

Abstract
Antiviral drug resistance is a matter of great clinical importance that, historically, has been investigated mostly from a virological perspective. Although the proximate mechanisms of resistance can be readily uncovered using these methods, larger evolutionary trends often remain elusive. Recent interest by population geneticists in studies of antiviral resistance has spurred new metrics for evaluating mutation and recombination rates, demographic histories of transmission and compartmentalization, and selective forces incurred during viral adaptation to antiviral drug treatment. We present up-to-date summaries on antiviral resistance for a range of drugs and viral types, and review recent advances for studying their evolutionary histories. We conclude that information imparted by demographic and selective histories, as revealed through population genomic inference, is integral to assessing the evolution of antiviral resistance as it pertains to human health.
Keywords: antiviral resistance, genetic barrier, mutagenesis, fluctuating selection, compensatory mutation, cost of adaptation
1. Resistance as an evolutionary process
Viral evolution, with its many public health consequences, is increasingly investigated within a population genetic framework. The growing availability of high quality molecular data has changed the scale at which these evolutionary processes can be monitored. Parameters such as nucleotide diversity, the strength of selection, and effective population size (Ne) can be monitored almost in real time using experimental evolution, and can be measured at increasingly frequent intervals in patients, informing treatment decisions (Chevillotte et al. 2010; Newman et al. 2013; Renzette et al. 2014). New mutations can also be observed at a fine scale, and of particular interest are those conferring drug resistance (Moya et al. 2004; zur Wiesch et al. 2011).
An increasing number of viral infections that impair host health are treated using antiviral drugs, typically targeting mechanisms of viral replication (Fig. 1). If the treatment is robust and viral fitness is impaired sufficiently, no viral genomes will be successfully replicated, but if treatment is not as effective and some genomes replicate, selective pressure may result in rapid adaptation toward resistance. This is exacerbated by the large population sizes and high rates of mutation characterizing many viruses: if resistance-conferring polymorphisms do not already exist in the population at the onset of treatment, they will likely arise soon thereafter. This problem has forced antiviral drug development to remain innovative, including combining existing drugs, and establishing new drug classes.
Figure 1.

Depictions of viral replication and protein synthesis. Representative replication mechanisms for DNA viruses HSV and HCMV, RNA viruses HCV and IAV, lentivirus HIV, and HBV. Bright blue strands represent viral DNA, green strands represent viral RNA, pink shapes represent virally produced enzymes, and purple shapes represent host-produced enzymes. When necessary, positive and negative-sense RNAs are designated with (+) and (−), respectively; note that only positive-sense RNA can be directly translated into proteins. Arrows indicate transcription, translation, replication, or integration activity, as denoted either by descriptive grey text or by the nearest enzyme. Bold, italicized text indicates drug classes for which known resistance mutations occur; the nearest enzyme (or replicative process) indicates the target of that drug class.
We here present a brief review of resistance mechanisms and their evolution, with a focus on viruses relevant to human health; we continue by describing evolutionary forces that are uniquely exemplified by viral resistance, and thus merit special attention from both the population genetics and virology fields.
2. Variation in viral biology and resistance mechanisms
Differences in viral replication biology drive the generation of new drug classes targeting different parts of the viral life cycle. Understanding these differences is critical not only for synthesizing new drugs, but also for predicting and countering evolution towards resistance, Below, we briefly review common modes of resistance (and relevant underlying biology) in six exemplary viruses: RNA viruses hepatitis C and influenza A virus (IAV), DNA viruses herpes simplex virus (HSV) and human cytomegalovirus (HCMV), retrovirus HIV, and unconventionally replicating hepatitis B virus (HBV).
Hepatitis C virus (HCV) is characterized by a high mutation rate and subsequently high genomic diversity (Simmonds 2004) (Table 1). This mutation rate is facilitated by frequent replication and poor proofreading function of the virally encoded RNA polymerase. In fact, the virus likely exists as a quasispecies, or a group of genomes forming a structured ‘cloud’ in sequence space, with replication near the maximum error rate allowed before genomic integrity is lost (Sanjuán et al. 2004; Eigen 2002). Within a host, HCV replicates in different compartments, such as different organs or bodily fluids (Halfon and Locarnini 2011). Bottlenecks are common during both inter-host transmission and intra-host compartmentalization (Bull et al. 2011); these bottlenecks reduce population size, thus amplifying the role of genetic drift in HCV evolution (Table 1). The most common antiviral drugs used against HCV are direct-acting antiviral agents (DAAs), which usually inhibit either protease or polymerase activity. Those inhibiting protease have a low genetic barrier to resistance, meaning that resistance is easily achieved through only one or a few mutations (see Section 4). Indeed, given the high rate of mutation, it is likely that these polymorphisms may already exist in a given population (Bull et al. 2011). However, new combinations of the DAAs, such as ledipasvir and sofosbuvir (marketed as Harvoni), appear to have a high genetic barrier to resistance, with very little (if any) cross-resistance between the two drugs, and are thus promising for future treatment (Gritsenko and Hughes 2015).
Table 1.
Estimates of population parameters for various viruses
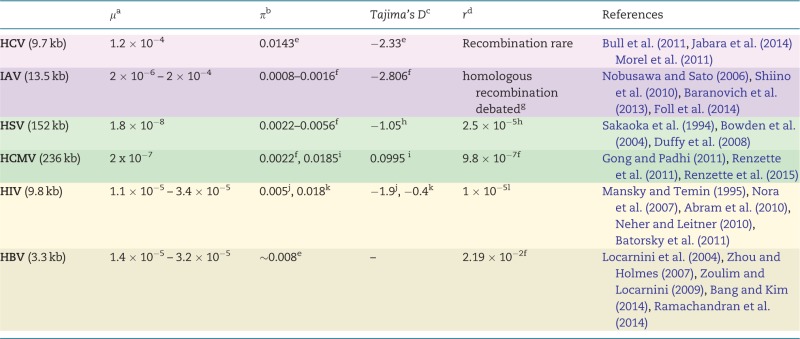 |
Estimates for several population genetic parameters for the six viruses highlighted here. Shading indicates viral type: purple represents RNA viruses, green represents DNA viruses, and beige represents those that utilize both DNA and RNA. Genome sizes are given under virus names. Most studies were based on clinical samples, but some relied on databases such as GenBank or the Los Alamos HIV database. Estimates of Tajima’s D in Hepatitis B have not been found to be reported. References are listed with respect to the order of entries per row; those within a single pair of brackets represent a single table entry. We emphasize that this table serves as a guide only, and that the original studies should always be consulted for technical details.
aμ, mutation rate: given as nucleotide substitutions/base/replication (genome-wide), from experimental measures.
bπ, nucleotide diversity: between-host estimates either from regions of interest with regards to resistance or from the full-genome.
cTajima’s D, a test-statistic to distinguish neutrally evolving populations from those evolving under non-random process(es); between-host estimates from either specific regions or the full-genome.
dr, recombination rate: given as recombination events/site/generation, based on population-level sequence diversity.
eProtease (0.13 kb, n = 20).
fGenome-wide.
gSee Han and Worobey (2011) for discussion.
hUL (2 kb, n = 42).
iUS28 (2.5 kb, n = 103).
jR(everse)T(ranscriptase) (0.6 kb, n = 28).
kenvelope (1.3 kb, n = 28).
lpol (3 kb, n = 9), effective recombination rate.
IAV is an RNA virus whose small genome codes for at least eleven proteins including two proteins on the virion surface: hemagglutinin (HA) and neuraminidase (NA). HA and NA allow viral envelopes to fuse with and separate from the cell membrane, respectively. As surface antigens, HA and NA are assumed to be under diversifying selection, and also tend to evolve relatively rapidly, with dN/dS ratios much higher than in the other viral proteins (Chen and Holmes 2010; Bhatt et al. 2011). The most common antivirals used to treat IAV are NA-inhibitors, which prevent detachment of viral envelopes from the cell membrane. The popular NA inhibitor oseltamivir has been used since the early 2000s to treat seasonal IAV cases, but around 2007 became associated with the mutation H274Y, which confers very high levels of resistance at very little fitness cost (see Section 5) (Moscona 2009). It is thought that other compensatory mutations (see Section 6) alleviated these costs, allowing it to circulate uncurtailed in the population (Bloom et al. 2010). Another NA inhibitor, zanamivir, has been successful in IAV treatment and has not been observed to readily develop resistance, but is not prescribed as frequently due to difficulty of administration; this may contribute to the relative rarity of resistance mutations in vivo (Thorlund et al. 2011). Finally, a newer antiviral called favipiravir is thought to act by inducing mutagenesis in the IAV genome (see Section 7). Thus far, resistance to favipiravir has yet to be observed in vivo, and has only been generated in the laboratory under high selection pressure (Baranovich et al. 2013; Furuta et al. 2013; Cheung et al. 2014).
HSV is a herpesvirus containing a large genome with low diversity as compared with RNA viruses, promoted in part by high replication fidelity and low recombination rates (Andrei et al. 2013) (Table 1). Infections are lifelong, cycling between periods of latency and viral shedding, though the virus is thought to be transmittable even in asymptomatic hosts (James and Prichard 2014). Systemic antiviral therapy is usually only necessary in immune-compromised patients, where it is relatively successful. The most commonly used drugs are Acyclovir and its related analogs, all nucleoside inhibitors, for which resistance mutations are known, affecting either a thymidine kinase necessary for prodrug activation by phosphorylation or the DNA polymerase (Griffiths 2011).
HCMV is also a herpesvirus for which infections are lifelong and generally asymptomatic. However, as with HSV, health is impaired when hosts are immuno-compromised including congenitally infected infants. Infection begins in a single compartment, such as plasma or saliva, before spreading to others; these subsequent divisions act as strong bottleneck events, curtailing population size. After compartmentalization, differentiation continues to the extent that populations from differing compartments within a single host are as diverse as those from between hosts (Renzette et al. 2013). The HCMV genome is generally diverse within-host, with levels of polymorphism comparable to RNA viruses, even though the DNA polymerase exhibits higher fidelity than RNA virus polymerases (Lurain and Chou 2010; Renzette et al. 2011) (Table 1). Approximately 5% of open reading frames show signatures of positive selection (Renzette et al. 2011). In particular, loci associated with envelope proteins tend to have elevated dN/dS, as diverse envelopes help to evade host immune defense, while loci for replicative proteins tend to be conserved. If treatment is necessary for HCMV, it usually involves antivirals acting as nucleoside analogs, including ganciclovir and cidofovir. Resistance mainly occurs either in a viral kinase necessary for phosphorylation of the prodrug or in the DNA polymerase (Gilbert and Boivin 2005). As mentioned earlier, resistance mutations are usually only observed in immune-compromised hosts, such as tissue transplant recipients or those co-infected with HIV. This observation could however be biased considering the population treated with antivirals in the first place: (usually) those with impaired immunity who show symptoms of viral infection. The causality in this relationship deserves further study.
HIV is a retrovirus encoding an RNA genome within the virion, but after infecting a host cell, it replicates its genome using reverse transcription, generating a copy of DNA and eventually double-stranded DNA that is inserted in the host genome and can then be transcribed back to RNA. Reverse transcriptase is highly error prone, leading to a high rate of nucleotide substitutions, increased population diversity, and frequent resistance mutations when drug treatment is ongoing (Götte 2012) (Table 1). In fact, at the onset of drug treatment, it is estimated that every single-point mutation in the viral genome is likely to exist in some infected cell (Goldberg et al. 2012). This affords the possibility for selection on standing variation: when selection is introduced (i.e. drug treatment), a standing polymorphism may become immediately adaptive and rise to fixation (Pennings et al. 2014). The most common antiviral therapy for HIV today is a multi-drug regimen; these often combine drugs from different classes, preferably without known cross-resistance mutations (Menéndez-Arias 2013). Such a regimen increases the genetic barrier by requiring several mutations in order for resistance to be conferred. The most common combinations include nucleoside reverse-transcriptase inhibitors, non-nucleoside reverse-transcriptase inhibitors, protease inhibitors, and/or integrase inhibitors. Several resistance mutations are known within each class, but combination treatment requires these to occur simultaneously for full resistance to be conferred (though fitness can be partially restored by only one mutation; see Pennings 2012).
HBV is an enveloped DNA virus whose replication involves transcription to RNA intermediates that are then reverse transcribed back to DNA. Despite its DNA genome, HBV is thought to exist as a quasispecies; high levels of diversity are maintained through lack of proofreading during reverse transcription (Fig. 1, Table 1). This means that, like HIV and many RNA viruses, HBV infections maintain polymorphism at almost all nucleotide positions within a host at a given time (Khudyakov 2010; Bang and Kim 2014), paving the way for resistance mutations to increase in frequency upon drug treatment. HBV is most commonly treated with reverse transcriptase inhibitors, particularly lamivudine. However, several resistance mutations against lamivudine have been observed, many of which have low genetic barriers. Given this, a second reverse transcriptase inhibitor is often supplemented (Bang and Kim 2014). This method of drug-switching, if done within a single class, is potentially problematic: it encourages more robust resistance mutations that are either effective across many drugs and/or suffer lower fitness costs when drug treatment (selection) ceases (Locarnini et al. 2004; Shaw et al. 2006; Bang and Kim 2014). Initial combination therapy is becoming more frequent in clinical trials, but is not usually the standard treatment.
3. Inferring population genetic parameters in viruses
Before discussing the evolutionary mechanisms and consequences of antiviral resistance, it is important to consider the interpretation of standard parameters in light of viral biology. For instance, the standard definition of Ne is the size of an imagined population that would experience the same rate of genetic drift as the population in question. However, this can be inferred using several methods (see Charlesworth 2009, for a synopsis), and can represent intra-host or global populations (Moya et al. 2004), be given as genome-wide or region-specific estimates (Rambaut et al. 2008), or given as short-time scale estimates versus those over deeper time (Pennings et al. 2014). Importantly, there is a growing literature suggesting the importance of incorporating multiple merger coalescent models in analysis of viral populations, given their biology; these models will also alter estimates of Ne (Neher and Hallatschek 2013; Tellier and Lemaire 2014). Similarly, recombination can be measured and/or reported in several ways (Schlub et al. 2010), which may require different interpretation based on the type of recombination event (i.e. homologous vs. non-homologous vs. reassortment). Patterns of nucleotide diversity and linkage disequilibrium will differ according to the coding density of viral genomes. Finally, it is unclear how latent virus copies affect estimates of these parameters, and debated whether they should be included at all. Therefore, while it is evident that these parameters are best evaluated on a population-by-population basis (especially in cases of direct clinical relevance), we attempt here to arrive at some generalizations for their application in various viruses (as seen in Table 1). In the future, more universal procedures for measuring these parameters across viruses would be of great value to the field.
4. Genetic barriers to resistance
The number and type of substitution(s) necessary to confer resistance often differ according to antiviral target and class; this is often quantified as the genetic barrier to resistance (Götte 2012). Many older classes of antivirals are now known to have low genetic barriers: viruses usually only require one or two substitutions to gain resistance (e.g. first generation reverse-transcriptase inhibitors used for HIV infections, Perno et al. 2008; Menéndez-Arias 2013). A common goal of new drug development is to increase this genetic barrier by developing new drugs or combinations of drugs such that resistance does not arise so readily.
Genetic barriers affect the rates at which resistance mutations are observed. If a certain pathway to resistance requires only one substitution, but another requires more, we would expect the former to occur more often. These pathways can even differ by viral genotype; for example, in HCV, the common resistance-conferring mutation R155K requires only a single nucleotide change in genotype 1a, but two in genotype 1b, where it is rarely observed (Götte 2012). Genetic barriers involve not only the substitution quantity, but also type; for instance, resistance mutations requiring transitions should be more common than those requiring transversions (Powdrill et al. 2011).
These step-wise mutations can be conceptualized as an adaptive walk within the context of Fisher’s geometric model (Bank et al. 2014, 2015; Foll et al. 2014; Tenaillon 2014). Upon environment change (i.e. when selective pressure changes), the distance to the optimum phenotype increases. When new mutations arise, they can move the phenotype closer to the optimum (i.e. a beneficial mutation), maintain the same distance relative to the optimum (i.e. a neutral mutation), or move the phenotype farther from the optimum (i.e. a deleterious mutation). Which of these occurs depends on the effect size of the mutation and the population’s current position in phenotypic space, but we know from the distribution of fitness effects of new mutations that beneficial mutations are less probable than neutral or deleterious ones (though this is a function of distance from the optimum, see Hietpas et al. 2013). We thus know that a greater proportion of multiple step combinations will be deleterious compared with single steps. This suggests that when selection is introduced, resistance is more likely to come about through a small number of mutations (low barrier) than through multiple mutations (high barrier). Therefore, rather than tracking already-known mutations, the model’s utility would lie in exploring new, potential paths on fitness landscapes that could be taken in response to the environmental changes induced by drug treatment. Questions when deriving new antiviral drugs could include: for a given target, or viral trait, what is the shape of the distribution of fitness effects? Can we predict the likelihood of single step versus multi-step adaptive walks? Considering genetic barriers through the lens of Fisher’s geometric model can help inform which viral traits should be targeted through selection in order to avoid rapidly developing resistance, though some natural extensions, such as accounting for the genetic code (Lourenço et al. 2013) may be required.
5. Fitness costs
Resistance mutations are often not maintained in the population after drug treatment ceases. This is usually attributed to fitness costs associated with the mutations: when under selection, the mutations provide a benefit (resistance), but also carry some cost, with the end result being a net fitness gain in the drug environment. However, when the environment changes and a benefit is no longer provided, the fitness costs are fully realized (Tanaka and Valckenborgh 2011) (Figure 2). These cost/benefit tradeoffs can take the form of, for example, binding site alteration resulting in decreased enzymatic production (e.g. TK mutations in HSV, Piret and Boivin 2014), binding site alteration resulting in failed binding of intended partners (e.g. mutations in HCV affecting protease inhibitor activity, Halfon and Locarnini 2011), and removal of chain-terminating analogs rendering lower replication rates (e.g. thymine analog mutations in HIV, [Hu et al. 2006]).
Figure 2.
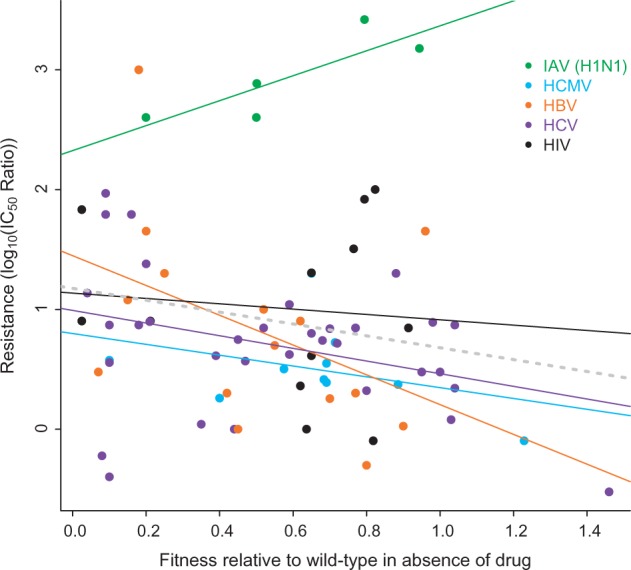
Resistance and associated fitness costs in drug absence. A meta-analysis of antiviral resistance mutations, particularly the level of resistance conferred in the presence of a drug, and viral fitness in the absence of drug. The figure is composed of metadata from studies that reported both (i) the IC50 ratio between wild-type and resistant viral strains measured with a common (non-experimental) antiviral, and (ii) the replication rates of both of those strains in a drug-free environment. A total of 76 observations were recorded from 18 studies involving five viral types (all of those reviewed here except HSV, for which there was no data available fitting the above criteria). Resistance mutations to the following drugs are included: oseltamivir (H1N1), ganciclovir (HCMV), lamivudine (HBV), boceprevir (HCV), telaprevir (HCV), raltegravir (HIV), elvitegravir (HIV), L-708906 (HIV), L-731988 (HIV), lamivudine (HIV), adefovir, (HIV), efavirenz, (HIV), and rilpivirine (HIV). However, neither drug nor target was a significant predictor of fitness costs according to a generalized linear model (P > 0.05). Data were sourced from Cihlar et al. (1998), Hazuda et al. (2000), Naeger et al. (2001), Ives et al. (2002), Chou et al. (2003), Brunelle (2005), Springer et al. (2005), Chou et al. (2007), Kobayashi et al. (2008), Baz et al. (2010), Martin et al. (2010), Abed et al. (2011), Shimakami et al. (2011), Wong et al. (2012), Jiang et al. (2013), Mesplède et al. (2013), Zhang 2013), Hu and Kuritzkes (2014) and can be found in the Supplementary Table.
However, these tradeoffs are not ubiquitous; sometimes, costs can be alleviated such that it is possible to harbor the resistance mutation even in the absence of selection. For example, the H274Y mutation in influenza (H1N1) confers resistance to oseltamivir, the most popular NA inhibitor. Until fairly recently, H274Y was associated with the usual fitness costs in the absence of the drug; however, since the 2008–09 flu season, these costs have diminished greatly in seasonal influenza, such that the majority of H1N1 strains can now carry H274Y at little or no cost (Renzette et al. 2014; Hauge et al. 2009; Poland et al. 2009; Baz et al. 2010). This fitness restoration is thought to be due to the development of compensatory mutations (see Section 6) (Bloom et al. 2010; Duan et al. 2014).
Fitness costs also co-vary with the degree of resistance conferred. Usually, mutations providing greater resistance carry higher fitness costs in the absence of drug, and vice-versa (Croteau et al. 1997; De Luca 2006). To illustrate this frequently cited correlation, previously published data on resistance (measured as IC50 ratio) and fitness (replication rate relative to the wild-type in a drug-free environment) were aggregated (Fig. 2). A linear model showed a non-significant relationship across all viruses. Within a given virus, though non-significant correlations between resistance and fitness were found for many, a significant negative correlation was seen for HCV (P = 0.0499). Surprisingly, for seasonal H1N1 (a strain of IAV), a positive relationship between fitness and resistance was seen. This uniquely positive correlation may be influenced by the inclusion of only H274Y mutations, as these were the only data available fitting inclusion criteria (see Fig. 2 legend; no mutants were from pandemic strains and none were on compensatory backgrounds). As mentioned above, H274Y has been found to have little or no fitness costs in the absence of drug owing to compensatory mutations. Since H274Y is indeed a unique resistance mutation in this respect, we re-analyzed the correlation between fitness cost and resistance when those data are omitted; indeed, an analysis of the remaining data indicates a significant negative correlation among all viruses (P = 0.0088). However, these results should be interpreted with caution, as they are aggregated from studies with various experimental procedures, using various viral strains with different genetic backgrounds. A single experiment assessing this relationship would be much more informative, and would indeed be an asset to the field in the future.
6. Compensatory mutations
As discussed above, resistance mutations often incur a fitness cost in the absence of selection. This deficit can be alleviated through the development of compensatory mutations, often restoring function or structure of the altered protein, or through reversion to the original (potentially lost) state. Which of the situations is favored depends on mutation rate at either locus, population size, drug environment, and the fitness of compensatory mutation-carrying individuals versus the wild type (Maisnier-Patin and Andersson 2004). Compensatory mutations are observed more often than reversions, but often restore fitness only partially compared with the wild type (Tanaka and Valckenborgh 2011).
In a fluctuating environment (e.g. incomplete adherence, see Section 8), the fate of compensatory mutations is uncertain. These mutations are generally either neutral or deleterious in the absence of the target mutation, and are thus likely to be lost if the primary resistance mutation is removed. However, if the compensatory mutations are antagonistically epistatic with the primary ones, then they may be maintained: by conferring a relatively higher fitness than if both the primary and compensatory mutations were absent, they may be conserved in the population (Khudyakov 2010; zur Wiesch et al. 2011). These compensatory mutations could then act as permissive (secondary) mutations, allowing the frequency of the primary resistance mutation to fluctuate (Bloom et al. 2010).
7. Mutagenesis
Efforts to prevent drug resistance, however promising, sometimes prove futile. An alternative approach to antiviral action is to target mutational processes, rather than replicative ones, in order to lower population fitness. More specifically, as population mutation rates increase—and particularly as the majority of new mutations are deleterious—the influx may have a number of effects. First, particularly in non-recombining viruses, Muller’s Ratchet (the process by which the most fit class is lost via genetic drift) is expected to operate, reducing fitness through time (Muller 1964). Secondly, Hill-Robertson interference (the process by which, owing to linkage, the probability of fixation of beneficial mutations and the probability of loss of deleterious mutations are reduced) will become stronger (Hill and Robeterson 1966). Notably, both effects may compound through time—with reduced fitness leading to reduced effective population size, which further speeds the Ratchet; this leads to compounding reductions in fitness, which may ultimately lead to mutational meltdown (i.e. population extinction).
Many RNA viruses replicate under conditions very close to the error threshold, or the error rate beyond which meltdown is likely (e.g. IAV and HCV, see Holmes 2003). New drug designs take advantage of this by acting to increase the base mutation rate such that populations exceed this threshold (Crotty et al. 2001; Mullins et al. 2011; Pauly and Lauring 2015). The most promising of these drugs is favipiravir, and while its exact mode of action is debated, it is thought to act on the RNA polymerase to decrease fidelity in nucleoside incorporation (Baranovich et al. 2013; Furuta et al. 2013). This would lead to a higher rate of mutant virion production and a decrease in specific infectivity (Arias et al. 2014). Though resistance to other mutagenic drugs has been observed before (usually through mutation to restore fidelity of the polymerase, e.g. Pfeiffer and Kirkegaard 2003), it is thus far uncommon in favipiravir. In fact, for IAV (a virus against which favipiravir shows great potential), no significant in vivo resistance has yet been detected in clinical trials, making favipiravir a promising new antiviral candidate. Finally, though the in vivo favipiravir resistance studies are encouraging, it should be noted that NA inhibitors like oseltamivir were once thought to be less likely to select for resistance mutations than previous generations of antivirals (Moscona 2005). It is therefore not implausible that resistance against mutagenic therapies could likewise arise in the future.
8. Adherence and fluctuating selection
As with antibiotics, interruptions to antiviral drug regimens pose a risk to both personal and public health as they allow viral populations that are not completely eliminated to repopulate within a host (Rong et al. 2007; Gardner et al. 2010; Chotiyaputta et al. 2012). The initial treatment creates a bottleneck in the viral population, through which the drug-resistant individuals are most likely to survive. When drug use is interrupted, repopulation will stem from these individuals, population size will increase dramatically, and the new population will likely have higher resistance and be more difficult to cull (e.g. Wang et al. 2011). In a clinical sense, the occurrence and frequency of such interruptions is summarized as adherence (Fox et al. 2008; Sethi et al. 2003). Non-adherence includes not only complete interruption of treatment, but also dosage and timing non-compliance. These can lead to treatment failure, often accompanied by novel resistance mutations (Fox et al. 2008).
The dynamic intra-host drug concentrations resulting from non-adherence can create a case of fluctuating selection in which the magnitude or even sign of selection can change as the environment fluctuates (though changes in population size may be equally important; see Pennings 2012). Population genetic models of fluctuating selection are not new, and several empirical examples are known (see Bell 2010, for review). Previously, estimation of fluctuating selection coefficients was based on phenotypic, labor-intensive, time-series data sets, but methods now exist based on polymorphism data from a single collection (e.g. Miura et al. 2013), and traditional tests for selection have been updated to accommodate such estimates (e.g. MK test, Gossmann et al. 2014). Therefore, the fate of an allele that fluctuates from being beneficial to neutral to deleterious, such as a resistance mutation with changing drug adherence, can be tracked using current methods.
In the context of antimicrobial drug resistance, fluctuating selection has been examined previously (zur Wiesch et al. 2010; Hall et al. 2011; Tanaka and Valckenborgh 2011), but application of these methods to antiviral resistance is lacking. Also, selection on resistance alleles or the allele frequencies themselves are rarely treated as parameters of interest; rather, whole-organism fitness is tracked. Therefore, the population genetic methods alluded to above could be applied to antiviral resistance, though not without some modification. One potential avenue would be to tailor future studies to known resistance mutations, in order to characterize their frequencies through periods of adherence and non-adherence.
9. Genetic screening
Before the onset of treatment with a new-to-host drug, screens for pre-existing resistance mutations are frequently administered, with respect to the drug in question (particularly in HIV and HCV). The presence of such mutations pre-treatment may be due to previous exposure to similar drugs, transmission of a strain already containing resistance mutations, or simply due to a high rate of mutation. Given the genomic data collected in these screens, a further goal may be to detect new, not yet reported resistance mutations in addition to scanning for already known mutants. This is analogous to searches for signatures of positive selection commonly employed in studies of adaption. Many methods exist currently that could be adopted by clinical researchers for purposes of improving mutation lists (Beerenwinkel et al. 2005; Nielsen 2005; Jensen et al. 2007); methods could also be extended to unveil novel adaptive events as viruses adapt to new environments (e.g. Pennings 2012; Foll et al. 2014).
10. Conclusion
As reviewed here, the application of evolutionary theory or metrics to the study of antiviral drug resistance is often illuminating. Tests for positive selection can potentially identify resistance mutations, fluctuating selection models may predict non-adherence outcomes, and phenomena such as mutational meltdown can guide the development of promising mutagenic drugs. Though evolutionary studies are increasingly addressing viral populations, the fields of virology and evolutionary biology could mutually benefit from further collaboration: the former through more elaborate methods for predicting and preventing resistance (and associated clinical outcomes), and the latter through better model systems for testing new theory or methods, including parameter ranges rarely observed in other taxa, such as large effective population sizes and high mutation rates (Table 1).
The clinical implications of antiviral resistance must not be underestimated. Both human and monetary costs of resistance remain high, particularly for IAV (Howard and Scott 2005; Moscona 2009). From a public health perspective, unpredictability of viral evolution and drug resistance means that antiviral treatments also remain costly (Lipsitch et al. 2012). Addressing clinical and public health questions with the tests and metrics mentioned here may indeed alleviate some of these costs by better illuminating resistance patterns and facilitating prediction of epidemics or pandemics.
Though we have reviewed here only a handful of viruses, and focused on commonly applied antivirals and associated mutations, the consequences of resistance are of course much further reaching. Viruses detailed here are those with well-studied resistance patterns, usually due to their serious human health effects; however, antiviral failure certainly occurs elsewhere, including in viruses with more minor effects (for which failure is not perilous to health and therefore not thoroughly studied) and in animal and plant viruses with agricultural impact. We have also only briefly summarized the drugs employed for treatment and their associated mutations, but reports of all known resistance mutants for a given virus are readily available, as are growth or fitness data (Chevillotte et al. 2010; Nguyen et al. 2012; Wyles 2013; Wensing et al. 2014) (Fig. 2).
Finally, while we here make some generalizations about antiviral resistance, it is imperative to note that the process differs, sometimes dramatically, according to viral biology, and should be accounted for in any analysis of resistance conferral. It is our aim that the review presented here will motivate further research on resistance evolution, and highlight the fact that clinically motivated and evolutionary-motivated studies often have overlapping and complementary goals which ought to be embraced.
Supplementary Material
Acknowledgements
We thank members of the Jensen lab for helpful comments on earlier versions of this article. This work was funded by grants from the Swiss National Science Foundation and a European Research Council (ERC) grant to J.D.J.
Conflict of interest: None declared.
Footnotes
The original version of this article contained formatting errors in figure 1. This has now been corrected.
Supplementary data
Supplementary data are available at Virus Evolution online.
References
- Abed Y. et al. (2011) ‘Role of Permissive Neuraminidase Mutations in Influenza A/Brisbane/59/2007-like (H1N1) Viruses’, PLoS Pathogens, 7: e1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abram M. E. et al. (2010) ‘Nature, Position and Frequency of Mutations Made in a Single Cycle of HIV-1 Replication’, Journal of Virology, 84: 9864–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei G. et al. (2013) ‘Heterogeneity and Evolution of Thymidine Kinase and DNA Polymerase Mutants of Herpes Simplex Virus Type 1: Implications for Antiviral Therapy’, Journal of Infectious Diseases, 207: 1295–305. [DOI] [PubMed] [Google Scholar]
- Arias A. et al. (2014) ‘Favipiravir Elicits Antiviral Mutagenesis During Virus Replication In Vivo’, eLife, 3: e03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang K. B, Kim H. J. (2014) ‘Management of Antiviral Drug Resistance in Chronic Hepatitis B’, World Journal of Gastroenterology, 20: 11641–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank C. et al. (2014) ‘A Bayesian MCMC Approach to Assess the Complete Distribution of Fitness Effects of New Mutations: Uncovering the Potential for Adaptive Walks in Challenging Environments’, Genetics, 196: 841–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank C. et al. (2015) ‘A Systematic Survey of An Intragenic Epistatic Landscape’, Molecular Biology and Evolution, 32: 229–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranovich T. et al. (2013) ‘T-705 (Fapiravir) Induces Lethal Mutagenesis in Influenza A H1N1 Viruses In Vitro’, Journal of Virology, 87: 3741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batorsky R. et al. (2011) ‘Estimate of Effective Recombination Rate and Average Selection Coefficient for HIV in Chronic Infection’, Proceedings of the National Academy of Sciences, 108: 5661–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baz M. et al. (2010) ‘Effect of the Neuraminidase Mutation H274Y Conferring Resistance to Oseltamivir on the Replicative Capacity and Virulence of Old and Recent Human Influenza A (H1N1) Virus’, The Journal of Infectious Diseases, 201: 740–5 [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N. et al. (2005) ‘Computational Methods for the Design of Effective Therapies Against Drug Resistant HIV Strains’, Bioinformatics, 21: 3943–50 [DOI] [PubMed] [Google Scholar]
- Bell G. (2010) ‘Fluctuating Selection: The Perpetual Renewal of Adaptation in Variable Environments’, Philosophical Transactions of the Royal Society B, 365: 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S. et al. (2011) ‘The Genomic Rate of Molecular Adaptation of the Human Influenza A Virus’, Molecular Biology and Evolution, 28: 2443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J. D. et al. (2010) ‘Permissive Secondary Mutations Enable the Evolution of Influenza Oseltamivir Resistance’, Science, 328: 1272–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden R. et al. (2004) ‘High Recombination Rate in Herpes Simplex Virus Type 1 Natural Populations Suggests Signficiant Co-Infection’, Infection, Genetics, and Evolution, 4: 115–23. [DOI] [PubMed] [Google Scholar]
- Brunelle M. (2005) ‘Susceptibility to Antivirals of a Human HBV Strain with Mutations Conferring Resistance to Both Lamivudine and Adefovir’, Hepatology, 41: 1391–8. [DOI] [PubMed] [Google Scholar]
- Bull R. A. et al. (2011) ‘Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection’, PLoS Pathogens, 7: e1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. (2009) ‘Effective Population Size and Patterns of Molecular Evolution and Variation’, Nature Reviews Genetics, 10: 195–205 [DOI] [PubMed] [Google Scholar]
- Chen R., Holmes E. C. (2010) ‘Hitchhiking and the population genetic structure of avian influenza virus’, Journal of Molecular Evolution, 70: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P. P. H. et al. (2014) ‘Generation and Characterization of Influenza A Viruses with Altered Polymerase Fidelity’, Nature Communications, 5: 4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevillotte M. et al. (2010) ‘A New Tool Linking Human Cytomegalovirus Drug Resistance Mutations to Resistance Phenotypes’, Antiviral Research, 85: 318–27. [DOI] [PubMed] [Google Scholar]
- Chotiyaputta W. et al. (2012) ‘Adherence to Nucleos(t)ide Analogues for Chronic Hepatitis B in Clinical Practice and Correlation with Virological Breakthroughs’, Journal of Viral Hepatitis, 19: 205–12 [DOI] [PubMed] [Google Scholar]
- Chou S. et al. (2003) ‘Viral DNA Polymerase Mutations Associated with Drug Resistance in Human Cytomegalovirus’, The Journal of Infectious Diseases, 188: 32–9 [DOI] [PubMed] [Google Scholar]
- Chou S. et al. (2007) ‘Growth and Drug Resistance Phenotypes Resulting from Cytomegalovirus Dna Polymerase Region III Mutations Observed in Clinical Specimens’, Antimicrobial Agents and Chemotherapy, 51: 4160–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihlar T. et al. (1998) ‘A Point Mutation in the Human Cytomegalovirus DNA Polymerase Gene Selected In Vitro by Cidofovir Confers a Slow Replication Phenotype in Cell Culture’, Virology, 248: 382–93 [DOI] [PubMed] [Google Scholar]
- Croteau G. et al. (1997) ‘Impaired Fitness of Human Immunodeficiency Virus Type 1 Variants with High-Level Resistance to Protease Inhibitors’, Journal of Virology, 71: 1089–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S. et al. (2001) ‘RNA Virus Error Catastrophe: Direct Molecular Test by Using Ribavirin’, Proceedings of the National Academy of Sciences of the United States of America, 98: 6895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A. (2006) The Impact of Resistance on Viral Fitness and Its Clinical Implications’, in Geretti A.M. (ed.) Antiretroviral Resistance in Clinical Practice, Mediscript [PubMed] [Google Scholar]
- Duan S. et al. (2014) ‘Epistatic interactions Between Neuraminidase Mutations Facilitated the Emergence of the Oseltamivir-Resistant H1n1 Influenza Viruses’, Nature Communications, 5: 5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S. et al. (2008) ‘Rates of Evolutionary Changes in Viruses: Patterns and DETERMINANTS’, Nature Reviews Genetics, 9: 267–76 [DOI] [PubMed] [Google Scholar]
- Eigen M. (2002) ‘Error catastrophe and antiviral strategy’, Proceedings of the National Academy of Sciences, 99: 13374–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll M. et al. (2014) ‘Influenza Virus Drug Resistance: A Time-Sampled Population Genetic Perspective’, PLoS Genetics, 10: e1004185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox A. et al. (2008) ‘Viral Re-suppression and Detection of Drug Resistance Following Interruption of a Suppressive NNRTI-based Regimen’, Aids, 22: 2279–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y. et al. (2013) ‘Favipiravir (T-705), A Novel Viral RNA Polymerase Inhibitor’, Antiviral Research, 100: 446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner E. M. et al. (2010) ‘Antiretroviral Medication Adherence and Class-Specific Resistance in a Large Prospective Clinical Trial’, Aids, 24: 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C., Boivin G. (2005) ‘Human Cytomegalovirus Resistance to Antiviral Drugs’, Antimicrobial Agents and Chemotherapy, 49: 873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg D. E. et al. (2012) ‘Outwitting Evolution: Fighting Drug-Resistant TB, Malaria, and HIV’, Cell, 148: 1271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Padhi A. (2011) ‘Evidence for Positive Selection in the Extracellular Domain of Human Cytomegalovirus Encoded G Protein-Coupled Receptor US28’, Journal of Medical Virology, 83: 1255–61 [DOI] [PubMed] [Google Scholar]
- Gossmann T. I. et al. (2014) ‘Fluctuating Selection Models and Mcdonald-Kreitman Type Analyses’, PLoS One, 9: e84540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götte M. (2012) ‘The Distinct Contributions of Fitness and Genetic Barrier to the Development of Antiviral Drug Resistance’, Current Opinion in Virology, 2: 644–50. [DOI] [PubMed] [Google Scholar]
- Griffiths A. (2011) ‘Slipping and Sliding: Frameshift Mutations in Herpes Simplex Virus Thymidine Kinase and Drug-Resistance’, Drug Resistance Updates, 14: 251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsenko D., Hughes G. (2015) ‘Ledipasvir/Sofosbuvir (harvoni): improving options for hepatitis C virus infection’, Pharmacy and Therapeutics, 40: 256. [PMC free article] [PubMed] [Google Scholar]
- Halfon P., Locarnini S. (2011) ‘Hepatitis C virus resistance to protease inhibitors’, Journal of Hepatology, 55: 192–206. [DOI] [PubMed] [Google Scholar]
- Hall A. R. et al. (2011) ‘Host-Parasite Coevolutionary Arms Races Give Way to Fluctuating Selection’, Ecology Letters, 14: 635–42 [DOI] [PubMed] [Google Scholar]
- Hauge S. H. et al. (2009) ‘Oseltamivir-Resistant Influenza Viruses A (H1N1), Norway, 2007-2008’, Emerging Infectious Diseases, 15: 155–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazuda D. J. et al. (2000) ‘Inhibitors of Strand Transfer that Prevent Integration and Inhibit HIV-1 Replication in Cells’, Science, 287: 646–50 [DOI] [PubMed] [Google Scholar]
- Hietpas R. T. et al. (2013) ‘Shifting Fitness Landscapes in Response to Altered Environments’, Evolution, 67: 3512–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W. G., Robeterson A. (1966) ‘The Effect of Linkage on Limits to Artificial Selection’, Genetical Research, 8: 269–94 [PubMed] [Google Scholar]
- Holmes E. C. (2003) ‘Error Thresholds and the Contraints to RNA Virus Evolution’, Trends in Microbiology, 11: 543–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard D. H., Scott R. D. (2005) ‘The Economic Burden of Drug Resistance’, Clinical Infectious Diseases, 41: S283–6 [DOI] [PubMed] [Google Scholar]
- Hu Z. et al. (2006) ‘Fitness Comparison of Thymidine Analog Resistance Pathways in Human Immmunodeficiency Virus Type 1’, Journal of Virology, 80: 7020–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z., Kuritzkes D. R. (2014) ‘Altered Viral Fitness and Drug Susceptibility in HIV-1 Carrying Mutations that Confer Resistance to Nonnucleoside Reverse Transcriptase and Integrase Strand Transfer Inhibitors’, Journal of Virology, 88: 9268–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives J. A. L. et al. (2002) ‘The H274Y Mutation in the Influenza A/H1N1 Neuraminidase Active Site Following Oseltamivir Phosphate Treatment Leave Virus Severely Compromised Both In Vitro and In Vivo’, Antiviral Research, 55: 307–17 [DOI] [PubMed] [Google Scholar]
- Jabara C. B. et al. (2014) ‘Hepatitis C Virus (HCV) NS3 Sequence Diversity and Antiviral Resistance-Associated Variant Frequency in HCV/HIV Coinfection’, Antimicrobial Agents and Chemotherapy, 58: 6079–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James S. H., Prichard M. N. (2014) ‘Current and Future Therapies for Herpes Simplex Virus Infections: Mechanism of Action and Drug Resistance’, Current Opinion in Virology, 8: 54–61. [DOI] [PubMed] [Google Scholar]
- Jensen J. D. et al. (2007) ‘Approaches for Identifying Targets of Positive Selection’, Trends in Genetics, 23: 568–77 [DOI] [PubMed] [Google Scholar]
- Jiang M. et al. (2013) ‘In Vitro Phenotypic Characterization of Hepatitis C Virus N23 Protease Variants Observed in Clinical Studies of Telaprevir’, Antimicrobial Agents and Chemotherapy, 57: 6236–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khudyakov Y. (2010) ‘Coevolution and HBV Drug Resistance’, Antiviral Therapy, 15: 505–15 [DOI] [PubMed] [Google Scholar]
- Kobayashi M. et al. (2008) ‘Selection of Diverse and Clinically Relevant Integrase Inhibitor-Resistant Human Immunodeficiency Virus Type 1 Mutants’, Antiviral Research, 80: 213–22 [DOI] [PubMed] [Google Scholar]
- Lipsitch M. et al. (2012) ‘Evolution, Safely, and Highly Pathogenic Influenza Viruses’, Science, 336: 1529–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locarnini S. et al. (2004) ‘Management of Antiviral Resistance in Patients with Chronic Hepatitis B’, Antiviral Therapy, 9: 679–93 [PubMed] [Google Scholar]
- Lourenço J. M. et al. (2013) ‘The Rate of Molecular Adpatation in a Changing Environment’, Molecular Biology and Evolution, 30: 1292–301 [DOI] [PubMed] [Google Scholar]
- Lurain N. S., Chou S. (2010) ‘Antiviral Drug Resistance of Human Cytomegalovirus’, Clinical Microbiology Reviews, 23: 689–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisnier-Patin S., Andersson D. I. (2004) ‘Adaptation to the Deleterious Effects of Antimicrobial Drug Resistance Mutations by Compensatory Evolution’, Research in Microbiology, 155: 360–9 [DOI] [PubMed] [Google Scholar]
- Mansky L. M, Temin H. M. (1995) ‘Lower In Vivo Mutationr Ate of Human Inmmunodeficiency Virus Type 1 than that Predicted from the Fidelity of Purified Reverse Transcriptase’, Journal of Virology, 69: 5087–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. et al. (2010) ‘Opposite Effect of Two Cytomegalovirus DNA Polymerase Mutations on Replicative Capacity and Polymerase Activity’, Antiviral Therapy, 15: 579–86 [DOI] [PubMed] [Google Scholar]
- Menéndez-Arias L. (2013) ‘Molecular Basis of Human Immunodeficiency Virus Type 1 Drug Resistance: Overview and Recent Developments’, Antiviral Research, 98: 93–120 [DOI] [PubMed] [Google Scholar]
- Mesplède T. et al. (2013) ‘Viral Fitness Cost Prevents HIV-1 from Evading Dolutegravir Drug Pressure’, Retrovirology, 10: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura S. et al. (2013) ‘Random Fluctuation of Selection Coefficients and the Extent of Nucleotide Variation in Human Populations’, Proceedings of the National Academy of Sciences of the United States of America, 110: 10676–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel V. et al. (2011) ‘Genetic Recombination of the Hepatitis C Virus: Clinical Implications’, Journal of Viral Hepatitis, 18: 22–83 [DOI] [PubMed] [Google Scholar]
- Moscona A. (2005) ‘Oseltamivir Resistance - Disabling our Influenza Defenses’, The New England Journal of Medicine, 353: 2633–6 [DOI] [PubMed] [Google Scholar]
- Moscona A. (2009) ‘Global Transmission of Oseltamivir-Resistant Influenza’, The New England Journal of Medicine, 360: 953–6. [DOI] [PubMed] [Google Scholar]
- Moya A. et al. (2004) ‘The Population Genetics and Evolutionary Epidemiology of RNA Viruses’, Nature Reviews Microbiology, 2: 279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H. J. (1964) ‘The Relation of Recombination to Mutational Advance’, Mutation Research, 1: 2–9. [DOI] [PubMed] [Google Scholar]
- Mullins J. I. et al. (2011) ‘Mutation of HIV-1 Genomes in a Clinical Population Treated with the Mutagenic Nucleoside KP1461’, PLoS One, 6: e15135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeger L. K. et al. (2001) ‘Increased Drug Susceptibility of HIV-1 Reverse Transcriptase Mutants Containing M194V and Zidovudine-Associated Mutations: Analysis of Enzyme Processivity, Chain-Terminator Removal and Viral Replication’, Antiviral Therapy, 6: 115–27 [PubMed] [Google Scholar]
- Neher R. A, Hallatschek O. (2013) ‘Genealogies of Rapidly Adapting Populations’, Proceedings of the National Academy of Sciences, 110: 437–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher R. A, Leitner T. (2010) ‘Recombination Rate and Selection Strength in HIV-Intrapatient Evoltuion’, PLoS Computaitonal Biology, 6: e1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman R. M. et al. (2013) ‘Whole Genome Pyrosequencing of Rare Hepatitis C Virus Genotypes Enhances Subtype Classification and Identification of Naturally Occurring Drug Resistance Variants’, The Journal of Infectious Diseases, 208: 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. T. et al. (2012) ‘Neuraminidase Inhibitor Resistance in Influenza Viruses and Laboratory Testing Methods’, Antiviral Therapy, 17: 159–73 [DOI] [PubMed] [Google Scholar]
- Nielsen R. (2005) ‘Molecular Signatures of Natural Selection’, Annual Reviews in Genetics, 39: 197–218 [DOI] [PubMed] [Google Scholar]
- Nobusawa E, Sato K. (2006) ‘Comparison of the Mutation Rates of Human Influenza A and B Viruses’, Journal of Virology, 80: 3675–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora T. et al. (2007) ‘Contribution of Recombination to the Evolution of Human Immunodeficiency Virus Expressing Resistance to Antiretroviral Treatment’, Journal of Virology, 81: 7620–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly M. D., Lauring A. S. (2015) ‘Effective Lethal Mutagenesis of Influenza Virus by Three Nucleoside Analogs’, Journal of Virology, 89: 3584–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings P. S. et al. (2014) ‘Loss and Recovery of Genetic Diversity in Adapting Populations of HIV’, PLoS Genetics, 10: e1004000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings P. S. (2012) ‘Standing Genetic Variation and The Evolution of Drug Resistance in HIV’, PLoS Computaitonal Biology, 8: e1002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perno C. et al. (2008) ‘Overcoming Resistance to Existing Therapies in HIV-Infected Patients: the Role of New Antiretroviral Drugs’, Journal of Medical Virology, 80: 565–76 [DOI] [PubMed] [Google Scholar]
- Pfeiffer J. K., Kirkegaard K. (2003) ‘A Single Mutation in Poliovirus RNA-Dependent RNA Plymerase Confers Resistance to Mutagenic Nucleotide Analogs Via Increased Fidelity’, Proceedings of the National Academy of Sciences of the United States of America, 100: 7289–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret J., Boivin G. (2014) ‘Antiviral Drug Resistance in Herpesviruses Other Than Cytomegalovirus’, Reviews in Medical Virology, 24: 186–218 [DOI] [PubMed] [Google Scholar]
- Poland G. A. et al. (2009) ‘Influenza Virus Resistance to Antiviral Agents: A Plea for Rational Use’, Clinical Infectious Diseases, 48: 1254–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powdrill M. H. et al. (2011) ‘Contribution of a Mutational Bias in Hepatitis C Virus Replication to the Genetic Barrier in the Development of Drug Resistance’, Proceedings of the National Academy of Sciences, 108: 20509–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S. et al. (2014) ‘Recent population expansions of hepatitis B virus in the United States’, Journal of Virology, 88: 13971–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2008) ‘The Genomic and Epidemiological Dynamics of Human Influenza A Virus’, Nature, 453: 615–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N. et al. (2011) ‘Extensive Genome-Wide Variability of Human Cytomegalovirus in Congenitally Infected Infants’, PLoS Pathogens, 7: e1001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N. et al. (2013) ‘Rapid Intrahost Evolution of Human Cytomegalovirus is Shaped by Demography and Positive Selection’, PLoS Genetics, 9: e1003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N. et al. (2014) ‘Evolution of the Influenza A Virus Genome During Development of Oseltamivir Resisitance In Vitro’, Journal of Virology, 88: 272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N. et al. (2015) ‘Limits and Patterns of Cytomegalovirus Genomic Diversity in Humans’, Proceedings of the National Academy of Sciences, 112: E4120–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong L. et al. (2007) ‘Emergence of HIV-1 Drug Resistance During Antiretroviral Treatment’, Bulletin of Mathematical Biology, 69: 2027–60 [DOI] [PubMed] [Google Scholar]
- Sakaoka H. et al. (1994) ‘Quantitative Analysis of Genomic Polymorphism of Herpes Simplex Virus Type 1 Strains From Six Countries: Studies of Molecular Evolution and Molecular Epidemiology of the Virus’, Journal of General Virology, 75: 513–27 [DOI] [PubMed] [Google Scholar]
- Sanjuán R. et al. (2004) ‘The Distribution of Fitness Effects Caused by Single-Nucleotide Substitutions in an RNA Virus’, Proceedings of the National Academy of Sciences, 201: 8396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlub T. E. et al. (2010) ‘Accurately Measuring Recombination Between Closely Related HIV-1 Genomes’, PLoS Computaitonal Biology, 6: e1000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A. K. et al. (2003) ‘Association Between Adherence to Antiretroviral Therapy and Human Immunodeficiency Virus Drug Resistance’, Clinical Infectious Diseases, 37: 1112–8 [DOI] [PubMed] [Google Scholar]
- Shaw T. et al. (2006) ‘HBV Drug Resistance: Mechanisms, Detection and Interpretation’, Journal of Hepatology, 44: 593–606 [DOI] [PubMed] [Google Scholar]
- Shiino T. et al. (2010) ‘Moleuclar Evoluationary Analysis of the Influenza A(H1N1)pdm, May-September 2009: Temporal and Spatial Spreading Profile of the Viruses in Japan’, PLoS One, 5: e11057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakami T. et al. (2011) ‘Protease Inhibitor-Resistant Hepatitis C Virus Mutants with Reduced Fitness Forms Impaired Production of Infectious Virus’, Gastroenterology, 140: 667–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. (2004) ‘Genetic Diversity and Evolution of Hepatitis C Virus - 15 Years on’, Journal of General Virology, 85: 3173–88. [DOI] [PubMed] [Google Scholar]
- Springer K. L. et al. (2005) ‘How Evolution of Mutations Conferring Drug Resistance Affects Viral Dynamics and Clinical Outcomes of Cytomegalovirus-Infected Hematopoietic Cell Transplant recipients’, Journal of Clinical Microbiology, 43: 208–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M. M., Valckenborgh F. (2011) ‘Escapting an Evolutionary Lobster Trap: Drug Resistance and Compensatory Mutation in a Fluctuating Environment’, Evolution, 65: 1376–87 [DOI] [PubMed] [Google Scholar]
- Tellier A, Lemaire C. (2014) ‘Coalescence 2.0: A Multiple Branching of Recent Theoretical Developments and Their Applications’, Molecular Ecology, 23: 2637–52 [DOI] [PubMed] [Google Scholar]
- Tenaillon O. (2014) ‘The Utility of Fisher’s Geometric Model in Evolutionary Genetics’, Annual Reviews in Ecology, Evolution, and Systematics, 45: 179–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorlund K. et al. (2011) ‘Systematic Review of Influena Resistance to the Neuraminidase Inhibitors’, BMC Infectious Diseases, 11: 134–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. et al. (2011) ‘Evolution of Drug-Resistant Viral Populations During Interruption of Antiretroviral Therapy’, Journal of Virology, 85: 6403–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensing A. M. et al. (2014) ‘2014 Update of the Drug Resistance Mutations in HIV-1’, Topics in Antiviral Medicine, 22: 642–50 [PMC free article] [PubMed] [Google Scholar]
- Wong D. D. Y. et al. (2012) ‘Comparable Fitness and Transmissibility Between Oseltamivir-Resistant Pandemic 2009 and Seasonal H1N1 Influenza Viruses with the H275Y Neuraminidase Mutation’, Journal of Virology, 86: 10558–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyles D. L. (2013) ‘Antiviral Resistance and the Future Landscape of Hepatitis C Virus Infection Therapy. The Journal of Infectious Diseases, 207: S33–9 [DOI] [PubMed] [Google Scholar]
- Zhang K. (2013) ‘Comprehensive Analysis of Hepatitis B Virus Antiviral Drug Resistance Mutations Based on a New High-Throughput Phenotypic Assay’, PhD thesis, Technical University of Munich
- Zhou Y, Holmes E. C. (2007) ‘Bayesian Estimates of the Evolutionary Rate and Age of Hepatitis B Virus’, Journal of Molecular Evolution, 65: 197–205 [DOI] [PubMed] [Google Scholar]
- Zoulim F, Locarnini S. (2009) ‘Reviews in Basic and Clinical Gastroenterology’, Gastroenterology, 137: 1593–608 [DOI] [PubMed] [Google Scholar]
- zur Wiesch P. et al. (2010) ‘Compensation of Fitness Costs and Reversibility of Antibiotic Resistance Mutations’, Antimicrobial Agents and Chemotherapy, 54: 2085–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Wiesch P. et al. (2011) ‘Population Biological Principles of Drug-Resistance Evolution in Infectious Diseases’, The Lancet Infections Diseases, 11: 236–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.