

Abstract
In this study, the effect of particle size of genistein-loaded solid lipid particulate systems on drug dissolution behavior and oral bioavailability was investigated. Genistein-loaded solid lipid microparticles and nanoparticles were prepared with glyceryl palmitostearate. Except for the particle size, other properties of genistein-loaded solid lipid microparticles and nanoparticles such as particle composition and drug loading efficiency and amount were similarly controlled to mainly evaluate the effect of different particle sizes of the solid lipid particulate systems on drug dissolution behavior and oral bioavailability. The results showed that genistein-loaded solid lipid microparticles and nanoparticles exhibited a considerably increased drug dissolution rate compared to that of genistein bulk powder and suspension. The microparticles gradually released genistein as a function of time while the nanoparticles exhibited a biphasic drug release pattern, showing an initial burst drug release, followed by a sustained release. The oral bioavailability of genistein loaded in solid lipid microparticles and nanoparticles in rats was also significantly enhanced compared to that in bulk powders and the suspension. However, the bioavailability from the microparticles increased more than that from the nanoparticles mainly because the rapid drug dissolution rate and rapid absorption of genistein because of the large surface area of the genistein-solid lipid nanoparticles cleared the drug to a greater extent than the genistein-solid lipid microparticles did. Therefore, the findings of this study suggest that controlling the particle size of solid-lipid particulate systems at a micro-scale would be a promising strategy to increase the oral bioavailability of genistein.
Keywords: Genistein, Solid lipid particles, Particle size, Oral bioavailability, Dissolution behavior, Glyceryl palmitostearate
INTRODUCTION
Genistein, an isoflavone mainly found in species of the Leguminosae family (Fig. 1), has received considerable attention as a phytoestrogen that can prevent hormone-related cancers and bone loss (Zhang et al., 1999). Despite its attractive therapeutic effects, the low solubility and bioavailability of genistein have limited its potential as a medicine (Dixon and Ferreira, 2002). Diverse pharmaceutical formulations such as self-emulsifying drug delivery systems, mixed micelles, polymeric particulate systems, and liposomes (Takeuchi et al., 2003; Mathot et al., 2006; Munish et al., 2016; Pankaj et al., 2016) have been investigated as strategies to increase the solubility and bioavailability of poorly soluble drugs such as genistein. In particular, solid lipid particulate systems (SLPS) are very promising because they increase the solubility of poorly soluble drugs, improve drug stability by protecting them from enzymatic or chemical degradation, and release incorporated drugs at a controlled rate, thereby enhancing drug bioavailability (Luo et al., 2006; Muller et al., 2006). Furthermore, SLPS can be prepared with physiologically tolerated lipids, which decrease the possibility of undesirable toxicities that can occur with other synthetic materials. SLPS can also be manufactured simply on a large scale using the appropriate equipment.
Fig. 1.
Chemical structure of genistein.
Particle size is one of the physicochemical properties of SLPS that acts as a crucial factor significantly affecting the oral bioavailability of any incorporated drug (Luo et al., 2006; Desai and Thakkar, 2016). The particle size of SLPS determines their surface area and diffusion length for the incorporated drugs to be released from the lipid matrix, which are closely related to the drug dissolution rate from the particles (Muller et al., 2000; Horter and Dressman, 2001). However, the optimal particle size of SLPS can differ depending on the physicochemical properties of the drugs (Harde et al., 2011) and, therefore, optimizing the particle size of SLPS is critical for the successful enhancement of the bioavailability of incorporated drugs. Therefore, in this study, genistein-loaded solid lipid micro- and nanoparticles (genistein-SLMs and -SLNs, respectively) were prepared with glyceryl palmitostearate (GPS), and the effect of particle size on the drug release behavior and resulting oral bioavailability in rats was investigated. The same lipid material was used to prepare SLPS with different particle sizes to primarily explore the effect of particle size on the bioavailability of genistein.
MATERIALS AND METHODS
Materials
Genistein (>98% purity) was obtained from Lexgene Biotech (Seoul, Korea). GPS and vitamin E D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) were provided by Gattefosse (Nanterre, France) and Eastman Chemical Co. (Kingsport, TN, USA), respectively. L-α-Lecithin, Tween 80, and sulfatase (from Helix pomatia) were purchased from Sigma-Aldrich Corp (St. Louis, MO, USA). Poloxamer 188 was obtained from BASF (Ludwigshafen, Germany). High-performance liquid chromatography (HPLC)-grade acetonitrile and methanol (MeOH) were purchased from JT Baker (Phillipsburg, NJ, USA). Ethyl acetate, sodium carboxymethylcellulose (Na-CMC), dimethyl sulfoxide (DMSO), formic acid, and other reagents were purchased from Junsei Chemical Co., (Tokyo, Japan). Male Sprague-Dawley rats (250–270 g, 7-week-old) were purchased from Hanlim Experimental Animals Co., Ltd (Hwasung, Korea). Distilled and deionized water was used for preparing all solutions. All other chemicals used were of analytical grade.
Methods
Preparation of genistein-SLMs and -SLNs: Genistein-SLMs were prepared using a melt dispersion technique (Reithmeier et al., 2001). Briefly, 1 g of GPS was melted at 80°C, followed by the addition of 500 mg genistein. The lipid phase was dispersed using a homogenizer for 2 min at 8,000 rpm in 10 mL distilled water containing Tween 80 (3%, w/w) at 60°C to prepare an oil-in-water (O/W) emulsion. The warm O/W emulsion was added to cold distilled water of which temperature was adjusted to 3°C with mechanical stirring at 500 rpm to produce the genistein-SLMs, which were then freeze-dried for 48 h.
Genistein-SLNs were prepared using a hot homogenization method (Mehnert and Mader, 2001). The same amount of genistein and GPS used to prepare the SLMs was melted in a water bath maintained at 90°C. The surfactants for the lipid phase, L-α-lecithin and vitamin E TPGS, were also dissolved to concentrations of 5% (w/w) and 0.05% (w/v), respectively in the melted mixture. Then, 10 mL distilled water containing Poloxamer 188 (2%, w/w), as a surfactant, was heated under the same conditions. The organic phase was subsequently added to the aqueous phase, and the mixture was sonicated using a probe-type sonicator. The resulting emulsion was processed using a high-pressure homogenizer (Nano Debee, BEE International Inc., MA, USA) at 15,000 psi and 10–20 cycles to produce genistein-SLNs, which were cooled at the ambient temperature and stored in a refrigerator at 4°C until used.
To prepare a suspension of genistein, 300 mg of genistein powder was sieved to a particle size range of 150–200 μm and added to 10 mL distilled water containing 0.2% (w/v) Na-CMC, a suspending agent, followed by vigorous vortexing.
Characterization of colloidal properties of genistein-SLMs and -SLNs: The size distribution of the genistein-SLMs was evaluated by analyzing images captured using an optical microscope (Model DM-LS, Leica Microsystems, Wetzlar, Germany). For the genistein-SLNs, the mean diameter and polydispersity index were determined using a zetasizer Nano ZS (Malvern Instrument, Worcestershire, UK) at a fixed angle of 173° and 25°C. All samples were measured in triplicate, and each run was performed at least 10 consecutive times. In addition, samples were equilibrated for 30 s before evaluation, and the equipment was optimized prior to every measurement. The zeta potential of the SLMs and SLNs was also determined using the same instrument and the measurements were performed in triplicate.
Evaluation of drug loading amount (LA) and efficiency (LE) in SLMs and SLNs: To evaluate the drug loading amount (LA) and efficiency (LE) in the SLPS, genistein-SLMs and -SLNs were dispersed in MeOH, and placed in a water bath at 80°C for 30 min to further disrupt the lipid matrix. After disruption, both formulations were cooled at −20°C for 30 min. The cold dispersion was then centrifuged at 3500 rpm for 10 min to precipitate the lipids. The supernatant was filtered through a 0.45 μm syringe filter, and drug levels in the filtrate were analyzed using an HPLC system (Thermo Finnigan LLC, CA, USA) under the analytical condition described in detail in the proceeding section. The following equations were used to calculate the drug LA and LE.
In vitro drug release study: The in vitro drug dissolution study was performed in accordance with the United States Pharmacopeia (USP) paddle method using a dialysis bag (molecular weight cut-off [MWCO] 12000–14000) containing genistein-SLMs or -SLNs dispersed in pH 1.2 or pH 6.8 buffer (5 mL). The dialysis bag was submerged in a bottle of dissolution tester 3 h prior to the measurements to ensure equilibrium. The dialysis bag was then immersed in a bottle of dissolution tester containing the same buffer solution (900 mL) at 37 ± 0.5°C. At predetermined times, samples were withdrawn, and the drug levels were analyzed using the HPLC system.
To determine the overall mechanism of drug release from the genistein-SLMs and -SLNs, the drug release data were analyzed according to zero-order, first-order, and Higuchi models using following equations.
In vivo bioavailability study: In vivo animal studies were conducted to evaluate the pharmacokinetic properties of genistein suspension, -SLMs and -SLNs. The animal experiments were performed in accordance with the guideline of the Institutional Animal Care and Use Committee (IACUC) at Chung-Ang University, and the protocol was reviewed and approved by the IACUC (approval number: 2017-00002). Male Sprague-Dawley rats (250–270 g, 7-week-old) were quarantined in the animal care room where the temperature and relative humidity were maintained at 21–25°C and 50–60%, respectively, for 1 week before the experiments. The rats were fasted overnight with free access to water. Genistein suspension, -SLMs and -SLNs were administered to the rats at doses equivalent to 4 mg/kg genistein by oral gavage. The rats were then anesthetized with diethyl ether, followed by cannulation of their femoral artery with a polyethylene tube (i.d. 0.5 mm, o.d. 0.58 mm) for blood sampling. For intravenous administration, the same amount of genistein used in the oral administration was solubilized in 50% DMSO and injected via cannulas into the femoral artery of the rats. Blood samples (300 μL) were collected from the rats at predetermined time points (0.083, 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, 12, and 24 h) and centrifuged at 12,000 rpm for 10 min. The plasma samples obtained were then stored at −20°C until analyzed using the HPLC system. The details of the experimental procedure for analyzing the plasma genistein content are provided in the following section.
Preparation of plasma samples: Sulfatase from H. pomatia, which exhibits glucuronidase and sulfatase activities on genistein conjugates, was used to determine the total genistein concentration in plasma samples (Shelnutt et al., 2002). Sulfatase (500 U) was added to the plasma samples (50 μL), which were incubated in a shaking incubator (Jeio Tech Co., Ltd., Seoul, Korea) at 37°C for 5 h. 4-Hydroxybenzophenone and ethyl acetate used as the internal standard and extraction solvent, respectively (1 mL) were then added to the samples, followed by vigorous vortexing for extraction. The samples were centrifuged at 3000 rpm for 15 min. The organic phase was transferred to a glass tube and evaporated under a gentle stream of nitrogen at 40°C. The residue was reconstituted with 70% MeOH (200 μL) for the HPLC analysis.
HPLC analysis of genistein: The quantitative measurement of genistein was carried out using an HPLC system equipped with an L2130 HPLC pump, L2200 autosampler, and L2400 ultraviolet (UV)-visible detector (Hitachi, Ltd., Japan). For drug analysis in the in vitro studies, a Capcell Pak C18 UG 120 column (4.6 mm×150 mm, 5 μm, Shiseido, Japan) and a mobile phase consisting of acetonitrile and 20 mM phosphate buffer (70:30, v/v, pH 4.5) were used. The mobile phase was run at a flow rate of 1.0 mL/min, and the column was maintained at 35°C. A 0.45 μm syringe filter was used to filter samples, the injection volume was 50 μL, and the UV detection of genistein was performed at 227 nm.
For the In vivo study, genistein and 4-hydroxybenzophenone were separated using the same column described above. The mobile phase was run at a gradient mode. Two different mobile phases were used for each mobile phase line (A and B). For A line, the mobile phase was composed of acetonitrile and 50 mM ammonium formate buffer solution (pH 3.5) at 6:4 (v/v) while for the B line, the mobile phase consisted of acetonitrile and 50 mM ammonium formate buffer solution (pH 3.5) at 2:8 (v/v). The gradient mode was set as follows: From 0 to 4 min, the mobile phase for the A line changed from 65% to 51%, B line 35% to 49%; from 4 to 8 min, A line 51% to 65%, B line 49% to 35%; and from 8 to 10 min, A line 65%, B line 35%. The flow rate was 1.5 mL/min, and the column oven temperature was adjusted to 40°C. The eluent was monitored at an absorption wavelength of 268 nm.
Pharmacokinetic analysis: The plasma pharmacokinetic parameters of genistein suspension, -SLMs and -SLNs were calculated using the BA Calc 2007, a pharmacokinetic analysis software (ver. 1.0.0, KFDA, Korea). The maximum plasma drug concentration (Cmax) and time to reach Cmax (Tmax) were directly obtained from the plasma concentration versus time profiles. The area under the concentration-time curve from 0 h to infinity (AUC0–∞), elimination half-life (t1/2), and elimination rate constant (Ke) were obtained. The percentage absolute bioavailability (AB%) of genistein was calculated using the following equation.
Where, i.v.=intravenous.
Statistical analysis: All the data are presented as the mean ± standard deviation (SD). The pharmacokinetic parameters were statistically analyzed using a one-way analysis of variance (ANOVA) and the Student’s t-test. p-values <0.05 were considered to be significantly different.
RESULTS
Particle characteristics, drug LA, and LE of genistein-SLMs and SLNs
The particle size distribution of the the genistein-SLNs was varied by changing the homogenization cycle and increasing the cycle decreased the particle size, although the particle size ranges (0–100, 100–200, and 200–400 nm) were not significantly different among the various cycles (Fig. 2). The average particle size range of 100–200 nm was not considerably changed after 15 cycles and, therefore, we homogenized the SLNs for 15 cycles in preparing the SLNs for further studies.
Fig. 2.

Effect of cycles of homogenization on particle size distribution of genistein-loaded solid lipid nanoparticles.
The mean particle size of the genistein-SLMs and -SLNs was 6.010 ± 0.975 μm and 120.6 ± 16.91 nm, respectively while their zeta potential values were similar and not significantly different (range, −25 to −30 mV, Fig. 3). The LE values of the SLMs and SLNs were 71.15 ± 3.00% and 61.20 ± 5.51%, respectively, and the LA of the SLMs and SLNs was 30.41 ± 4.79% and 19.52 ± 5.27%, respectively (Fig. 4). The LE and LA of the SLMs and SLNs were significantly different (p<0.05).
Fig. 3.

Zeta potential value and mean particle size (A) and distribution of zeta potential (B) measured from genistein-loaded solid lipid microparticles (SLMs) and -solid lipid nanoparticles (SLNs) (n=3).
Fig. 4.
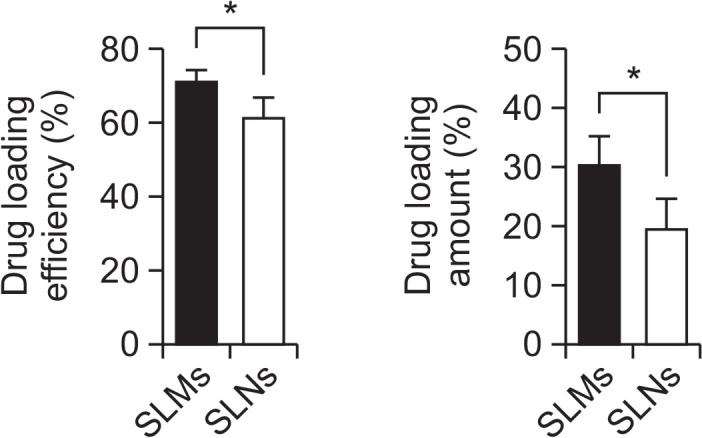
Drug loading efficiency and drug loading amount of genistein-loaded solid lipid microparticles (SLMs) and –solid lipid nanoparticles (SLNs). Asterisk (*) indicates p-value less than 0.05.
In vitro drug release study
Fig. 5 shows the results of the in vitro drug release study performed with the genistein bulk powders, suspensions, -SLMs, and -SLNs. Similar drug release patterns with the same rank order of rate (bulk powder<suspension<SLMs<SLNs) were observed in the pH 1.2 and pH 6.8 release media. Among the samples, the genistein bulk powders showed the slowest drug release rate while the suspension displayed an initial burst release, followed by a sustained release behavior. The genistein-SLMs gradually released the loaded genistein as a function of time. Although the drug release rate from the SLMs was slower than that of the genistein suspension until 4 h, it became faster than that of the suspension after 6 h.
Fig. 5.

In vitro release profile of genistein obtained from genistein bulk powder, genistein suspension, genistein-loaded solid lipid microparticles (SLMs) and -solid lipid nanoparticles (SLNs) at (A) pH 1.2 and (B) pH 6.8.
The SLNs exhibited a biphasic drug release pattern and had the fastest drug dissolution rate during the entire experimental period. An initial burst release was observed within 1 hour after the in vitro drug release study commenced and a prolonged release pattern followed. The overall release behavior of genistein from the SLMs and SLNs was more fitted to the first-order and Higuchi models, respectively, than to the other models and exhibited correlation coefficient (R2) values of 0.8740 and 0.8784, respectively (Fig. 6).
Fig. 6.

Drug release kinetics of genistein-loaded solid lipid microparticles (SLMs) and –solid lipid nanoparticles (SLNs) analysed by zero-order, first-order, and Higuchi models. R2 indicates correlation coefficient.
In vivo pharmacokinetic study
An In vivo pharmacokinetic study of genistein-SLMs and -SLNs was performed in comparison with the genistein suspension using rats to evaluate the effect of SLPS particle size on the pharmacokinetic behavior of genistein. The plasma drug concentration of the rat plasma samples was monitored using HPLC system. The assay exhibited a good linearity ranging from 100 to 4000 ng/mL with a standard regression equation of peak area (y) to drug concentration (x) of y=0.4469x+ 0.0356 (r2=0.9998).
The plasma drug concentration-time curves and the pharmacokinetic parameters obtained are shown in Fig. 7 and 8, respectively. At all the time points examined, the drug plasma concentrations measured from the genistein-SLMs and -SLNs were considerably higher than that of the genistein suspension. The Cmax values of genistein-SLMs and -SLNs were 1.20 and 1.55 μg/mL, respectively, which were higher than that of the genistein suspension (1.12 μg/mL). However, only the SLNs exhibited a statistically different Cmax value compared to that of the genistein suspension (p<0.05). The difference in the Cmax values of the SLMs and SLNs were statistically significant (p<0.05). In addition, the drug plasma concentrations of the SLMs and SLNs were more maintained than that of the suspension during the entire test period (Fig. 7). The small peaks observed at 4–6 h appeared to be likely due to the enterohepatic circulation, which has also been reported in previous studies (Yang et al., 2012).
Fig. 7.

Mean concentration-time profile of genistein in plasma after oral administration obtained with genistein suspension, and genistein-loaded solid lipid microparticles (SLMs) and -solid lipid nanoparticles (SLNs). Error bars represent the standard deviation of means (n=5).
Fig. 8.

Comparison of pharmacokinetic parameters among intravenous injection, genistein suspension, genistein-loaded solid lipid microparticles (SLMs) and -solid lipid nanoparticles (SLNs). Single asterisk (*) indicates p-value less than 0.05 and double asterisk (**) indicates p-value less than 0.01.
The Tmax of the genistein-SLMs and -SLNs was 15 and 5 min after the administration, respectively, which was significantly faster than that of the suspension (30 min, p<0.01). The t1/2 of the genistein incorporated in SLMs was 5.94 ± 0.92, which was higher than that in the drug suspension. In contrast, the t1/2 of the genistein incorporated in the SLNs was shorter than that in the drug suspension, and the difference in the Tmax values between the genistein-SLMs and -SLNs was statistically significant (p<0.01).
The t1/2 value of the SLMs was 5.94 ± 0.92 h, which increased compared to that of the genistein suspension (4.53 ± 1.40 h). However, the SLNs exhibited a decreased t1/2 at 3.61 ± 0.91 h. In comparison to the t1/2 of genistein suspension, statistical significance (p<0.05) was observed only from the SLMs. The t1/2 values of the SLMs and SLNs was also statistically different, exhibiting p value less than 0.01.
AUC0–∞ values of the genistein-SLMs and -SLNs were 7.49 ± 0.73 and 6.33 ± 0.43 μg·h−1·mL−1, respectively, which were 1.91- and 1.61-fold greater than that of the drug suspension (3.92 ± 1.53 μg·h−1·mL−1). The difference in AUC0–∞ between the SLPS (genistein-SLMs and -SLNs) and genistein suspension was statistically significant (p<0.01).
Furthermore, the AUC0–∞ of the SLMs was also significantly greater than that of the SLNs (p<0.01). When the AB% of genistein was calculated from the AUC0–∞ values obtained after oral and intravenous administration of the drug, the values of the suspension, -SLMs, and -SLNs were 38.58, 73.61, and 62.21%, respectively. The AB% of the SLMs and SLNs was significantly higher than that of the suspension was (p<0.01). Furthermore, the difference in AB% between the SLMs and SLNs was also statistically significant (p<0.05) and, therefore, the oral absorption of genistein appeared to be considerably enhanced by incorporating the drug into the SLPS.
DISCUSSION
Various pharmaceutical formulations such as polymeric micro-/nano-particles (Tang et al., 2011), β-cyclodextrin complexes (Lee et al., 2007), micellar systems (Kwon et al., 2007), and self-nanoemulsifying drug delivery systems (Tripathi et al., 2016) have been explored to improve dissolution behavior and oral bioavailability of genistein. Among them, SLPS have been suggested as a promising drug delivery system for enhancing the oral bioavailability of genistein because they can be fabricated to release the incorporated drugs in a controlled manner by varying the composition of the solid matrix, which is a critical determinant of oral bioavailability (Harde et al., 2011). In addition, SLPS can be prepared with the lipids and surfactants used in common oral dosage forms (Mehnert and Mader, 2001) at comparatively low amounts. SLPS can also be manufactured on a large scale using diverse instruments such as high-pressure homogenizers (Dingler and Gohla, 2002).
The diverse characteristics of SLPS including the physicochemical properties of the lipids and zeta potential of their surfaces have been known to affect drug dissolution behavior and the resulting oral bioavailability (Scalia et al., 2015). The particle size of the SLPS is considered one of the most crucial properties affecting the oral bioavailability of the incorporated drugs because it largely determines the surface area of the SLPS and diffusion length of the entrapped drugs to be released from the lipid matrix. In general, decreasing the particle size of the SLPS increases the drug dissolution rate and enhances the bioavailability. However, unexpected phenomena such as the precipitation of released drugs could occur depending on physicochemical properties of the drug incorporated in the SLPS, thereby changing the drug absorption behavior in the gastrointestinal tract and the resulting oral bioavailability (Muller et al., 2006). Thus, to maximally enhance the oral bioavailability of drugs loaded into SLPS, the particle size of the SLPS needs to be optimized depending on the individual drug. In the present study, SLPS with different particle sizes at a micro- and nano-scale were loaded with genistein, and resulting drug dissolution rate and oral bioavailability were evaluated.
As presented in Fig. 3, genistein-SLMs and SLNs exhibited good colloidal characteristics such as narrow particle size distribution and similar zeta potential values, and their LE and LA were also comparable, which was desirable for evaluating the effect of different particle sizes of the SLPS on the resulting dissolution rate and pharmacokinetic behavior of genistein (Fig. 4).
The in vitro drug release study was performed using genistein bulk powders, genistein suspension with a smaller particle size than that of the bulk powders, and genistein-SLMs and -SLNs. The drug release rate from the genistein bulk powders was the slowest, which could be attributed to their small surface area and the low aqueous solubility of the drug. The genistein suspension showed an initial burst release followed by a sustained release behavior. The behavior was largely due to the comparatively large surface area of the drug particles in the suspension, and the precipitation of genistein that occurred possibly because of the drug release rate was faster than that of the bulk powders.
The genistein-SLMs and -SLNs exhibited considerably higher drug release rates than those of the bulk powders and suspension. However, the drug release behavior of the SLMs was highly different from that of the SLNs. The SLMs displayed a gradual drug release pattern during the entire test period while that of the SLNs was biphasic and characterized by an initial burst release followed by a prolonged release. The relatively slow drug release rate of the SLMs at the early stage might be attributable to the smaller surface area and the longer diffusion length of the genistein from its lipid matrix than from that of the SLNs (Emami et al., 2015). The initial burst drug release exhibited by the genistein-SLNs might have been due to their large surface area and the short diffusion length of the incorporated drug into the release media (Uner and Yener, 2007).
The prolonged drug release observed following the initial burst release could be attributed to the precipitation of genistein likely caused by the rapid drug dissolution rate (Luo et al., 2006), similar to that of the genistein suspension. When the overall mechanism of drug release from the genistein-SLMs and -SLNs was examined, the first-order and Higuchi models appropriately interpreted the kinetics of drug release from the SLMs and SLNs, respectively (Fig. 6). The drug release from the SLMs showed an excellent fit to the Higuchi model, indicating that genistein was released from the SLMs by Fickian diffusion from the lipid matrices. The good correlation of the drug release from SLNs and first-order model implies that the drug was released from the SLNs in a drug concentration-dependent manner.
The increased drug dissolution rates of genistein from the SLMs and SLNs might increase the drug absorption and bioavailability compared to that of the genistein suspension. This result is in good agreement with that of other studies demonstrating that rapid drug dissolution increased the drug absorption and oral bioavailability (Li et al., 2009). However, the genistein-SLMs unexpectedly increased the oral bioavailability of genistein to a greater extent than the genistein-SLNs did although the SLNs exhibited a faster drug dissolution rate than the SLMs did. This result could be explained by the difference in the t1/2 of genistein in the SLMs and SLNs. The t1/2 of the genistein-SLMs and -SLNs significantly increased and decreased, respectively, compared to that of the genistein suspension. Therefore, the genistein absorbed from the SLMs and SLNs might have been eliminated from the rats at considerably different rates.
Prolonged drug release has been reported to induce slow drug clearance, thereby increasing oral bioavailability (Kumar and Pandit, 1997; Li et al., 2016). Therefore, the genistein loaded in the SLMs was gradually released from the particles and might have been slowly eliminated from the systemic circulation, thereby increasing the AUC and oral bioavailability (Boyd et al., 2007). In contrast, the rapid absorption of genistein from the SLNs might have enhanced its elimination more than that from the SLMs, thereby resulting in a lower bioavailability of the drug from the SLNs than from the SLMs. The close relationship between the particle size of the SLPS and the t1/2 and AB% of genistein was also clear when the parameter values were plotted (Fig. 9).
Fig. 9.
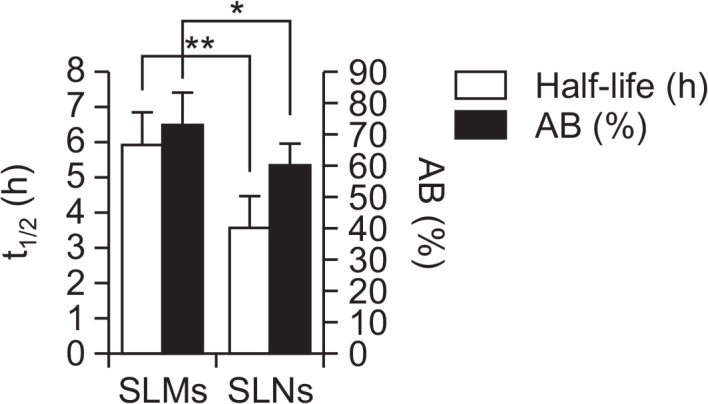
Relationship between elimination half-life and absolute bioavailability of genistein-loaded solid lipid microparticles (SLMs) and -solid lipid nanoparticles (SLNs). Single asterisk (*) indicates p-value less than 0.05 and double asterisk (**) indicates P value less than 0.01.
In addition, the prolonged drug release from the SLMs might have increased the bioavailability of genistein by avoiding the first pass effect. The rapidly released genistein from the SLNs could mainly be absorbed in the upper part of the intestine, pass through the mesenteric vessels, and subsequently undergo extensive first-pass metabolism in the liver of the rats. In contrast, the genistein-SLMs might have slowly disintegrated in the gastrointestinal tract and, thereby, some of them reached the colon. This phenomenon might increase the bioavailability of genistein because the drug absorbed in the colon avoids the first pass effect (Drover et al., 2002). Therefore, the SLMs were more efficient in enhancing the oral bioavailability of genistein than the SLNs were, although both formulations increased the bioavailability of genistein considerably more than the suspension did. However, it should also be considered that the optimal particle size of SLPS for enhancing the oral availability of poorly soluble drugs could change depending on the physicochemical properties of the drugs because of variable drug release rates from the lipid matrix and possible drug precipitation.
In conclusion, the changes in particle size of the SLPS considerably affected the drug release behavior and the resulting oral bioavailability. The large surface area of SLPS has been known to cause rapid drug dissolution rates and enhanced drug absorption. However, in this study, the SLMs with a smaller surface area enhanced the oral bioavailability of genistein more than the SLNs did mainly because the prolonged, gradual absorption of the genistein incorporated in the SLMs delayed the drug clearance. The findings of this study suggest that controlling the particle size of SLPS at a micro-scale could be a promising strategy for increasing the oral bioavailability of genistein.
Acknowledgments
This work was supported by a grant from the National Research Foundation (NRF) of Korea funded by the Korea government (MSIP) (No. 2015R1A5A1008958). This work was also supported by the Basic Science Research Program through the NRF of Korea funded by the Ministry of Education (No. 2015R1D1A1A02062278).
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
REFERENCES
- Boyd BJ, Khoo SM, Whittaker DV, Davey G, Porter CJ. A lipid-based liquid crystalline matrix that provides sustained release and enhanced oral bioavailability for a model poorly water soluble drug in rats. Int J Pharm. 2007;340:52–60. doi: 10.1016/j.ijpharm.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Desai J, Thakkar H. Effect of particle size on oral bioavailability of darunavir-loaded solid lipid nanoparticles. J Microencapsul. 2016;33:669–678. doi: 10.1080/02652048.2016.1245363. [DOI] [PubMed] [Google Scholar]
- Dingler A, Gohla S. Production of solid lipid nanoparticles (SLN): scaling up feasibilities. J Microencapsul. 2002;19:11–16. doi: 10.1080/02652040010018056. [DOI] [PubMed] [Google Scholar]
- Dixon RA, Ferreira D. Genistein. Phytochemistry. 2002;60:205–211. doi: 10.1016/S0031-9422(02)00116-4. [DOI] [PubMed] [Google Scholar]
- Drover DR, Angst MS, Valle M, Ramaswamy B, Naidu S, Stanski DR, Verotta D. Input characteristics and bioavailability after administration of immediate and a new extended-release formulation of hydromorphone in healthy volunteers. Anesthesiology. 2002;97:827–836. doi: 10.1097/00000542-200210000-00013. [DOI] [PubMed] [Google Scholar]
- Emami J, Mohiti H, Hamishehkar H, Varshosaz J. Formulation and optimization of solid lipid nanoparticle formulation for pulmonary delivery of budesonide using Taguchi and Box-Behnken design. Res Pharm Sci. 2015;10:17–33. [PMC free article] [PubMed] [Google Scholar]
- Harde H, Das M, Jain S. Solid lipid nanoparticles: an oral bioavailability enhancer vehicle. Expert Opin Drug Deliv. 2011;8:1407–1424. doi: 10.1517/17425247.2011.604311. [DOI] [PubMed] [Google Scholar]
- Horter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 2001;46:75–87. doi: 10.1016/s0169-409x(00)00130-7. [DOI] [PubMed] [Google Scholar]
- Kumar DS, Pandit JK. Relationship between dissolution rate and bioavailability of sustained-release ibuprofen capsules. Drug Dev Ind Pharm. 1997;23:987–992. doi: 10.3109/03639049709149151. [DOI] [Google Scholar]
- Kwon SH, Kim SY, Ha KW, Kang MJ, Huh JS, Im TJ, Kim YM, Park YM, Kang KH, Lee S, Chang JY, Lee J, Choi YW. Pharmaceutical evaluation of genistein-loaded pluronic micelles for oral delivery. Arch Pharm Res. 2007;30:1138–1143. doi: 10.1007/BF02980249. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kim YH, Yu HJ, Cho NS, Kim TH, Kim DC, Chung CB, Hwang YI, Kim KH. Enhanced bioavailability of soy isoflavones by complexation with beta-cyclodextrin in rats. Biosci Biotechnol Biochem. 2007;71:2927–2933. doi: 10.1271/bbb.70296. [DOI] [PubMed] [Google Scholar]
- Li HL, Zhao XB, Ma YK, Zhai GX, Li LB, Lou HX. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J. Control. Release. 2009;133:238–244. doi: 10.1016/j.jconrel.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Li Y, Sun D, Palmisano M, Zhou S. Slow drug delivery decreased total body clearance and altered bioavailability of immediate- and controlled-release oxycodone formulations. Pharmacol Res Perspect. 2016;4:e00210. doi: 10.1002/prp2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Chen DW, Ren LX, Zhao XL, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J. Control. Release. 2006;114:53–59. doi: 10.1016/j.jconrel.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Mathot F, Van Beijsterveldt L, Preat V, Brewster M, Arien A. Intestinal uptake and biodistribution of novel polymeric micelles after oral administration. J. Control. Release. 2006;111:47–55. doi: 10.1016/j.jconrel.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Mehnert W, Mader K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196. doi: 10.1016/S0169-409X(01)00105-3. [DOI] [PubMed] [Google Scholar]
- Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. doi: 10.1016/S0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- Muller RH, Runge S, Ravell V, Mehnert W, Thunemann AF, Souto EB. Oral bioavailability of cyclosporine: solid lipid nanoparticles (SLN) versus drug nanocrystals. Int J Pharm. 2006;317:82–89. doi: 10.1016/j.ijpharm.2006.02.045. [DOI] [PubMed] [Google Scholar]
- Munish A, Meenakshi B, Komal S. Sodium alginate-arabinoxylan composite microbeads: preparation and characterization. J Pharm Investig. 2016;46:645–653. doi: 10.1007/s40005-016-0244-1. [DOI] [Google Scholar]
- Pankaj VD, Ritu MG, Shashikant ND. Formulation and development of solid self micro-emulsifying drug delivery system (S-SMEDDS) containing chlorthalidone for improvement of dissolution. J Pharm Investig. 2016;46:633–644. doi: 10.1007/s40005-016-0243-2. [DOI] [Google Scholar]
- Reithmeier H, Herrmann J, Gopferich A. Lipid microparticles as a parenteral controlled release device for peptides. J. Control. Release. 2001;73:339–350. doi: 10.1016/S0168-3659(01)00354-6. [DOI] [PubMed] [Google Scholar]
- Scalia S, Young PM, Traini D. Solid lipid microparticles as an approach to drug delivery. Expert Opin Drug Deliv. 2015;12:583–599. doi: 10.1517/17425247.2015.980812. [DOI] [PubMed] [Google Scholar]
- Shelnutt SR, Cimino CO, Wiggins PA, Ronis MJ, Badger TM. Pharmacokinetics of the glucuronide and sulfate conjugates of genistein and daidzein in men and women after consumption of a soy beverage. Am J Clin Nutr. 2002;76:588–594. doi: 10.1093/ajcn/76.3.588. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Matsui Y, Yamamoto H, Kawashima Y. Mucoadhesive properties of carbopol or chitosan-coated liposomes and their effectiveness in the oral administration of calcitonin to rats. J. Control. Release. 2003;86:235–242. doi: 10.1016/S0168-3659(02)00411-X. [DOI] [PubMed] [Google Scholar]
- Tang J, Xu N, Ji H, Liu H, Wang Z, Wu L. Eudragit nanoparticles containing genistein: formulation, development, and bioavailability assessment. Int. J. Nanomedicine. 2011;6:2429–2435. doi: 10.2147/IJN.S24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Kushwah V, Thanki K, Jain S. Triple antioxidant SNEDDS formulation with enhanced oral bioavailability: Implication of chemoprevention of breast cancer. Nanomedicine. 2016;12:1431–1443. doi: 10.1016/j.nano.2016.03.003. [DOI] [PubMed] [Google Scholar]
- Uner M, Yener G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspectives. Int. J. Nanomedicine. 2007;2:289–300. [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Kulkarni K, Zhu W, Hu M. Bioavailability and Pharmacokinetics of Genistein: Mechanistic Studies on its ADME. Anticancer Agents Med Chem. 2012;12:1264–1280. doi: 10.2174/187152012803833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Song TT, Cunnick JE, Murphy PA, Hendrich S. Daidzein and genistein glucuronides in vitro are weakly estrogenic and activate human natural killer cells at nutritionally relevant concentrations. J Nutr. 1999;129:399–405. doi: 10.1093/jn/129.2.399. [DOI] [PubMed] [Google Scholar]