

Abstract
Dexmedetomidine is an α2-adrenergic receptor agonist that exhibits a protective effect on ischemia-reperfusion injury of the heart, kidney, and other organs. In the present study, we examined the neuroprotective action and potential mechanisms of dexmedetomidine against ischemia-reperfusion induced cerebral injury. Transient focal cerebral ischemia-reperfusion injury was induced in Sprague-Dawley rats by middle cerebral artery occlusion. After the ischemic insult, animals then received intravenous dexmedetomidine of 1 μg/kg load dose, followed by 0.05 μg/kg/min infusion for 2 h. After 24 h of reperfusion, neurological function, brain edema, and the morphology of the hippocampal CA1 region were evaluated. The levels and mRNA expressions of interleukin-1β, interleukin-6 and tumor nevrosis factor-α as well as the protein expression of inducible nitric oxide synthase, cyclooxygenase-2, nuclear factor-κBp65, inhibitor of κBα and phosphorylated of κBα in hippocampus were assessed. We found that dexmedetomidine reduced focal cerebral ischemia-reperfusion injury in rats by inhibiting the expression and release of inflammatory cytokines and mediators. Inhibition of the nuclear factor-κB pathway may be a mechanism underlying the neuroprotective action of dexmedetomidine against focal cerebral I/R injury.
Keywords: Cerebral ischemia-reperfusion, Dexmedetomidine, Inflammation, Inducible nitric oxide synthase, Cyclooxygenase-2, Nuclear factor-κB
INTRODUCTION
Transient ischemic hypoperfusion can cause severe acute brain injury, especially in the hippocampus, which is highly susceptible to ischemic hypoxia. During the process of reperfusion, induction of a series of complex mechanisms was also reported to initiate neuronal apoptosis and cause delayed neurological damage (Lee et al., 2000). Clinically, acute cerebral ischemia-reperfusion (I/R) injury can occur during the perioperative period for nerve surgery and operations on the heart or great vessels, particularly in elderly or critically ill patients. Therefore, there is strong interest in reducing or preventing perioperative I/R-induced brain damage worldwide.
The mechanisms underlying the pathogenesis of ischemic brain injury are complex, although local tissue inflammation plays a key role (Basu et al., 2005; Fujita et al., 2010). The tissue damage after cerebral ischemia and a large number of oxygen free radicals generated during reperfusion would induce to generate vast cytokines, such as interleukin, tumor necrosis factor, and so on. In turn, these cytokines stimulate expression of adhesion molecules in cerebrovascular endothelial cells, attracting circulating neutrophils that can migrate and adhere to the central ischemic tissues, thus propagating the local inflammatory response. Increased levels of arachidonic acid metabolites and other inflammatory mediators can further aggravate local brain inflammation, eventually leading to neuronal apoptosis and necrosis. Inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) are two main mediators of this inflammatory cascade (Dimagl et al., 1999; Culmsee and Krieqlstein, 2007).
As a member of the transcription factor protein family, nuclear factor (NF)-κB plays a key regulatory role in inflammation and immune responses. Indeed, production of all inflammatory molecules is modulated by the NF-κB signaling pathway, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1β, iNOS, and COX-2 (Berti et al., 2002; Hseu et al., 2005). The NF-κB pathway can be activated or regulated through the nuclear translocation of the p65 subunit and the mutual inhibition between inhibitor of kappa B (IκB) proteins. In most inactive cells, NF-κB compounds containing IκBa constantly shuttle between the cytoplasm and the nucleus, while those containing IκBb and IκBe are located mainly in the cytoplasm. Upon stimulation, IκB is phosphorylated by IκB kinase (IκK), which is ubiquitinated and subsequently degraded, leading to translocation of NF-κBp65 to the nucleus, binding to specific target genes, and increased expression of pro-inflammatory factors (Baldwin, 1996).
Many anesthetics applied during the perioperative period have been shown to exhibit neuroprotective properties against cerebral ischemia, including sevoflurane, lidocaine, and diprivan (Cui et al., 2013; Zhao et al., 2014). As a new type of anesthetic adjuvant drug, dexmedetomidine is a potent and highly selective α2 adrenoreceptor agonist, which can reduce the central sympathetic tension and possess the pharmacological features of sedation, analgesia, diuresis, anti-anxiety without respiratory depression (Kamibayashi and Maze, 2000). Although several studies have reported a neuroprotective action of dexmedetomidine in various animal models of cerebral injury, there are contrasting findings regarding its efficacy (Hoffman et al., 1991; Kuhmonen et al., 2001; Laudenbach et al., 2002).
Thus, the aim of the present study was to investigate the neuroprotective action and potential molecular mechanisms of dexmedetomidine following transient focal cerebral I/R.
MATERIALS AND METHODS
Animals
Male Sprague Dawley rats (16–18 weeks old, 300–360 g body weight) were purchased from Experimental Animal Center of Shandong University (Jinan, China) and were housed with free access to food and water at a constant temperature of 22 ± 2°C, humidity of 55 ± 5%, and a 12 h light/dark cycle. The recommendations in the Guidefor the Care and Use of Laboratory Animals of the National Institutes of Health (Bethesda, MD, USA) was strictly performed during this study. All experimental protocols were approved by the Ethics Committee of Animal Experiments of the Provincial hospital Affiliated to Shandong University.
Materials
Dexmedetomidine was purchased from Heng Rui Medicine Co., Ltd. (Jiangsu, China). The following primary antibodies (Santa Cruz Biotechnology Inc., CA, USA) were used for Western blot: anti-β-actin (sc-130656), anti-iNOS (sc-649), anti-COX-2 (sc-23983), anti-NF-κBp65 (sc-372), anti-IκBα (sc-371), and anti-p-IκBα (sc-101713). TNF-α (H052), IL-1β (H002), and IL-6 (H007) ELISA kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Primers were synthetized by Kangcheng Biotechnology (Guangzhou, China).
Establishment of model and groups
Forty-five rats were divided randomly into the sham operation (Sham), middle cerebral artery occlusion (MCAO), and MCAO rats with DEX administration groups (D+MCAO). Focal cerebral ischemia was induced by intraluminal filament (18.5 ± 0.5 mm internal diameter) occlusion of right middle artery for 60 min. While rats in Sham group only underwent surgical exposure of the right common carotid artery rather than occlusion. In D+MCAO group, intravenous infusion of DEX was performed with a load dosage of 1 ug/kg at once after ischemia insult and then 0.05 ug/kg/min for 2 h. After reperfusion for 24 h, all rats was immediately performed neurologic grades and elevated body swing test for assessment of neural function. Thereafter all rats were anesthetized with mebumal sodium (50 mg/kg) and thereafter decapitated. Brains were removed rapidly, five brains were for assessment of brain edema, five were for hematoxylin-eosin (HE) staining and immunohistochemical detection, five brains were carefully separated through cortex to remove hippocampus for western blot, real-time PCR and enzyme-linked immunosorbent assay.
Neurobehavioral grades
Neurobehavior was scored on a 5-point scale as previously reported (Longa et al., 1989); no neurological deficit=0, failure to extend left forepaw fully=1, circling to the left when walking= 2, falling to the left when walking=3, and failure to walk spontaneously=4.
Elevated body swing test
Each rat was hung by the tail approximately 5 cm from the ground, and the number of head deflections to the side (head deviation of approximately 10° from the center line) was recorded (Borlongan et al., 1998). This process was repeated 10 times for each rat. Animals with >75% of deflection to the left side were considered successful models.
Assessment of brain edema
The brains of five rats were rapidly removed and weighed (wet weight), and each brain was divided into six equal slices. The brain slices were then dried at 60°C for three days, and the dry weight determined. The degree of brain edema was calculated as: wet weight − dry weight / wet weight×100%.
Histological characterization of the hippocampal CA1 region
The brains of five rats per group were removed and dehydrated by gradient ethanol, cleared by xylene, embedded in paraffin, and then sectioned (5 μm thick) coronally backward from 2 mm before or after the optic chiasma and mounted onto glass slides. Four sections from each rat at the level of the hippocampus were stained for HE to assess neuron morphology in hippocampal CA1 region. Five paraffin sections from each rat were also used for immunohistochemical studies described below.
Immunohistochemistry for NF-κBp65
The NF-κBp65 rabbit polyclonal antibody and the immunohistochemical SP kit (Santa Cruz Biotechnology Inc.) were used to detect expression of NF-κBp65 in the hippocampal CA1 region. Acording to the manufacturer’s instructions, the brain sections were roasted, dewaxed and antigen repaired, then hydrated to dispose 3% H2O2 for 10 min at room temperature. Added NF-κBp65 antibody (1:200) to react for 2 h at 37°C and biotin labeled anti-rabbit antibody (1:200) to incubate at 37°C for 30 min, and incubate with horse reddish peroxidase labeled streptomycin avidin at 37°C for 2 h. Then dyeing via DAB/ H2O2 reaction. The NF-κBp65 positive cells appeared brown cytoplasm. Four views of each slide were observed randomly under a 400-fold microscope. The average of the integral optical density (IOD) of each view was detected by an Image-Pro plus 6.0 analytical systems. A negative control without primary antibody was included in the experiment to verify the antibody specificity.
Real-time PCR
Total RNA of the hippocampus was isolated using Trizol (Invitrogen, CA, USA) and reversed transcribed to cDNA using a reverse transcriptase kit (Takara, Dalian, China). Quantitative PCR was performed using the ABI 7500 system (Applied Biosystems, CA, USA) and a SYBR green kit (Takara). The PCR primers are described in Table 1.
Table 1.
Primers used in Real-time PCR
| Gene | Forward Primers (5′-3′) | Reverse Primers (5′-3′) |
|---|---|---|
| IL-1β | CACACTAGCAGGTCGTCATCATC | ATGAGAGCATCCAGCTTCAAATC |
| IL-6 | GCCCTTCAGGAACAGCTATGA | TGTCAACAACATCAGTCCCAAGA |
| TNFα | CAGAGCAATGACTCCAAAGTA | CAAGAGCCCTTGCCCTAA |
| β-actin | GTCAGGTCATCACTATCGGCAAT | AGAGGTCTTTACGGATGTCAACGT |
Levels of IL-1β, IL-6, and TNF-α in the hippocampus
Enzyme-linked immunosorbent assays were used to quantitate levels of IL-1β, IL-6, and TNF-α in the rat hippocampus. 10% hippocampal homogenate in saline solution was made using a glass homogenizer in an ice bath. The homogenate was centrifuged at 4°C and the supernatant collected and stored at −20°C until use. Levels of IL-1β, IL-6, and TNF-α in supernatants were measured with commercial ELISA kits (Abcam, Cambridge, England), according the manufacturer’s instructions. Breifly, cytokine beads were placed at the upper compartment of a 96-well transwell plate (3 μm pore size) while samples were placed at the bottom, incubate for 1 hour at room temperature, wash plate and stop the reaction. The concentrations of IL-1β, IL-6, and TNF-α were determined according to a standard curve.
Western blot
Briefly, the proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then electrophoretically transferred onto polyvinylidene fluoride membranes. Membranes were blocked with 5% skimmed milk for 1 h at room temperature, and then incubated overnight at 4°C with anti-iNOS, anti-COX2, anti-NF-κBp65, anti-IκBα, and anti-p-IκBα antibodies. Actin was used as a loading control. The membranes were then incubated with the corresponding secondary antibodies, and the reaction was visualized with chemiluminescence reagents provided with an ECL kit (Bioworld, MN, USA) and exposed to X-ray film. The intensity of the appropriate bands on the blots was quantified by densitometry.
Statistical analyses
Statistical analysis was performed using a standard software package (SAS 10.0 Software, SAS, NC, USA). All data are presented as means ± standard deviation. Two-way ANOVA followed by a Bonferroni multiple group comparison. Values of p<0.05 were considered statistically significant.
RESULTS
Effect of dexmedetomidine on neurological function and brain edema in MCAO rats
The neurobehavioral scores, left-deflection percent, and degree of brain edema in the MCAO group were significantly higher than those in the sham group (p<0.01). By contrast, neurological dysfunction of rats was alleviated in the D+MCAO group, with a significant decrease in the Longa’s scales, the left-deflection percent, and the degree of brain edema compared with the MCAO group (p<0.05; Table 2).
Table 2.
Neurological function and brain edema in rats of each group
| Parameter | Longa’s scales | Left-deflection percent (%) | Degree of brain edema (%) |
|---|---|---|---|
| Sham | 0 | 46 ± 8 | 54 ± 5 |
| MCAO | 2.75 ± 0.95* | 92 ± 19* | 74 ± 8* |
| D+MCAO | 2.05 ± 0.58*# | 80 ± 9%*# | 60 ± 4## |
p<0.01 vs. Sham group;
p<0.05,
p<0.01 vs. MCAO group.
Effect of dexmedetomidine on hippocampal morphology in MCAO rats
By HE staining, pyramidal cells and the neurons in the hippocampal CA1 region of sham group animals were arranged neatly, and the cellular structure was complete with a large and round nucleus and a clear nucleolus. By contrast, MCAO rats showed marked neuronal degeneration, with a triangular or irregular shape, concentrated cytoplasm and nucleus, and a damaged hippocampal structure. This pattern of injury was significantly alleviated in the D+MCAO group (Fig. 1).
Fig. 1.

Morphological changes of hippocampal CA1 by HE staining (×200). (A) Sham group: the cells arranged neatly, cellular structure was complete with a large and round nucleus and clear nucleolus (arrow). (B) MCAO group: nerve cells become triangular or irregular, with concentrated cytoplasm and nucleus (arrow). (C) D+MCAO group: the damages in the shape and arrangement of cells (arrow) were much better than MCAO group.
Effect of dexmedetomidine on hippocampal IL-1β, IL-6, and TNF-α levels and mRNA expression in MCAO rats
As shown in Table 3, for rats subjected to MCAO, the levels of IL-1β, IL-6 and TNF-α in hippocampus were all largely higher than those in Sham group (p< 0.01), while in DEX treatment group, the levels of IL-1β, IL-6 and TNF-α were obviously reduced compared to MCAO group (p<0.05). Meantime the expression of IL-1β, IL-6 and TNF-α mRNA were all obviously upregulated in MCAO group (p<0.01), while in DEX treatment rats, the mRNA levels of these cytokines significantly were lower than those in MCAO rats (p<0.05; Fig. 2).
Table 3.
levels of IL-1β, IL-6 and TNF-α in hippocampus of each group
| Parameter | IL-1β (ng/g) | IL-6 (ng/g) | TNF-α (ng/g) |
|---|---|---|---|
| Sham | 2.57 ± 0.34 | 1.59 ± 0.21 | 0.98 ± 0.13 |
| MCAO | 3.61 ± 0.27** | 2.47 ± 0.27** | 1.86 ± 0.15** |
| D+MCAO | 2.92 ± 0.23*## | 1.96 ± 0.19*# | 1.53 ± 0.11**# |
p<0.05,
p<0.01 vs. Sham group;
p<0.05,
p<0.01 vs. MCAO.
Fig. 2.

mRNA levels of IL-1β, IL-6 and TNFα in hippocampus of each group. The mRNA levels of IL-1β, IL-6 and TNFα were measured in each group by Real-time PCR. β-actin was used as an internal control. *p<0.05, **p<0.01 vs. Sham; #p<0.05, ##p<0.01 vs. MCAO.
Effect of dexmedetomidine on hippocampal iNOS and COX-2 protein expression in MCAO rats
By Western blot, the protein expression of iNOS and COX-2 was significantly increased by approximately 3.3 times and 2.5 times, respectively, in MCAO rats compared with the sham group (p<0.01). By contrast, iNOS and COX-2 protein expression in the D+MCAO group were markedly downregulated to approximately 57% and 62%, respectively, of those in the MCAO group (p<0.01; Fig. 3).
Fig. 3.

Effect of dexmedetomidine on hippocampal iNOS and COX-2 protein expression in MCAO rats. (A) Representative images of western blotting showing DEX inhibited the expression of iNOS and COX-2. (B) Quantitative analysis of iNOS expression. (C) Quantitative analysis of COX-2 expression. *p<0.05, **p<0.01 vs. Sham; #p<0.01, vs. MCAO.
Effect of dexmedetomidine on hippocampal NF-κB activity in MCAO rats
There was a significant increase in the number of NF-κBp65-positive cells in hippocampus of MCAO rats compared with the sham group (p<0.01). By contrast, the number of NFκBp65-positive cells was markedly decreased in the D+MCAO group (p<0.05; Fig. 4). The protein expression of p65 and P-IκBa were significantly increased, and expression of IκBa significantly decreased, in MCAO rats compared with the control group. By contrast, the expression of NF-κBp65 and P-IκBa was decreased (p<0.01), and expression of IκBa increased, in the hippocampus in the D+MCAO group (p<0.05; Fig. 5).
Fig. 4.

Effect of dexmedetomidine on hippocampal NF-κBp65 expression in MCAO rats. (A) Photomicrographs of NF-κBp65 immunoreactivity in hippocampal CA1 (200 amplification). There were slightly NF-κBp65-positive expression in Sham group (a), while in MCAO rats (b), abundant NF-κBp65-positive expression in cytoplasm and nucleus of hippocampal neurons with brown staining (arrow), whereas in D+MCAO group, positive expression of NF-κBp65 was decreased (c). (B) Quantification of NF-κBp65 IOD level. *p<0.05, **p<0.01 vs. Sham; #p<0.01 vs. MCAO.
Fig. 5.
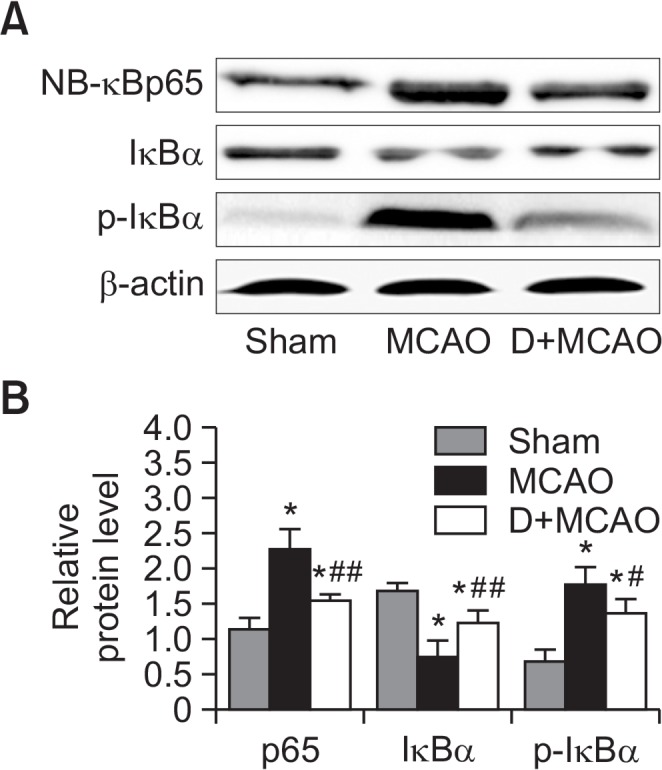
Effect of dexmedetomidine on hippocampal NF-κB activity in MCAO rats. (A) Representative image of western blotting of p65, p-IκBα and IκBa in hippocampus. (B) Quantitative analysis of Fig. 5A. *p<0.01 vs. Sham; #p<0.05, ##p<0.01 vs. MCAO.
DISCUSSION
The main finding of the present study was that dexmedetomidine treatment alleviated the neurological dysfunction, brain edema, and hippocampal morphological damage following I/R cerebral injury in rats. The unilateral MCAO is an excellent technique to establish a focal cerebral I/R injury model, as the operation is simple without craniotomy, and as the occlusion effect is stable and the ischemia/reperfusion time can be controlled accurately. In the present study, rats received right MCAO for 1 h and reperfusion for 24 h to induce focal cerebral I/R injury. Our model of transient focal cerebral ischemia resulted in acute nervous dysfunction in rats. Previous studies have confirmed that cerebral infarction reaches a maximum volume after transient cerebral ischemia and reperfusion for 24 h. Therefore, the majority of studies use 24 h of I/R as the time window to evaluate acute cerebral injury (Derugin et al., 2000). Morphological and functional damage of the hippocampus is the earliest and most common pathology induced by focal cerebral ischemia. Indeed, in the present study, we observed obvious pathological neuronal injury in the hippocampal CA1 region with HE staining at 24 h of focal cerebral I/R.
Inflammation plays an important role in the pathogenesis cerebral injury induced by I/R (Ritter et al., 2000). In the early stage of acute cerebral ischemia, necrotic cells and oxygen free radicals, as well as other related messenger molecules, can activate the NF-κB pathway to induce transcription and release of many inflammatory molecules, including the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α, which can aggravate endothelial cell injury in the cerebral vasculature (Yasuda et al., 2011). In the present study, focal cerebral I/R increased the levels and mRNA expression of IL-1β, IL-6, and TNFα in the hippocampus, suggesting that the inflammatory response was an important component of acute injury induced by cerebral I/R, consistent with previous reports (Ritter et al., 2000).
Dexmedetomidine was reported to protect the heart and kidney from I/R injury both in vitro and in vivo (Okada et al., 2007; Kocoglu et al., 2009). However, the mechanism underlying the protective effects of dexmedetomidine remain unclear. Dexmedetomidine treatment prior to an ischemic insult was reported to protect against I/R-induced intestinal injury via inhibition of the inflammatory response and suppression of capase-3 protein (Zhang et al., 2012). In the present study, dexmedetomidine alleviated hippocampal injury and dysfunction in rats exposed to cerebral I/R, which was associated with a significant decrease in mRNA and protein levels of pro-inflammatory factors. Thus, the neuroprotective action of dexmedetomidine may be related, at least in part, to inhibition of the inflammatory response.
The pro-inflammatory mediators iNOS and COX-2 are markedly elevated in the brain following cerebral ischemia, and contribute to the pathogenesis of brain damage (Iadecola et al., 1997; Koistinaho et al., 1999). Anti-inflammatory agents or inhibitors of iNOS and COX-2 were also reported to reduce ischemic brain injury and improve outcomes (Wang et al., 2008; Kim et al., 2009). Further, iNOS-deficient mice showed smaller infarcts than wild-type mice when exposed to cerebral ischemia (Ferriero et al., 1996). In the present study, iNOS and COX-2 expression in the hippocampus were markedly increased at 24 h of cerebral I/R, but were decreased by dexmedetomidine treatment. Thus, inhibition of iNOS and COX-2 expression may be a mechanism underlying the neuroprotective action of dexmedetomidine following cerebral I/R.
As an important nuclear transcription factor, NF-κB is widely expressed in neurons, glia, and vascular endothelial cells. NF-κB is located downstream of several signaling pathways, including toll-like receptors and the mitogen-activated protein kinases, and is responsible for the regulation of inflammation and immune responses (Guo et al., 2010; Qiao et al., 2012). The activity of NF-κB is mainly regulated by its interaction with the inhibitory IκB protein. Activation of the NF-κB requires the degradation of IκB protein and p65 translocation to the nucleus (Xie et al., 2011; Min et al., 2012). In resting state, IκBa-containing complexes constantly shuttle between the nucleus and the cytoplasm, whereas IκBb- and κBe-containing complexes are predominantly cytoplasmic. Upon stimulation, IκB is phosphorylated by IκB kinase, which is ubiquitinated and subsequent degraded, leading to rapid translocation of NF-κBp65 to the nucleus. IκB is then combined with a specific DNA sequence to activate downstream-associated factors, including pro-inflammatory molecules. Several studies have reported that NF-κB activation can induce an inflammatory response and neuronal apoptosis in the hippocampus following cerebral I/R injury (Lou et al., 2007; Boersma and Meffert, 2008). Further, IκB was suggested as a potential target to reduce cerebral I/R damage via suppression of NF-κB activation (Zhang et al., 2013; Jiang et al., 2014).
In the present study, expression of NF-κBp65 and p-IκBα were markedly elevated in the hippocampal CA1 region of cerebral I/R rats, indicating NF-κB activation. Interestingly, treatment with dexmedetomidine immediately after transient ischemia significantly alleviated this increase in hippocampal NF-κBp65 and Iκa phosphorylation, and reduced expression and release of pro-inflammatory cytokines. Further, dexmedetomidine treatment was associated with improved hippocampal cellular structure and neurobehavioral function of rats. Thus, these data suggest that the neuroprotective action of dexmedetomidine against cerebral I/R injury may involve inhibition of the NF-κB signaling pathway to alleviate the cerebral inflammatory response.
In conclusion, we demonstrated that treatment with dexmedetomidine immediately after cerebral ischemia alleviated brain edema and hippocampal neuron damage and improved function outcomes induced by focal cerebral I/R in rats. The neuroprotective actions of dexmedetomidine against cerebral I/R injury may involve inhibition of NF-κβ signaling and reduction of cerebral inflammation. Further studies are required to confirm the detailed mechanisms of dexmedetomidine against I/R cerebral injury.
Acknowledgments
The work was financially supported by Excellent Adult and Young Scientist Foundation of Shandong Province (BS2010YY056) and Science Technology Development Plan of Shandong Province (2014GSF118110).
REFERENCES
- Baldwin AS., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Basu A, Lazovic J, Krady JK, Mauger DT, Rothstein RP, Smith MB, Levison SW. Interleukin-1 and the interleukin-1 type 1 receptor are essential for the progressive neurodegeneration that ensues subsequent to a mild hypoxic/ischemic injury. J Cereb Blood Flow Metab. 2005;25:17–29. doi: 10.1038/sj.jcbfm.9600002. [DOI] [PubMed] [Google Scholar]
- Berti R, Williams AJ, Moffett JR, Hale SL, Velarde LC, Elliott PJ, Yao C, Dave JR, Tortella FC. Quantitative real-time RT-PCR analysis of inflammatory gene expression associated with ischemia-reperfusion brain injury. J Cereb Blood Flow Metab. 2002;22:1068–1079. doi: 10.1097/00004647-200209000-00004. [DOI] [PubMed] [Google Scholar]
- Boersma MC, Meffert MK. Novel roles for the NF-κB signaling pathway in regulating neuronal function. Sci Signal. 2008;1:pe7. doi: 10.1126/stke.16pe7. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Hida H, Nishino H. Early assessment of motor dysfunctions aids in successful occlusion of the middle cerebral artery. Neuroreport. 1998;9:3615–3621. doi: 10.1097/00001756-199811160-00012. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Krieqlstein J. Ischaemic brain damage after stroke: new insights into efficient therapeutic strategies. International Symposium on Neurodegeneration and Neuroprotection. EMBO Rep. 2007;8:129–133. doi: 10.1038/sj.embor.7400892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui DR, Wang L, Jiang W, Qi AH, Zhou QH, Zhang XL. Propofol prevents cerebral ischemia-triggered autophagy activation and cell death in the rat hippocampus through the NF-κB/p53 signaling pathway. Neuroscience. 2013;246:117–132. doi: 10.1016/j.neuroscience.2013.04.054. [DOI] [PubMed] [Google Scholar]
- Derugin N, Wendland M, Muramatsu K, Roberts TP, Gregory G, Ferriero DM, Vexler ZS. Evolution of brain injury after transient middle cerebral artery occlusion in neonatal rats. Stroke. 2000;31:1752–1761. doi: 10.1161/01.STR.31.7.1752. [DOI] [PubMed] [Google Scholar]
- Dimagl U, ladecola C, Moskowitz MA. Pathobiology of ischaemic stroke : an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/S0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Ferriero DM, Holtzman DM, Black SM, Sheldon RA. Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol Dis. 1996;3:64–71. doi: 10.1006/nbdi.1996.0006. [DOI] [PubMed] [Google Scholar]
- Fujita M, Tsuruta R, Kaneko T, Otsuka Y, Kutsuna S, Izumi T, Aoki T, Shitara M, Kasaoka S, Maruyama I, Yuasa M, Maekawa T. Hyperoxia suppresses excessive superoxide anion radical generation in blood, oxidative stress, early inflammation, and endothelial injury in forebrain ischemia/reperfusion rats: laboratory study. Shock. 2010;34:299–305. doi: 10.1097/SHK.0b013e3181ceeeec. [DOI] [PubMed] [Google Scholar]
- Guo Y, Xu X, Li Q, Li Z, Du F. Anti-inflammation effects of picroside 2 in cerebral ischemic injury rats. Behav Brain Funct. 2010;6:43. doi: 10.1186/1744-9081-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman WE, Kochs E, Werner C, Thomas C, Albrecht RF. Dexmedetomidine improves neurologic outcome from incomplete ischemia in the rat: Reversal by the α2-adrenergic antagonist atipamezole. Anesthesiology. 1991;75:328–332. doi: 10.1097/00000542-199108000-00022. [DOI] [PubMed] [Google Scholar]
- Hseu YC, Wu FY, Wu JJ, Chen JY, Chang WH, Lu FJ, Lai YC, Yang HL. Anti-inflammatory potential of Antrodia Camphorata through inhibition of iNOS, COX-2 and cytokines via the NF-κB pathway. Int Immunopharmacol. 2005;5:1914–1925. doi: 10.1016/j.intimp.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Xia YY, He JM, Guo ML, Li RP. Total bakkenolides protects neurons against cerebral ischemic injury through inhibition of nuclear factor-κB activation. CNS Neurol. Disord. Drug Targets. 2014;13:874–884. doi: 10.2174/18715273113129990104. [DOI] [PubMed] [Google Scholar]
- Kamibayashi T, Maze M. Clinical uses of α2-adrenergic agonists. Anesthesiology. 2000;93:1345–1349. doi: 10.1097/00000542-200011000-00030. [DOI] [PubMed] [Google Scholar]
- Kim DH, Kim S, Jung WY, Park SJ, Park DH, Kim JM, Cheong JH, Ryu JH. The neuroprotective effects of the seeds of Cassia obtusifolia on transient cerebral global ischemia in mice. Food Chem Toxicol. 2009;47:1473–1479. doi: 10.1016/j.fct.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Kocoglu H, Ozturk H, Ozturk H, Yilmaz F, Gulcu N. Effect of dexmedetomidine on ischemia-reperfusion injury in rat kidney: A histopathologic study. Ren Fail. 2009;31:70–74. doi: 10.1080/08860220802546487. [DOI] [PubMed] [Google Scholar]
- Koistinaho J, Koponen S, Chan PH. Expression of cyclooxygenase-2 mRNA after global ischemia is regulated by AMPA receptors and glucocorticoids. Stroke. 1999;30:1900–1905. doi: 10.1161/01.STR.30.9.1900. discussion 1905–1906. [DOI] [PubMed] [Google Scholar]
- Kuhmonen J, Haapalinna A, Sivenius J. Effects of dexmedetomidine after transient and permanent occlusion of the middle cerebral artery in the rat. J. Neural Transm. (Vienna) 2001;108:261–271. doi: 10.1007/s007020170071. [DOI] [PubMed] [Google Scholar]
- Laudenbach V, Mantz J, Lagercrantz H, Desmonts JM, Evrard P, Gressens P. Effects of α2-adrenoceptor agonists on perinatal excitotoxic brain injury: comparison of clonidine and dexmedetomidine. Anesthesiology. 2002;96:134–141. doi: 10.1097/00000542-200201000-00026. [DOI] [PubMed] [Google Scholar]
- Lee JM, Grabb MC, Zipfel GJ, Choi DW. Brain tissue responses to ischemia. J Clin Invest. 2000;106:723–731. doi: 10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.STR.20.1.84. [DOI] [PubMed] [Google Scholar]
- Lou HY, Wei XB, Zhang B, Sun X, Zhang XM. Hydroxyethylpuerarin attenuates focal cerebral ischemia-reperfusion injury in rats by decreasing TNF-α expression and NFκB activity. Yao Xue Xue Bao. 2007;42:710–715. [PubMed] [Google Scholar]
- Min KJ, Cho KH, Kwon TK. The effect of oxidized low density lipoprotein (oxLDL)-induced heme oxygenase-1 on LPS-induced inflammation in RAW 264.7 macrophage cells. Cell Signal. 2012;24:1215–1221. doi: 10.1016/j.cellsig.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Okada H, Kurita T, Mochizuki T, Morita K, Sato S. The cardioprotective effect of dexmedetomidine on global ischaemia in isolated rat hearts. Resuscitation. 2007;74:538–545. doi: 10.1016/j.resuscitation.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Qiao H, Zhang X, Zhu C, Dong L, Wang L, Zhang X, Xing Y, Wang C, Ji Y, Cao X. Luteolin downregulates TLR4, TLR5, NF-κB and p-p38MAPK expression, upregulates the p-ERK expression, and protects rat brains against focal ischemia. Brain Res. 2012;1448:71–81. doi: 10.1016/j.brainres.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Ritter LS, Orozco JA, Coull BM, McDonagh PF, Rosenblum WI. Leukocyte accumulation and hemodynamic changes in the cerebral microcirculation during early reperfusion after stroke. Stroke. 2000;31:1153–1161. doi: 10.1161/01.STR.31.5.1153. [DOI] [PubMed] [Google Scholar]
- Wang C, Liu JL, Sang HF, Lu Y, Dong HL, Xiong LZ. Therapeutic time window of flurbiprofen axetil’s neuroprotective effect in a rat model of transient focal cerebral ischemia. Chin Med J. 2008;121:2572–2577. [PubMed] [Google Scholar]
- Xie C, Kang J, Ferguson ME, Nagarajan S, Badger TM, Wu X. Blueberries reduce pro-inflammatory cytokine TNF-α and IL-6 production in mouse macrophages by inhibiting NF-κB activation and the MAPK pathway. Mol Nutr Food Res. 2011;55:1587–1591. doi: 10.1002/mnfr.201100344. [DOI] [PubMed] [Google Scholar]
- Yasuda Y, Shimoda T, Uno K, Tateishi N, Furuya S, Tsuchihashi Y, Kawai Y, Naruse S, Fujita S. Temporal and sequential changes of glial cells and cytokine expression during neuronal degeneration after transient global ischemia in rats. J Neuroinflammation. 2011;8:70. doi: 10.1186/1742-2094-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhen YF, Ci-Ren Pu-Bu-, Song LG, Kong WN, Shao TM, Li X, Chai XQ. Salidroside attenuates beta amyloid-induced cognitive deficits via modulating oxidative stress and inflammatory mediators in rat hippocampus. Behav Brain Res. 2013;244:70–81. doi: 10.1016/j.bbr.2013.01.037. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Liu ZM, Wen SH, Li YS, Li Y, Yao X, Huang WQ, Liu KX. Dexmedetomidine administration before, but not after, ischemia attenuates intestinal injury induced by intestinal ischemia-reperfusion in rats. Anesthesiology. 2012;116:1035–1046. doi: 10.1097/ALN.0b013e3182503964. [DOI] [PubMed] [Google Scholar]
- Zhao P, Ji G, Xue H, Yu W, Zhao X, Ding M, Yang Y, Zuo Z. Isoflurane postconditioning improved long-term neurological outcome possibly via inhibiting the mitochondrial permeability transition pore in neonatal rats after brain hypoxia-ischemia. Neuroscience. 2014;280:193–203. doi: 10.1016/j.neuroscience.2014.09.006. [DOI] [PubMed] [Google Scholar]