies) was then added to each reaction to a final dilution of 1:1000. The mixture was transferred to a MicroAmp Fast Optical 96-Well Reaction Plate (Life Technologies), and the plate was sealed with a MicroAmp Optical Adhesive Film (Life Technologies). Thermal denaturation curves were recorded in a QuantStudio 7 Flex Real-Time PCR System (Life Technologies) by raising the temperature from 25 to 99°C at a rate of 3°C per min. The fluorescent dye was excited at 470 nm and the fluorescence emission was measured at 587 nm. Fluorescence intensities for each protein were plotted as a function of increasing temperature and the sigmoidal thermal denaturation curve was fitted with the Boltzmann equation using Protein Thermal Shift Software (Life Technologies) to obtain the melting temperature. Data points after the maximum fluorescence intensity were excluded from fitting. Only minimal background fluorescence from the reaction buffer was observed with no-protein controls. Triplicates of each protein sample were performed.
Size exclusion chromatography
Protein samples at 10–30 μM were run at a flow rate of 0.5 ml min−1 in storage buffer (20 mM Tris at pH 8.0, 350 mM NaCl, 10% glycerol, 10 mM imidazole and 5 mM BME) on a Superdex 200 Increase 10/300 GL column (GE Healthcare) attached to an AKTA pure fast protein liquid chromatography purification system (GE Healthcare). Eluted proteins at a fixed volume of 300 μl per fraction were detected by UV absorbance at 280 nm. Molecular weights and void volume were determined using a Gel Filtration Markers Kit (Sigma Aldrich).
RESULTS
Mutation of ParB interaction hubs disrupts spreading
Guided by our interaction map (Figure 1F) and a sequence alignment between ParB homologs (Supplementary Figure S1), we systematically mutated both selected hubs and their interacting partners in BsSpo0J. Alanine mutations were introduced at these sites in addition to various mutations previously identified in genetic screens (Supplementary Table S1). We also introduced various non-conservative mutations, including charge-reversal (E52R, H57E, E78R and R105E), glutamine to arginine (Q61R and Q140R) and hydrophobic-to-charged (L60E and V75E) substitutions (Supplementary Table S1).
Chromosomal ParB proteins such as BsSpo0J fused to fluorescent proteins form compact foci that require origin-proximal parS sites (13,26,29,36). BsSpo0J mutants that fail to spread in vivo are defective in foci formation (19,26,37). As a proxy for in vivo spreading, we expressed mGFPmut3-tagged BsSpo0J (mGFPmut3-BsSpo0J) (26) in a spo0J deletion strain under the control of its native promoter in B. subtilis. Immunoblot analysis indicated that all of our mGFPmut3-BsSpo0J constructs were full length and expressed at similar levels (Supplementary Figure S2). Wild-type mGFPmut3-BsSpo0J appeared as distinctive condensed foci localized toward the poles of each bilobed nucleoid under vegetative growth conditions (Figure 2).
Figure 2.

In vivo characterization of BsSpo0J spreading (see colors online). Localization of mGFPmut3-BsSpo0J variants. See Supplementary Figures S3–5 for images of all characterized mutants. Nucleoid (false-colored red) was labeled with HBsu-mCherry. Scale bar = 5 μm.
Previously identified spreading-deficient mutants in ParB Box II (Supplementary Figure S1) including G77S (19), R79A (26) and R80A (37) abolished foci formation and appeared diffuse but nucleoid-associated (Figure 2 and Supplementary Figure S3). Strikingly, we found several additional point mutants in our systematic mutagenesis including R39A, L60E, V75E, Q61R and N112S that also failed to form foci and appeared diffuse in the cell (Figure 2 and Supplementary Figures S3–5), suggesting they are also defective in spreading. These mutated residues (except N112) are predicted to be hubs for multiple protein–protein interactions in the HpSpo0J–parS tetrameric complex (Figure 1F). The mutant N112S was previously found to be impaired in localizing SMC to the origin (12).
In addition to these no-foci mutants, we also identified mutants that showed an incomplete loss of foci, suggesting that they are partially impaired in spreading. These partial loss-of-function mutants can be subcategorized qualitatively based on the fluorescence intensities of the green fluorescent protein (GFP) foci: The mutants I74A, E78R and H57E formed very weak foci, which we termed ‘fuzzy’ foci (Figure 2; Supplementary Figures S3 and 4). Interestingly, in the HpSpo0J–parS structure, H67 (H57 in BsSpo0J) interacts with E88 (E78 in BsSpo0J) in trans (Figure 1F and Supplementary Figure S6). By contrast, the mutants R105E, Y72A, Q140R and P62A formed what we term ‘faint’ foci, which were more distinct than fuzzy foci but still less intense than those formed by wild-type mGFPmut3-BsSpo0J (Figure 2; Supplementary Figures S3 and 4). Faint foci are similar to those formed by the previously identified partial loss-of-function mutation R82A (26) (Supplementary Figure S3), suggesting that these mutations weaken but do not eliminate the intermolecular interactions required for ParB spreading.
Spreading-deficient mutants are abnormal in DNA bridging
To further investigate whether the newly identified spreading-deficient mutants of BsSpo0J are impaired in DNA bridging, which is necessary for the formation of a higher order partition complex, we purified the mutants (see ‘Materials and Methods’ section and Supplementary Figure S7) and tested their ability to compact DNA in single-molecule experiments. We used a recently developed single-molecule method to detect association of unlabeled protein to DNA while simultaneously monitoring changes in DNA conformation (35). In this assay, 20-kb dsDNAs that were sparsely labeled with Cy3 dyes were immobilized at one end to a functionalized glass coverslip in a microfluidic flow cell (Figure 3A). DNAs were stretched by buffer flow and visualized using total internal reflection fluorescence microscopy. The integrated fluorescence intensity of each Cy3-labeled DNA molecule increased dramatically up to 4.5-fold on average on association of unlabeled wild-type BsSpo0J as a result of a chemical effect known as protein-induced fluorescence enhancement (PIFE) (Figure 3B). At the same time, DNAs were compacted all the way to the tether point as wild-type BsSpo0J can bridge distal segments of DNA to facilitate DNA looping (26,28). In addition, there is a significant lag time between protein association and the initiation of DNA compaction (Figure 3B; Supplementary Figure S8 and Table 1), consistent with the idea that cooperative interactions between ParB dimers are required for DNA bridging (26,28).
Using the single-molecule PIFE assay, we found that the non-spreading mutants R79A, R80A, R39A and L60E (Figure 2 and Supplementary Figure S3) abolished DNA compaction in vitro (Figure 3C and Supplementary Figure S9), even when tested at a higher protein concentration (Supplementary Figure S10). Importantly, association of these BsSpo0J mutants with DNA, as measured by PIFE, occurred with the same rate as the wild-type protein. A higher fold change in the fluorescence intensity of Cy3-labeled DNAs was observed at equilibrium for the wild-type protein (black curves in Figure 3C, Supplementary Figures S9 and 10) as compared to the DNA compaction deficient mutants (red curves in Figure 3C; Supplementary Figures S9 and 10). This difference in the magnitude of the PIFE effect is likely caused by higher order interactions that stabilize wild-type BsSpo0J on DNA, thus increasing its fractional occupancy. The H57E, R82A and R105E mutants, which are partially impaired in spreading (Figure 2 and Supplementary Figure S3), also failed to compact DNA (Figure 3C; Supplementary Figures S9 and 10), while R82A compacted DNA only at a reduced salt concentration (26) (Supplementary Figure S11). We also identified several BsSpo0J mutants, such as M104A and Q140A, that appeared to form wild-type-like GFP foci in cells (Figure 2 and Supplementary Figure S3), but compacted DNA with slower kinetics in vitro (Figure 3C; Supplementary Figures S9 and 10; Table 1). In the HpSpo0J–parS crystal structure, both residues M114 (M104 in BsSpo0J) and E150 (Q140 in BsSpo0J) interact with residue R89 (R79 in BsSpo0J) in cis (Figure 1F), and E150 (Q140 in BsSpo0J) is also a hub that coordinates with multiple residues in cis (Figure 1F). Taken together, we identified a group of point mutants (Group I in Table 2) with a modest to severe defect in spreading in vivo that also showed an inability to bridge DNA in vitro.
Table 2. Summary of phenotypes for BsSpo0J mutants.
| Group | Subgroup | Mutants | parS binding | In vitro DNA compaction | In vivo foci formation |
|---|---|---|---|---|---|
| I | A | R39A, H57E, L60E, R79A, R80A | Normal | No compaction | No foci or fuzzy foci |
| B | R82A, R105E | Normal | No compaction (salt dependent) | Faint foci | |
| C | Q61A, V75A, F81A, M104A, R105A, Q140A, Q140R | Normal | Slower | WT like | |
| II | P62A, Y72A, I74A, V75E, G77S, E78R | Abnormal | Faster | No foci or faint foci or fuzzy foci | |
| III | A | Q61R, N112S | Normal | Faster | No foci |
| B | E52R | Normal | Faster | WT like |
Mutants were classified into three groups as described in the main text.
Our single-molecule characterization also allowed us to identify two other groups of non-spreading mutants (Group II and Group III in Table 2) that are unexpectedly hyperactive in DNA compaction. Specifically, mutants such as G77S, V75E and Q61R that abolished foci formation (Figure 2; Supplementary Figures S4 and 5) showed a more than 3-fold faster rate of DNA compaction compared to the wild-type protein (Figure 3C, Supplementary Figure S12 and Table 1). Such hyperactivity in DNA compaction was also observed for mutants such as I74A, E78R and P62A (Figure 3C, Supplementary Figure S12 and Table 1) that appeared to have fuzzy or faint GFP foci (Figure 2 and Supplementary Figure S4). In fact, we identified a consecutive series of conserved residues in ParB Box II (Supplementary Figure S1) of BsSpo0J from Y72 to E78 that when mutated (see Table 2 and Supplementary Table S1 for specific mutations) caused defective foci formation (Supplementary Figure S4) but hyperactive DNA compaction (Supplementary Figure S12 and Table 1). In addition, we observed that the mutant N112S, which exhibited a complete loss-of-foci phenotype in cells (Supplementary Figure S5), had a faster rate of DNA compaction than the wild-type protein (Supplementary Figure S12A and Table 1). Moreover, N112S failed to compact DNA all the way to the tether point compared to wild-type BsSpo0J (Supplementary Figure S12A) although nearly complete compaction was achieved with slower kinetics at a higher protein concentration (Supplementary Figure S12B).
Hyperactive mutants in DNA compaction bind abnormally to parS
To investigate the paradox of loss of foci in vivo despite hyperactive DNA compaction in vitro, we used an electrophoretic mobility gel shift assay (EMSA) to test the ability of the newly identified mutants in specific binding to parS. We used either a 39-bp or a 24-bp DNA duplex, in which a central 16-bp parS site is flanked by non-specific DNA sequence. Similar to the previously characterized spreading-deficient mutant R79A (26), all of Group I mutants (Table 2) such as R39A, L60E and H57E that displayed a varied degree of deficiency in spreading (Figure 2 and Supplementary Figure S3) and were either completely or partially defective in DNA bridging (Figure 3C, Supplementary Figures S9–11 and Table 1), were indistinguishable from the wild-type protein in EMSAs (Figure 4A and B; Supplementary Figures S13 and 14). To measure non-specific DNA binding, we performed EMSA experiments using labeled 24-bp or 39-bp DNA duplexes with a scrambled parS site. Under the same binding conditions, we did not observe any non-specific binding for wild-type or mutant BsSpo0J to the 24-bp scrambled parS DNA (data not shown). All of Group I mutants demonstrated wild-type ability in non-specific binding to the 39-bp scrambled parS DNA (Supplementary Figure S15). These results are consistent with previous studies using size exclusion chromatography with multi-angle light scattering (SEC-MALS), in which non-specific binding of wild-type BsSpo0J was only observed by increasing the protein concentrations to 80 μM when using a 24-bp substrate and by extending the length of the DNA substrate beyond 24-bp (28).
In contrast, Group II mutants (Table 2) such as Y72A, V75E and E78R that were hyperactive in DNA compaction (Figure 3C, Supplementary Figure S12 and Table 1) produced an abnormal pattern of bands in both specific and non-specific binding compared to the wild-type protein (Figure 4C–F; Supplementary Figures S13–15). In particular, in EMSAs with the 24-bp parS DNA duplex, which only allows for binding of a single ParB dimer (28), Group II mutants failed to produce a discrete shifted band (Figure 4D and F; Supplementary Figure S13). At higher protein concentrations the mutants either retained labeled DNA in the well or produced an indiscrete smear. Interestingly, Group III mutants (Table 2) behaved similarly to the wild-type protein in both specific and non-specific binding (Supplementary Figures S13–15), even though they were also hyperactive in DNA compaction (Supplementary Figure S12 and Table 1).
Group II mutations cause mis-folding in ParB
Group II mutants (Table 2), including the well-characterized spreading-deficient mutant G77S (19,26), had either a complete or partial loss-of-foci phenotype (Figure 2 and Supplementary Figure S4) but were hyperactive in DNA compaction (Figure 3C, Supplementary Figure S12 and Table 1). Notably, this group of mutants all demonstrated an abnormal shifting pattern with both parS and scrambled parS DNAs compared to the wild-type protein (Figure 4C–F; Supplementary Figures S13–15). Except for P62A, all of these mutants involve residues in the first half of ParB Box II (Y72 to E78 in BsSpo0J) (Supplementary Figure S1). Most of the residues in this region are located in one of the β-sheets that was proposed to stabilize ParB dimerization in solution at the N-terminus in the crystal structure of the C-terminally truncated TtSpo0J (Supplementary Figure S16) (31). Within the structure of the HpSpo0J–parS tetrameric complex (30), these residues interact with the helix-turn-helix domain on the nearby dimer in cis (between chains B and D in Supplementary Figure S17B) along with being involved in protein–protein interactions in trans (between chains A and B in Supplementary Figure S17C). Therefore, we hypothesized that Group II mutants are locally mis-folded, leading to abnormal dimerization and intermolecular interactions.
To test our hypothesis, we first performed differential scanning fluorimetry (see ‘Materials and Methods’ section) to examine the protein stability of all the mutants we constructed. While Group I and Group III mutants (Table 2) were very similar to wild-type BsSpo0J (Figure 5A and B; Supplementary Figures S18 and 19B), we observed that all of Group II mutants showed either a non-sigmoidal thermal denaturation curve with high initial fluorescence (Figure 5A and Supplementary Figure S19A) or a significantly lower melting temperature (Figure 5B), indicating that they were less thermally stable and thereby less well folded compared to the wild-type protein. Consistent with these results, we observed a significant fraction of aggregated proteins for the Group II mutants G77S and P62A in size exclusion chromatography (Figure 5C). In contrast, wild-type BsSpo0J and Group I mutants R79A and R105E were eluted as a single peak (Figure 5C), which we confirmed to be BsSpo0J dimers in solution using SEC-MALS (Supplementary Figure S7B). In addition, G77S displayed a second peak with a lower molecular weight running next to the dimer (purple curve in Figure 5C), consistent with an abnormal oligomeric state in solution.
Figure 5.

Thermal stability and oligomeric state of BsSpo0J (see colors online). (A) Thermal denaturation curves for wild-type BsSpo0J and mutants at a protein concentration of 100 μg ml−1 measured with differential scanning fluorimetry (see ‘Materials and Methods’ section). Fluorescence intensities were normalized to the maximum in each curve. Only one replicate of each protein is shown. See Supplementary Figures S18 and 19 for results of all characterized mutants. (B) Melting temperatures of wild-type BsSpo0J (highlighted by the red dashed line for reference) and mutants arranged in groups (see Table 2) determined from a Boltzmann fitting on the thermal denaturation curves (see ‘Materials and Methods’ section). Error bars are standard errors of the mean between three replicates. Group II mutants were significantly less stable compared to the wild-type protein (**P < 0.001, based on an unpaired t-test with unequal variances). (C) Chromatograms of wild-type BsSpo0J and mutants measured in size exclusion chromatography. All mutants showed a major peak at 12.7 ml overlapping with the wild-type protein, corresponding to a BsSpo0J dimer. Group II mutants (G77S and P62A) displayed a significant peak at 8.5 ml, corresponding to void volume (Vo). G77S also showed a second major peak at 13.4 ml with a lower molecular weight. All proteins showed a minor peak at 15.5 ml with a low molecular weight, likely corresponding to a monomeric protein. Absorbance at 280 nm was normalized to the maximum in each curve.
To test whether G77S compacts DNA through the same cis and trans interactions between ParB dimers as observed in the HpSpo0J–parS crystal structure, we combined G77S with the R79A mutation from Group I (Table 2), which on its own abolished DNA compaction. Strikingly, we observed that G77S + R79A behaved nearly identically to G77S in both in vivo and in vitro assays (Table 1; Supplementary Figure S20 compared to Figure 2–5), implying that G77S compacted DNA through an abnormal mode of protein–protein interactions likely mediated through an unfolded region of the protein. Collectively, these observations support our hypothesis that Group II mutations disrupt protein folding and parS binding rather than specific DNA bridging interactions.
Further investigation on Group III mutations
Our identification on the three point mutants Q61R, N112S and E52R, which exhibited properties in vivo and in vitro that were inconsistent with the other two groups, results in a new Group III classification (Table 2). All of the mutants in this group were hyperactive in DNA compaction (Table 1 and Supplementary Figure S12) but showed similar thermal stability (Figure 5B and Supplementary Figure S19B) and DNA binding ability as the wild-type protein (Supplementary Figures S13–15). The mutant Q61R was shown to be defective in ParB spreading as it abolished GFP foci formation in cells (Supplementary Figure S5). One possible explanation for the defect in Q61R is that ParB spreading requires a precisely tuned interaction strength between ParB dimers (38). Interactions between dimers with abnormally high affinity, as indicated by faster than wild-type DNA compaction in vitro, might prevent productive spreading in vivo. This hypothesis predicts that combining a hyperactive mutant with a hypoactive mutant would partially rescue ParB spreading compared to each mutant alone. To test this idea, we constructed double mutants, Q61R + R82A and Q61R + R105E, in which R82A and R105E are Group I mutants (Table 2) that were impaired in spreading (Supplementary Figure S3) and deficient in DNA compaction (Supplementary Figures S9–11). Combination with the Q61R mutation partially rescued the ability of R82A and R105E to compact DNA, albeit only at a higher protein concentration and with much slower kinetics than wild-type BsSpo0J (Supplementary Figure S20B and C compared to Supplementary Figures S9 and 10). The double mutants still bound to parS and non-specific DNA like the wild-type protein (Supplementary Figure S20D–F). They also exhibited wild-type-like thermal stability (Figure 5B and Supplementary Figure S20G). However, combining Q61R with R82A and R105E did not rescue the formation of GFP foci in cells (Supplementary Figure S20A). Taken together, these results argue that the defect in the Q61R mutant does not arise from the enhancement of interactions between ParB dimers, but is possibly due to the formation of non-native protein–protein interactions.
DISCUSSION
Using the structure of H. pylori ParB (HpSpo0J) co-crystallized with parS DNA (30), we generated a 2D network map (Figure 1F) that captures residues acting as hubs for multiple cis and trans interactions between ParB dimers. We systematically mutated these residues in B. subtilis ParB (BsSpo0J) and performed a number of assays to characterize their effects on ParB spreading: (i) we determined whether the BsSpo0J mutants were able to form foci in cells when fused to GFP, which we used as a proxy for ParB spreading in vivo; (ii) we determined whether purified mutants could compact DNA in an in vitro single-molecule assay, indicating whether they were still able to bridge DNA that is required for forming a higher order partition complex; and (iii) we determined binding of the mutants to parS and non-specific DNA in EMSAs. Through this characterization, we have identified a number of new spreading-deficient ParB point mutants that we assigned into three major groups based on their phenotypes (Table 2).
Group I mutants (Table 2), which include the previously reported arginine-rich patch in ParB Box II (26,30) (Supplementary Figure S1), all behaved identically to the wild-type protein in DNA binding (Figure 4A and B; Supplementary Figures S13–15), protein stability and oligomerization assays (Figure 5 and Supplementary Figure S18). However, they showed a variable defect in their ability to form mGFPmut3-BsSpo0J foci in cells (Figure 2 and Supplementary Figure S3) that correlated well with their inability to compact flow-stretched DNA in single-molecule experiments (Figure 3C, Supplementary Figures S9–11 and Table 1). Therefore, Group I mutants were deficient in spreading because they are defective in cooperative interactions between ParB dimers that enable the trapping of DNA loops (26,28).
Earlier structural work on a C-terminally truncated T. thermophilus ParB (TtSpo0J) in the absence of DNA speculated that the N-terminal domains of ParB could act as a secondary dimerization interface upon binding to parS, enabling the C-terminal domains to interact with neighboring ParB dimers to spread linearly along DNA (31). In contrast to this proposed model, none of the spreading-deficient Group I mutations is in the C-terminal domain (Supplementary Figure S16) or disrupts ParB dimerization (Figure 5C). Residues affected by Group I mutations, such as R89 (R79 in BsSpo0J) and R115 (R105 in BsSpo0J), are hubs that coordinate a large number of residues both in cis and in trans (Figure 1F) in the HpSpo0J–parS tetrameric complex (30). Therefore, our results are most consistent with the HpSpo0J–parS crystal structure and strongly suggest that a network of both cis and trans interactions between ParB dimers is required for forming a higher order partition complex as well as for ParB spreading.
In addition to Group I mutants, our comprehensive mutagenesis studies allowed us to identify a series of spreading-deficient mutants that are hyperactive in DNA compaction but exhibited different phenotypes in DNA binding and protein stability, resulting in the classification of two additional groups (Table 2). Group II mutations (Table 2) act to inhibit spreading by disrupting global folding and dimerization of BsSpo0J (Figure 5 and Supplementary Figure S19A) instead of attenuating the specific protein–protein interactions between ParB dimers required for tetramerization and spreading. Given these observations, our work argues against using Group II mutants in future cellular experiments, including the well-studied G77S (13,19,26,29,36,39,40).
On the other hand, Group III mutants (Table 2) retained wild-type ability in DNA binding (Supplementary Figures S13–15) and protein folding (Figure 5 and Supplementary Figure S19B). Our double mutant analysis on Q61R (Supplementary Figure S20) did not support the hypothesis that a fine-tuned interaction strength between ParB dimers is required for spreading (38). The mutant N112S, which was previously found to be defective in recruiting SMC to the origin (12), failed to compact DNA all the way to the tether point with the same kinetics as the wild-type protein (Supplementary Figure S12A) even at a higher protein concentration (Supplementary Figure S12B). Future experiments beyond the scope of this study are necessary to elucidate why these mutants are defective in spreading.
Interestingly, we noted that in contrast to Q61R and N112S, the Group III mutant E52R (Table 2) formed wild-type GFP foci in cells (Supplementary Figure S5) even though it showed hyperactivity in DNA compaction (Table 1 and Supplementary Figure S12). In the HpSpo0J–parS crystal structure, E62 (E52 in BsSpo0J) is one of the three conserved residues besides M114 (M104 in BsSpo0J) and E150 (Q140 in BsSpo0J) that interact with the hub at R89 (R79 in BsSpo0J) in cis (Figure 1F). The fact that the BsSpo0J mutants E52R, M104A and Q140A or Q140R were either normal or only mildly deficient in spreading (Figure 2; Supplementary Figures S3 and 5) suggests that the network of interactions between ParB dimers, rather than individual interactions, is essential for ParB spreading.
In conclusion, we demonstrated that a highly cooperative network of cis and trans protein–protein interactions revealed in the HpSpo0J–parS tetrameric complex (30) is required for ParB spreading. This model is supported by the results from mutation of residues, such as R115 (R105 in BsSpo0J) and L70 (L60 in BsSpo0J), that showed severe defects in DNA bridging in vitro and ParB spreading in vivo (Table 2) and are hubs for exclusively cis and trans interactions in the network, respectively (Supplementary Figure S21). It remains unclear whether cis interactions can be strictly interpreted as nearest-neighbor interactions between ParB dimers (26,29) (Figure 6B), given that each pair of the four parS molecules located in cis in the crystal structure were not part of a continuous DNA duplex (30) (Figure 1A). We propose that interactions through this interface may also occur between ParB dimers that are far apart but brought into close proximity through DNA looping (Figure 6B). Taken together, we envision that ParB molecules that dimerize through their C-terminal domains bind both specifically to parS and non-specifically to distant chromosomal DNA through their central DNA-binding domain (Figure 6A). ParB dimers nucleated at parS can interact with other dimers both in cis and in trans (Figure 6B) to form a tetramer (Figure 6C); this tetrameric ParB complex is the fundamental unit for the assembly of a higher order partition complex that can bridge and trap large DNA loops over multiple kilobases.
Figure 6.
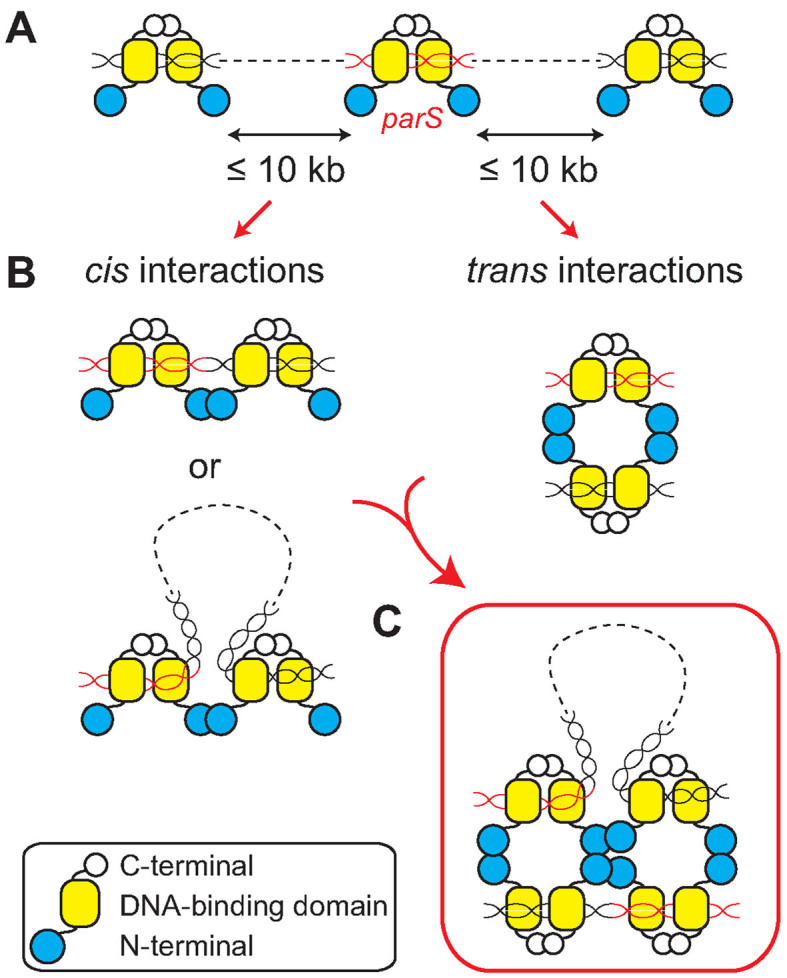
Model of ParB spreading (see colors online). (A) ParB proteins dimerized through their C-terminal domains bind specifically to parS (red) and non-specifically to DNA that can be up to tens of kilobases away from parS. (B) ParB dimers bound to DNA can interact with each other through their N-terminal domains both in cis and in trans. Interactions in cis may occur between ParB dimers that are bound directly adjacent to each other on DNA or far apart but brought into close proximity through DNA looping. (C) ParB dimers nucleated at parS oligomerize into a tetrameric structure through interactions both in cis and in trans. A higher-order partition complex is assembled through DNA bridging that connects distant DNA loci and traps large DNA loops over multiple kilobases.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Xindan Wang and David Rudner for support on strain construction and cellular imaging. We acknowledge the Nikon Imaging Center at Harvard Medical School for providing the epifluorescence microscope. We thank Kelly Arnett and the Center for Macromolecular Interactions at Harvard Medical School for experimental support on differential scanning fluorimetry and size exclusion chromatography with multi-light angle scattering. Sequencing reactions were carried out with an ABI3730xl DNA analyzer at the DNA Resource Core of Dana-Farber/Harvard Cancer Center.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation CAREER Award [MCB-1148818 to J.J.L.]; Molecular Biophysics Training Grant, Harvard University, National Institutes of Health [NIGMS 5T32 GM008313 to D.S.]; Harvard-Amgen Scholars Program and BCMP Summer Scholars Program at Harvard Medical School [to K.R.]; National Science Foundation Graduate Research Fellowship Program (to T.G.W.G). Funding for open access charge: National Science Foundation [MCB-1148818 to J.J.L.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Badrinarayanan A., Le T.B., Laub M.T.. Bacterial chromosome organization and segregation. Annu. Rev. Cell Dev. Biol. 2015; 31:171–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baxter J.C., Funnell B.E.. Plasmid partition mechanisms. Microbiol. Spectr. 2014; 2:6. [DOI] [PubMed] [Google Scholar]
- 3. Schumacher M.A. Structural biology of plasmid partition: uncovering the molecular mechanisms of DNA segregation. Biochem. J. 2008; 412:1–18. [DOI] [PubMed] [Google Scholar]
- 4. Bartosik A.A., Lasocki K., Mierzejewska J., Thomas C.M., Jagura-Burdzy G.. ParB of Pseudomonas aeruginosa: interactions with its partner ParA and its target parS and specific effects on bacterial growth. J. Bacteriol. 2004; 186:6983–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fogel M.A., Waldor M.K.. A dynamic, mitotic-like mechanism for bacterial chromosome segregation. Genes Dev. 2006; 20:3269–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee M.J., Liu C.H., Wang S.Y., Huang C.T., Huang H.. Characterization of the Soj/Spo0J chromosome segregation proteins and identification of putative parS sequences in Helicobacter pylori. Biochem. Biophys. Res. Commun. 2006; 342:744–750. [DOI] [PubMed] [Google Scholar]
- 7. Lin D.C., Grossman A.D.. Identification and characterization of a bacterial chromosome partitioning site. Cell. 1998; 92:675–685. [DOI] [PubMed] [Google Scholar]
- 8. Livny J., Yamaichi Y., Waldor M.K.. Distribution of centromere-like parS sites in bacteria: insights from comparative genomics. J. Bacteriol. 2007; 189:8693–8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Minnen A., Attaiech L., Thon M., Gruber S., Veening J.W.. SMC is recruited to oriC by ParB and promotes chromosome segregation in Streptococcus pneumoniae. Mol. Microbiol. 2011; 81:676–688. [DOI] [PubMed] [Google Scholar]
- 10. Mohl D.A., Gober J.W.. Cell cycle-dependent polar localization of chromosome partitioning proteins in Caulobacter crescentus. Cell. 1997; 88:675–684. [DOI] [PubMed] [Google Scholar]
- 11. Nardmann J., Messer W.. Identification and characterization of the dnaA upstream region of Thermus thermophilus. Gene. 2000; 261:299–303. [DOI] [PubMed] [Google Scholar]
- 12. Gruber S., Errington J.. Recruitment of condensin to replication origin regions by ParB/SpoOJ promotes chromosome segregation in B. subtilis. Cell. 2009; 137:685–696. [DOI] [PubMed] [Google Scholar]
- 13. Sullivan N.L., Marquis K.A., Rudner D.Z.. Recruitment of SMC by ParB-parS organizes the origin region and promotes efficient chromosome segregation. Cell. 2009; 137:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lim H.C., Surovtsev I.V., Beltran B.G., Huang F., Bewersdorf J., Jacobs-Wagner C.. Evidence for a DNA-relay mechanism in ParABS-mediated chromosome segregation. Elife. 2014; 3:e02758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ptacin J.L., Lee S.F., Garner E.C., Toro E., Eckart M., Comolli L.R., Moerner W.E., Shapiro L.. A spindle-like apparatus guides bacterial chromosome segregation. Nat. Cell Biol. 2010; 12:791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shebelut C.W., Guberman J.M., van Teeffelen S., Yakhnina A.A., Gitai Z.. Caulobacter chromosome segregation is an ordered multistep process. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:14194–14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Montero Llopis P., Rudner D.Z.. Bacillus subtilis chromosome organization oscillates between two distinct patterns. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:12877–12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oliva M.A. Segrosome complex formation during DNA trafficking in bacterial cell division. Front. Mol. Biosci. 2016; 3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Breier A.M., Grossman A.D.. Whole-genome analysis of the chromosome partitioning and sporulation protein Spo0J (ParB) reveals spreading and origin-distal sites on the Bacillus subtilis chromosome. Mol. Microbiol. 2007; 64:703–718. [DOI] [PubMed] [Google Scholar]
- 20. Kusiak M., Gapczynska A., Plochocka D., Thomas C.M., Jagura-Burdzy G.. Binding and spreading of ParB on DNA determine its biological function in Pseudomonas aeruginosa. J. Bacteriol. 2011; 193:3342–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murray H., Ferreira H., Errington J.. The bacterial chromosome segregation protein Spo0J spreads along DNA from parS nucleation sites. Mol. Microbiol. 2006; 61:1352–1361. [DOI] [PubMed] [Google Scholar]
- 22. Bingle L.E., Macartney D.P., Fantozzi A., Manzoor S.E., Thomas C.M.. Flexibility in repression and cooperativity by KorB of broad host range IncP-1 plasmid RK2. J. Mol. Biol. 2005; 349:302–316. [DOI] [PubMed] [Google Scholar]
- 23. Lynch A.S., Wang J.C.. SopB protein-mediated silencing of genes linked to the sopC locus of Escherichia coli F plasmid. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:1896–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rodionov O., Lobocka M., Yarmolinsky M.. Silencing of genes flanking the P1 plasmid centromere. Science. 1999; 283:546–549. [DOI] [PubMed] [Google Scholar]
- 25. Rodionov O., Yarmolinsky M.. Plasmid partitioning and the spreading of P1 partition protein ParB. Mol. Microbiol. 2004; 52:1215–1223. [DOI] [PubMed] [Google Scholar]
- 26. Graham T.G., Wang X., Song D., Etson C.M., van Oijen A.M., Rudner D.Z., Loparo J.J.. ParB spreading requires DNA bridging. Genes Dev. 2014; 28:1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanchez A., Cattoni D.I., Walter J.C., Rech J., Parmeggiani A., Nollmann M., Bouet J.Y.. Stochastic self-assembly of ParB proteins builds the bacterial DNA segregation apparatus. Cell Syst. 2015; 1:163–173. [DOI] [PubMed] [Google Scholar]
- 28. Taylor J.A., Pastrana C.L., Butterer A., Pernstich C., Gwynn E.J., Sobott F., Moreno-Herrero F., Dillingham M.S.. Specific and non-specific interactions of ParB with DNA: implications for chromosome segregation. Nucleic Acids Res. 2015; 43:719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broedersz C.P., Wang X., Meir Y., Loparo J.J., Rudner D.Z., Wingreen N.S.. Condensation and localization of the partitioning protein ParB on the bacterial chromosome. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:8809–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen B.W., Lin M.H., Chu C.H., Hsu C.E., Sun Y.J.. Insights into ParB spreading from the complex structure of Spo0J and parS. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:6613–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonard T.A., Butler P.J., Lowe J.. Structural analysis of the chromosome segregation protein Spo0J from Thermus thermophilus. Mol. Microbiol. 2004; 53:419–432. [DOI] [PubMed] [Google Scholar]
- 32. Yamaichi Y., Niki H.. Active segregation by the Bacillus subtilis partitioning system in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:14656–14661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Youngman P.J., Perkins J.B., Losick R.. Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proc. Natl. Acad. Sci. U.S.A. 1983; 80:2305–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harwood C.R., Cutting S.M.. Molecular Biological Methods for Bacillus. 1990; Chichester; NY: Wiley. [Google Scholar]
- 35. Song D., Graham T.G., Loparo J.J.. A general approach to visualize protein binding and DNA conformation without protein labelling. Nat. Commun. 2016; 7:10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X., Le T.B., Lajoie B.R., Dekker J., Laub M.T., Rudner D.Z.. Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev. 2015; 29:1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Autret S., Nair R., Errington J.. Genetic analysis of the chromosome segregation protein Spo0J of Bacillus subtilis: evidence for separate domains involved in DNA binding and interactions with Soj protein. Mol. Microbiol. 2001; 41:743–755. [DOI] [PubMed] [Google Scholar]
- 38. Funnell B.E. ParB Partition Proteins: complex formation and spreading at bacterial and plasmid centromeres. Front. Mol. Biosci. 2016; 3:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adams D.W., Wu L.J., Errington J.. Nucleoid occlusion protein Noc recruits DNA to the bacterial cell membrane. EMBO J. 2015; 34:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilhelm L., Burmann F., Minnen A., Shin H.C., Toseland C.P., Oh B.H., Gruber S.. SMC condensin entraps chromosomal DNA by an ATP hydrolysis dependent loading mechanism in Bacillus subtilis. Elife. 2015; 4:e06659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.