


Abstract
Transcription can be quite disruptive for chromatin so cells have evolved mechanisms to preserve chromatin integrity during transcription, thereby preventing the emergence of cryptic transcripts from spurious promoter sequences. How these transcripts are regulated and processed remains poorly characterized. Notably, very little is known about the termination of cryptic transcripts. Here, we used RNA-Seq to identify and characterize cryptic transcripts in Spt6 mutant cells (spt6-1004) in Saccharomyces cerevisiae. We found polyadenylated cryptic transcripts running both sense and antisense relative to genes in this mutant. Cryptic promoters were enriched for TATA boxes, suggesting that the underlying DNA sequence defines the location of cryptic promoters. While intragenic sense cryptic transcripts terminate at the terminator of the genes that host them, we found that antisense cryptic transcripts preferentially terminate near the 3΄-end of the upstream gene. This finding led us to demonstrate that most terminators in yeast are bidirectional, leading to termination and polyadenylation of transcripts coming from both directions. We propose that S. cerevisiae has evolved this mechanism in order to prevent/attenuate spurious transcription from invading neighbouring genes, a feature that is particularly critical for organisms with small compact genomes.
INTRODUCTION
Transcription initiation occurs at promoters, which in eukaryotes are defined by small DNA motifs that direct the assembly of a preinitiation complex containing an RNA polymerase and its associated factors (1). In recent years, it has become evident that, in addition to DNA sequence, chromatin structure plays critical roles in defining promoter regions. In Saccharomyces cerevisiae, where chromatin structure has been studied extensively, promoters are characterized by a nucleosome-free region flanked by well positioned nucleosomes carrying specific histone post-translational modifications and the histone variant H2A.Z (2). Outside promoters, different epigenetic signatures decorate nucleosomes, allowing for the prediction of regulatory regions based epigenetic signatures (3). These chromatin states are highly dynamic, notably during transcription elongation where histone chaperones, histone acetyltransferases, histone deacetylases, histone methyltransferases and chromatin remodelers coordinate with RNA polymerase II (RNAPII) to maintain proper chromatin structure and epigenetic information over genes (4,5). Tampering with these chromatin modifying enzymes leads to the emergence of cryptic transcription, which is initiated within genes (6). While this phenomenon is best described in yeast, recent studies suggest that cryptic transcription initiated within genes also occurs in cancer cells (7,8). Maintaining proper chromatin structure and epigenetic state during transcription is therefore critical for transcription initiation fidelity and may play direct role in human diseases.
The first evidence that histone chaperones are important for preventing intragenic cryptic transcription came from the Winston group (9). In the course of confirming microarray data by northern blots, Kaplan et al. made the surprising finding that a mutant for the transcription-associated histone chaperone Spt6 expresses short transcripts, initiated from within gene bodies (9). This was accompanied with hypersensitivity to nuclease, leading them to propose that Spt6 prevents cryptic transcription by maintaining proper chromatin structure during elongation (9). Subsequently, similar phenotypes were shown in mutants for other histone chaperones, for genes involved in proper expression of histone genes, for histone methyltrasferases, for demethylases and for chromatin remodelers (9–22). In essence, any mutant that contributes to making gene body chromatin looking like promoter chromatin may contribute to the emergence of intragenic cryptic transcription. This includes mutants that cause nucleosome loss (9), H2A.Z mislocalization (23,24), histone acetylation (12) and increased histone turnover (19).
While many of the factors involved in repressing cryptic transcription and their associated mechanisms are known, the repertoire of cryptic transcripts emerging in these mutants remains ill-defined. In addition, how these transcripts are terminated and processed has to our knowledge never been formally investigated. Here, we addressed these questions by RNA-Seq profiling of spt6-1004 mutant cells. Interestingly, we found that antisense cryptic transcription often terminates at the terminator of the adjacent gene, thanks to the previously underestimated bidirectionality of most yeast terminators.
MATERIALS AND METHODS
RNA-Seq
Cells from spt16-1004 and its respective wild type (WT) strain were grown to an OD600 of 0.5 at 30°C, shifted to 37°C for 80 min and harvested. Total RNA was extracted using the hot phenol method. Prior to library preparation, total RNA was either depleted for ribosomal RNA using the Ribo-zero Gold yeast kit (Epicentre-Illumina) or enriched for polyadenylated RNA using the NEBnext Poly(A) kit (New England Biolabs). Strand-specific RNA-Seq libraries were prepared using the KAPA stranded RNA-Seq library preparation kit (KAPA Biosystems) prior to paired-end sequencing on an Illumina Hi-Seq2000. Reads were mapped to the sacCer3 assembly of the S. cerevisiae genome using TopHat2 (25). Intron length range was set at 50–1000 bp and a reference annotation file was provided to guide the assembly. The number (between 10 million and 19 million) and percentage (between 90% and 99%) of mapped reads for each sample are listed in Supplementary Table S1. The replicates were highly correlated with Pearson correlation factor of 0.999 (WT biological duplicates) and 0.997 (spt6-1004 biological duplicates).
Identification of intragenic sense cryptic transcripts
Sense cryptic transcripts were detected from RNA-Seq data using a probabilistic method we developed and is embedded in the R package yCrypticRNAs available at (https://cran.r-project.org/web/packages/yCrypticRNAs/index.html). For each position of a gene, the cumulative RNA-Seq signal was calculated by summing the number of reads/fragments between the given position and the previous position, starting at the 5΄ end, in the WT and mutant samples. The cumulative values from the mutant were then subtracted from those of the WT. The resulting differential cumulative values were then used to calculate, for each position of the gene, the perpendicular distance (f value) between the cumulative values and a diagonal linking the first and last data points. The f score for a gene was then obtained by taking the maximum f value minus the minimum f value. In principle, a high f value should correlate with the presence of a cryptic transcript as it indicates the presence of excess RNA-Seq reads in the 3΄ end of the gene in the mutant compared to the WT. The f value, however, is also influenced by the expression level and the length of the gene. In order to eliminate these biases and assess the significance of f scores, the RNA-Seq values over the assessed genes were randomly permutated multiple times (10 000 permutations) and the f score re-calculated after each permutation. The resulting f score distribution was used to calculate a z score estimating the probability that cryptic transcription was initiated somewhere within the tested genes. In the current work, the z scores were calculated using values from spt6-1004 and WT cells for which values from replicates were merged together. As a control, we calculated the z score for each gene comparing the replicates in mutant (spt6-1004rep1/spt6-1004rep2) and WT (WTrep1/WTrep2) cells. Using the z scores obtained when comparing replicates, we determined a cutoff by allowing 1% false discovery. This enabled the identification of 1703 sense cryptic transcripts in spt6-1004 cells (See Supplementary Table S2).
For genes identified as harbouring a sense cryptic transcript based on the above method, we then determined the position of the cryptic transcription start sites (cTSS) as follow. For each position of a gene, an f value was calculated as described above. The position where the maximum f (f max) value is reached represents the position where the cryptic transcript is initiated (cTSS). The exact position of the f max, however, is influenced by local noise in the RNA-Seq data. In order to identify the position of cTSS in a probabilistic manner, the data were sampled with replacement (bootstrapped) multiple times to calculate the distribution of f max and its position. Here, 200 iterations were used, each time removing 10% of the data. This allowed for the identification of a cryptic zone, a region within the gene where a cryptic transcript is likely to have initiated. In the current implementation of our method, the cryptic zone was determined using the mean and standard deviation of all the positions for which the simulated f value was within the bootstrapped distribution. We identified a total of 1640 cryptic zones ranging in size between 4 bp and 1046 bp with a median size of 138 bp and an average size of 162 bp.
Identification of intragenic antisense cryptic transcripts
Cryptic transcripts running on the antisense strand were detected using StringTie (26). The minimum assembled transcript length was set to 100 (-m 100), the minimum reads per bp coverage to consider for transcript assembly was set to 2 (-c 2), the gap between read mappings triggering a new bundle was set to 5 (-g 5) and no reference annotation file was provided for guiding the assembly process. We next removed all the transcripts that overlapped with known annotations on the same strand to keep de novo antisense cryptic transcripts. The fragments per kilobase per million mapped reads (FPKM) for each antisense cryptic transcript was calculated in WT and in spt6-1004 cells and antisense transcripts having at least four FPKM and a log2 fold change of at least 1.25 were kept for further analysis. We identified 1616 antisense cryptic transcripts, overlapping 1491 genes, in spt6-1004 cells using this approach (see Supplementary Table S3).
Promoter sequence analyses
The yeast genome was scanned for the TATA box consensus sequence in S. cerevisiae (TATAWAWR) and a very close sequence with only one mismatch (TATATAAT) using HOMER (27). A score of 1 was set to each position where a motif was found. The resulting scores were used to look at the sequence distribution around canonical and cryptic promoters using VAP (28,29).
Terminator motif analyses
The yeast genome was scanned for the ‘efficiency’ (UAUAUA, UACAUA, UAUGUA) and ‘positioning/A-rich’ (AAUAAA, AAAAAA) motifs as described in (30). Because these A/T-rich motifs are frequently found in the genome but require a stereotypical organization to be functional (30), we required each positioning motif to be within 50 bp upstream of a positioning motif and vice-versa. A score of 1 was set to each position where motifs were found. The resulting scores were used to look at the motif distribution relative to genes on both sense and antisense strands using VAP (28,29).
Identification of bidirectional or unidirectional terminators
Gene terminators that are challenged by a cryptic transcript were classified as unidirectional or bidirectional as follows. Unidirectional terminators are defined as terminators that allow termination of their corresponding sense transcripts but are inefficient at terminating antisense transcription. Bidirectional terminators are defined as terminators which allow both sense and antisense transcription termination. Terminators were classified as bidirectional if they allow termination of antisense transcription within a 500 bp window around their annotated transcription termination sites (TTS). This means that an antisense transcript terminating 250 bp before or after a gene's TTS was considered to be terminated at that terminator. This method identified 579 bidirectional terminators. However, we noticed many antisense transcripts for which the RNA-Seq signal drastically decreased, but did not completely disappear, around TTS. These are indication of terminators that are bidirectional but with weaker activity. To identify those weak bidirectional TTS, we used a probabilistic method similar to that used to identify cryptic transcripts. For each gene, we calculated the probability of a termination event in the –250 and +250 bp region around their TTS on the antisense strand, using both the f score and the z score. We found that genes with an antisense cryptic transcript terminating in their 3΄-end tend to have very low f and z scores. We thus selected bidirectional promoters having f and z scores values smaller than –94.47 and –11.35 respectively. These cutoffs allowed 15% and 5% false discovery respectively, based on WT replicates. Using this approach, we identified 97 weak bidirectional terminators. Unidirectional terminators were identified by selecting TTS that overlap an antisense cryptic transcript, but are located at least 250 bp away from the cryptic TTS. We found 150 unidirectional terminators using this criterion. See Supplementary Table S4 for the directionality call for each terminator evaluated.
Termination assays
Putative terminator sequences were tested using a modified version of the assay developed by Carroll et al. to test snoRNA terminators (31). In this system, the HIS3 gene is expressed from a plasmid under the control of the ADH1 promoter and putative terminators are cloned between the promoter and the HIS3 gene. This allows termination to be monitored by growth on plates lacking histidine. To adapt this assay to protein-coding gene terminators, we inserted the 5΄ UTR (627 bp) from the YNR051C gene between the promoter and the HIS3 open reading frame. This creates space between the promoter and the cloning site for the putative terminators, therefore mimicking the promoter-terminator organization found in typical yeast protein-coding genes. Putative terminators (500 bp fragments) were then cloned downstream of this 5΄ UTR and termination was monitored by northern blotting using a single stranded RNA probe corresponding to the YNR051C 5΄ UTR. Northern blotting was used instead of growth on histidine minus plates since the terminators tested often contain ATG codons. Northern blots were performed using fluorescent probes as described previously (24).
RESULTS
A probabilistic method for the identification of intragenic sense cryptic transcripts from RNA-Seq
The genome-wide identification of intragenic sense cryptic transcripts is not trivial since these transcripts are embedded within genes. Previous studies have identified genes hosting cryptic transcripts by looking for genes with excessive signal in the 3΄ portion of the gene using either tiling arrays or RNA-Seq (10,32). This approach suffers from the fact that RNA-Seq signal can be quite ‘wavy’, leading to the introduction of randomness in the 3΄/5΄ ratio measurements. More recently, DeGennaro et al. looked at the cumulative RNA-Seq signal starting from the 3΄-end to identify genes with excessive signal in the 3΄-end in Schizosaccharomyces pombe spt6-1 cells relative to WT (33). This method generates fewer false positives than the 3΄/5΄ ratio method but suffers from a lack of sensitivity (see Supplementary Figure S1 and below). In addition, neither of these methods allows for the mapping of the 5΄-end of cryptic transcripts (i.e. cryptic promoters) as they only predict which genes are hosting a cryptic transcript.
To address these limitations, we developed a probabilistic method for the identification of intragenic cryptic transcripts from RNA-Seq experiments (Figure 1A). Briefly, for each position of a gene, the cumulative RNA-Seq signal is calculated by summing the number of reads per fragment between the given position and the previous position (from 5΄ to 3΄). The cumulative values from the WT are subtracted from those of the mutant and the perpendicular distance (f value) between the differential cumulative values and a diagonal linking the first and last data points is calculated. The f score for a gene is then obtained by taking the maximum f value minus the minimum f value. High f values correlate with the presence of a cryptic transcript as it indicates the presence of excess RNA-Seq reads in the 3΄-end of the gene in the mutant compared to the WT. In order to add a statistical score to the method, and to remove biases from gene length and gene expression levels, which can both impact the f score, the RNA-Seq values are randomly permutated several times and the f score re-calculated after each permutation. Those simulated f scores are used to calculate a z score representing the distance between the observed f score and the distribution of the permutated f scores. The z score represents the probability that cryptic transcription is initiated somewhere within the tested genes. In addition, the method allows for the identification of the position where the cryptic transcript is initiated by considering the position where the maximum f value (f max) is reached (Figure 1B). This f max value is computed several times by re-sampled the data (each time randomly eliminating a fraction of the values) allowing for the identification of a ‘cryptic zone’, a region within the gene where a cryptic transcript is likely to have initiated, as well as the most likely cTSS coordinate (see Material and Methods for more details). The method is embedded with the open-source R package yCrypticRNAs available at (https://cran.r-project.org/web/packages/yCrypticRNAs/index.html).
Figure 1.

Identification of sense and antisense cryptic transcripts in S. cerevisiae. (A) Schematic representation of the probabilistic method for the identification of genes harboring sense cryptic transcripts. (B) Schematic representation of the probabilistic method for the identification of the cryptic zone and the TSS (left) and the RNA-Seq signal over the CTF4 gene as an example (right). The f max is calculated 200 times, each time by deleting 10% of the data. This allows the determination of a cryptic zone (yellow), a region within the gene where a cryptic transcript is likely to have initiated. The figure also shows the most likely position of the cTSS (green). (C) Workflow of how antisense cryptic transcripts (aCT) were identified in this study.
In order to benchmark our method relative to previously published ones, we implemented the 3΄/5΄ ratio method developed by Cheung et al. (10), as well as the 3΄ enrichment method developed by DeGennaro et al. (33) and applied both methods, together with our probabilistic method, to our spt6-1004 RNA-Seq data. Note that the ‘transition point’ algorithm developed by Lickwar et al. (34) was not included in our analysis since the source code was not available. All methods successfully identified a set of 11 previously validated cryptic transcripts (except for the VPS72 gene which was not identified using the 3΄ enrichment method) (9,10) (Supplementary Table S5). Compared to the number of genes identified by the probabilistic method (1760 genes), the 3΄ enrichment method identified fewer genes (446 genes) while the 3΄/5΄ ratio method identified a larger number of genes (2151 genes) (Supplementary Figure S1A). To evaluate the accuracy and sensitivity of these methods, 200 genes were randomly selected from those identified by either method (also including negative controls). The gene list was randomized and submitted to three lab members who visually inspected the RNA-Seq data on a genome browser. For each of the 200 genes, each curator had to determine independently whether they considered the gene to contain a cryptic transcript or not (Supplementary Figure S1B). As expected, a combination of any two methods gives the best prediction with >70% of the identified genes being considered positives by the curators. When considering genes identified exclusively by one method, however, the probabilistic method outperformed the other two, with fewer false positives and better sensitivity. Thus, the probabilistic method provides a good compromise between false positives and false negatives.
Pervasive sense and antisense transcription in spt6-1004
Applying the method described above to RNA-Seq experiments generated from polyadenylated-enriched RNA preparations identified 1703 intragenic sense cryptic transcripts in spt6-1004 cells (with a false discovery rate of 1%; Figure 2A). Importantly, this list largely overlaps with the list of genes previously published by Cheung et al. (10) (Supplementary Figure S2) and includes all the previously confirmed cryptic transcripts for this mutant (9,10), namely FLO8, RAD18, SPB4, STE11, VPS72, APM2, DDC1, SYF1, OMS1, PUS4 and CHS6 (Figure 2A, red dots and Supplementary Table S5).
Figure 2.

Sense and antisense cryptic transcription in spt6-1004 cells. (A) Density of z scores obtained by comparing biological replicates as a control (black and gray) or spt6-1004 to WT cells (blue). Values for genes previously confirmed to have a cryptic transcript (CT) are represented by the red dots. n = 5695 genes, which exclude dubious and overlapping genes on the same strand. (B) Expression of transcripts in spt6-1004 cells for three groups of genes: All 5,695 annotated ORFs (ORFs); 642 sense cryptic transcripts (sCT); 1616 antisense cryptic transcripts (aCT). (C) Venn diagram comparing the number of genes having intragenic sense cryptic transcripts and overlapping with antisense cryptic transcripts. Expected-by-chance numbers are in parenthesis. (D) Aggregate profile of the starting positions of antisense cryptic transcripts over sense cryptic transcripts. (E) Aggregate profiles of TATAWAWR motif enrichment around canonical (grey), sense cryptic (blue) and antisense cryptic (gold) promoters. The dotted lines represent the enrichment of TATATAAT motif as a control.
Contrary to intragenic sense cryptic transcripts, antisense cryptic transcripts are more easily identified since very little signal is detected on the antisense strand of genes in WT cells. We therefore used a standard assembler (26) to identify de novo transcripts overlapping annotated genes on the antisense strand (Figure 1C). Using this approach, we identified 1616 antisense cryptic transcripts, overlapping 1491 genes, in spt6-1004 cells. This number is most likely an underestimation of the full set of antisense cryptic transcripts because our algorithm (as would be the case for any RNA-Seq-based method) is not efficient at identifying antisense cryptic transcripts that invade the neighboring gene on the same strand (due to merging of the RNA-Seq signal). Tampering with Spt6 therefore leads to widespread cryptic transcription running both sense and antisense relative to genes, consistent with previous studies in budding (10,11) and fission yeasts (33). Interestingly, antisense cryptic transcripts are globally expressed at lower levels than those on the sense strand (Figure 2B). It is not clear whether this is due to differences in transcription or RNA stability, although cryptic transcripts from both strands appear to be polyadenylated (see below). Sense and antisense transcripts emerge from the same gene slightly more often than expected by chance but we often detect genes with a cryptic transcript running only in one direction (Figure 2C). Mapping the 5΄ ends of antisense cryptic transcripts relative to the position of the 5΄ ends of the sense cryptic transcripts revealed that both sense and antisense cryptic transcripts tend to initiate from the same region within a given gene (Figure 2D). Also noteworthy, the expression levels (Supplementary Figure S3A) and the fold change (Supplementary Figure S3B; compared to WT cells) between the sense and antisense cryptic transcript are not correlated when they occur on the same gene. Finally, the level of sense cryptic transcripts generally correlates with the expression level of the gene that hosts them (Supplementary Figure S3C), corroborating previous observations (10). This is not the case for antisense cryptic transcripts, however (Supplementary Figure S3C).
Cryptic promoters are enriched for TATA box sequences
We next asked whether cryptic promoters share sequence attributes with canonical promoters. The best characterized core promoter motif is the TATA box. We therefore mapped the occurrence of the TATAWAWR sequence around predicted sense and antisense cryptic promoters. The motif was shown enriched 50–100 bp upstream of cTSS, a pattern similar to what is observed at the transcription start site (TSS) of annotated genes (Figure 2E). A control motif with a single mismatch did not show any enrichment, demonstrating the specificity of the signal. TATA box enrichment, however, was lower at cTSS than at the TSS of annotated genes, suggesting that cryptic promoters may preferentially use different types of promoter elements. Alternatively, this may reflect imperfect mapping of some cTSS, especially in the sense direction. We also performed de novo motif searches but these analyses failed to identify motifs other than TATA elements (data not shown). The presence of TATA elements within cryptic promoters suggests that these promoters may be regulated by the same mechanism as their canonical counterparts. This result is consistent with the fact that genes with an intragenic TATA box were shown to be three times more likely to express a sense cryptic transcript in spt6-1004 cells (10) and with mutational analyses which demonstrated that a cryptic transcript within the FLO8 gene requires an intragenic TATA box (9). Altogether, our data suggest that the underlying DNA sequence, such as the presence of TATA box motifs, contributes to the location of the cryptic promoters, notwithstanding the fact that nucleosome depletion likely drives their usage in spt6-1004 cells (9).
Antisense cryptic transcripts terminate at the 3΄-end of the adjacent gene via the polyadenylation-dependent termination pathway
Intragenic sense cryptic transcripts naturally terminate at the terminator of the gene hosting them, but where does antisense cryptic transcription terminate? Visual inspection of the data revealed that antisense cryptic transcripts usually run pass the promoter and invade the upstream intergenic region. For technical reasons (see above), very few antisense cryptic transcripts were identified that invade the upstream gene on the same strand (opposite strand to the gene where the antisense cryptic transcript has initiated), making it difficult to evaluate where these transcripts terminate. We surmise, however, that these transcripts would terminate at the terminator of the upstream gene. In cases where the upstream gene is in the same orientation as the gene where the antisense cryptic transcript has emerged, however, we noticed that the antisense cryptic transcripts terminate near the 3΄-end of the upstream gene (see Figure 3A for an example). In order to systematically test this unexpected observation, we mapped the 5΄ and 3΄-end of antisense cryptic transcripts relative to annotated genes. Only genes with an upstream neighbor in the divergent orientation and a downstream neighbor in the tandem orientation were used in order to limit signal from neighboring genes. Interestingly, while antisense cryptic transcripts initiate at random positions (Figure 3B; dashed trace), this analysis revealed that these transcripts tend to terminate in the 3΄ region of the gene (Figure 3B; solid trace). These regions contain terminators, but on the opposite strand. This prompted us to investigate whether known DNA motifs involved in polyadenylation-dependent termination were present on both strands in terminator regions. In silico and experimental analyses have defined yeast terminators as an array of motifs often referred to as the ‘efficiency’, ‘positioning/A-rich’, ‘near upstream/U-rich’, ‘polyadenylation site’ and ‘near downstream/U-rich’ motifs ((30) and references therein). In order to look for evidence of terminators on the antisense strand, we mapped the density of the ‘efficiency’ and ‘positioning’ motifs (the other motifs having poor information content on their own), relative to genes, on both strands. We only considered motifs occurring in the correct arrangement (‘efficiency’ being always upstream of ‘positioning’). As expected, when looking on the sense strand, we found enrichment for both motifs at the 3΄-end of annotated genes (Figure 3C and D, blue). Surprisingly, however, both motifs were also enriched on the antisense strand (Figure 3C and D, gold). The position of these termination motifs is consistent with antisense cryptic transcript coming from the downstream gene being terminated at these sites via the polyadenylation-dependent termination pathway.
Figure 3.
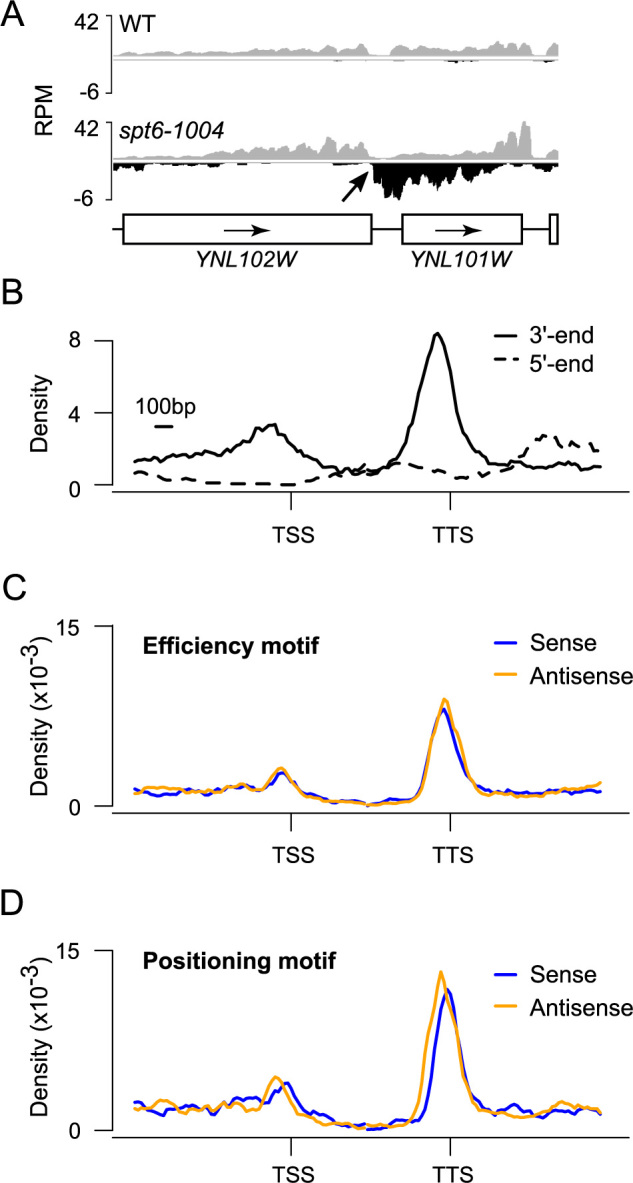
Antisense cryptic transcripts are polyadenylated and tend to terminate at the 3΄-end of adjacent genes. (A) A genome-browser snapshot illustrating a terminator (YNL102W) efficiently terminating an antisense cryptic transcript. RNA-Seq signal on the Watson (gray) and Crick (black) strands is shown. (B) Aggregate profile of antisense starting (dashed trace) and ending (solid trace) positions over genes. (C) Aggregate profile of the efficiency motif (UAUAUA, UACAUA, UAUGUA) enrichment on the sense (blue) and antisense (gold) strands over genes. (D) Aggregate profile of the positioning motif (AAUAAA, AAAAAA) enrichment on the sense (blue) and antisense (gold) strands over genes. For panels ‘B’, ‘C’ and ‘D’ the analyses were done over 1408 yeast genes that are oriented in a divergent, tandem manner (← → →) such that the gene upstream of the TSS is transcribed right to left, while the gene to the right of the TTS is transcribed left to right. This insures that terminators of adjacent genes are not adjacent to the gene of interest.
Our RNA-Seq experiments were performed on polyadenylated-enriched RNA, suggesting that antisense cryptic transcripts are indeed polyadenylated. We noticed, however, that the level of antisense cryptic transcripts is on average 1.6 (mean) fold less abundant than those on the sense strand (see Figure 2B), suggesting that they may be terminating via another pathway leading to less stable transcripts. In order to address this issue, we repeated the RNA-Seq experiments using ribosomal RNA depletion and polyadenylated-enrichment in parallel starting from the same total RNA preparations. Quite strikingly, the expression level of antisense cryptic transcripts in both datasets is very similar in all respects (Supplementary Figure S4). Notably, the expression level of antisense cryptic transcripts is similarly lower than that of sense cryptic transcripts using both methods (Supplementary Figure S5), further supporting the idea that both the sense and antisense cryptic transcripts terminate via the polyadenylation-dependent termination pathway.
Most yeast terminators are bidirectional
The data shown above implies that many, perhaps most, yeast terminators are functionally bidirectional. Terminator bidirectionality has been described anecdotally for a few yeast genes (35–37), but its prevalence was never thoroughly investigated. Among the eight terminators that were previously shown to be bidirectional (ARO4, TRP1, TRP4, ADH1, CYC1, GAL1, GAL7, GAL10), only one, ARO4, is facing a cryptic transcript coming from the antisense strand in spt6-1004 cells. Satisfyingly, this cryptic transcript indeed terminates around the ARO4 terminator (not shown) demonstrating that our data can capture terminator bidirectionality. Because hundreds of antisense cryptic transcripts emerge in spt6-1004 cells, we reasoned that this could be used as an opportunity to classify terminators as uni- or bidirectional. From the 1616 antisense cryptic transcripts identified in spt6-1004 cells, we could predict the directionality of 826 terminators. Of those, 676 were classified as bidirectional and 150 as unidirectional (see Materials and Methods) (Figure 4A). Figure 4B shows examples of bidirectional (left) and unidirectional (right) terminators identified in our analysis. Visual inspection of these putatively unidirectional terminators suggests that many of them are likely to be bidirectional but were not captured by our directionality prediction algorithm. These analyses predict that >80% of yeast promoters are functionally bidirectional.
Figure 4.

Yeast terminators are mostly bidirectional. (A) Pie charts displaying the terminators that are challenged by antisense cryptic transcription (aCT; left) and the number of bidirectional, weak bidirectional and unidirectional terminators (right). (B) Genome-browser snapshots illustrating examples of bidirectional (left) and unidirectional (right) terminators. RNA-Seq signal on the Watson (gray) and Crick (black) strand are shown. (C) Schematic representation of the terminator assay used in panels D and E. (D) RNA blot for the terminator assay testing terminators predicted to be bidirectional (s, terminator cloned in sense orientation; as, terminator cloned in antisense orientation). (E) Same as ‘D’ but for terminators predicted to be unidirectional. Note that the full length transcript is undetectable in this assay, most likely due to its instability. The ‘asterisk’ indicates the transcript from the endogenous YNR051C gene and is used as a loading control. These experiments have been performed three to seven times (depending on the terminator tested) and showed consistent results. The membranes shown here are representative results. More replicates are shown in Supplementary Figure S6.
In order to challenge this prediction, we tested bidirectionality of a set of terminators from each group. For this, we modified a previously developed genetic system where candidate terminators are cloned downstream of a strong promoter driving the expression of a HIS3 gene on a plasmid to allow terminator efficiency to be monitored by northern blotting (31) (see Figure 4C). As expected, all terminators tested were active in this assay when tested in their natural (sense) orientation, since all generated a short transcript detectable with a probe targeting the 5΄UTR (Figure 4D and E). When cloned in the inverted orientation (antisense), all terminators predicted to be bidirectional efficiently terminated transcription, confirming their bidirectionality (Figure 4D). Among the terminators predicted to be unidirectional, some (YNL058C, HIS1 and GCN4) did not generate a robust signal for short transcripts when cloned in the reverse orientation (Figure 4E). This is consistent with them being unidirectional terminators although we cannot exclude the possibility that short transcripts are generated but are unstable. The three other terminators predicted to be unidirectional (YGL101W, HMG2 and YJL136C), however, clearly behaved as bidirectional (Figure 4E), suggesting that our predictions are overestimating the number of unidirectional terminators. Taken together, our RNA-Seq analyses and Northern blot experiments establish bidirectionality as a prevalent characteristic of most yeast terminators. Importantly, these analyses clearly show that terminator bidirectionality allows the termination of antisense cryptic transcription from invading neighboring genes (Figure 5).
Figure 5.

A graphical representation of the full length and cryptic transcripts described in this study. Full length transcripts, as they normally occur in WT cells, are depicted in blue. Sense (red) and antisense (green) cryptic transcripts, initiated from within Gene B in spt6-1004 cells, are also depicted. The transcription start sites are depicted as solid arrows, the cryptic transcription start sites as dashed arrows and the terminators as ‘T’. The green transcript (cryptic antisense) terminates at the terminator of Gene A, despite the fact that Gene A is on the other strand. This is evidence that the terminator of Gene A is bidirectional. We estimate that more than 80% of terminators in S. cerevisiae are bidirectional. Assuming that cleavage and polyadenylation of the green transcript leads to termination of the associated polymerase a few base pairs later, this system would prevent the green transcript and the associated RNA polymerase from invading Gene A.
DISCUSSION
We provide a detailed analysis of cryptic transcription in spt6-1004 cells using RNA-Seq. Consistent with previous work, we found cryptic transcripts running both sense and antisense relative to annotated genes in this mutant (Figure 5, red and green transcripts). Cryptic promoters are enriched for TATA motifs suggesting that DNA sequence is the main determinant of cryptic promoters, despite chromatin structure disruption being the driving force for their usage. Surprisingly, we observed that antisense cryptic transcription tends to terminate near the 3΄-end of the upstream gene (Figure 5, green transcript). This prompted us to systematically predict the ability of yeast terminators to terminate transcription coming from the other direction. Quite strikingly, we found that most terminators (>80%) are efficient at inducing termination and polyadenylation of antisense transcripts. Consistently, we found that DNA motifs characteristic of yeast terminators are enriched on both strands in the 3΄-end of genes. We therefore conclude that most yeast terminators are functionally bidirectional. Assuming that these polyadenylated transcripts are coupled to the termination of their associated polymerases, the bidirectional nature of yeast terminators would prevent/attenuate antisense transcription from invading the upstream gene when it is in tandem. In Figure 5, for example, termination of the green cryptic antisense transcript at the Gene A terminator would prevent/attenuate transcription from running over Gene A, and eventually its promoter, which could lead to transcriptional interference through different mechanisms (38).
Promoters in S. cerevisiae are intrinsically bidirectional but divergent transcription is rapidly terminated via the Nrd1 pathway (39,40). This pathway prevents divergent transcription, initiated at canonical promoters, from invading the upstream gene. From our data, it appears that contrary to divergent transcription, cryptic antisense transcription is not efficiently terminated via this pathway. Indeed, these transcripts are not terminated in the promoter region of the gene that host them (as are divergent transcripts) but rather read through the intergenic region until they reach the terminator region of the upstream gene, where they terminate via the polyadenylation pathway (Figure 5, green transcript). Why the Nrd1 termination pathway is inefficient at terminating antisense cryptic transcription is not known but the status of the C-terminal domain of RNAPII may be part of the answer. Indeed, the Nrd1 pathway relies mainly on P-Ser5, while the polyadenylation pathway proceeds via P-Ser2 (41). When an RNAPII molecule transcribing an antisense cryptic transcript reaches the Nrd1 sites in the promoter region of the gene hosting it, it has already transcribed longer than an RNAPII molecule that would have initiated divergently from that promoter. The CTD phosphorylation status of this RNAPII is therefore likely not optimal for Nrd1-dependent termination (too low in P-Ser5 and too high in P-Ser2), perhaps explaining why termination via this pathway is inefficient in that context. Alternatively, it may be that some antisense cryptic transcription is actually terminating via the Nrd1 pathway but generating unstable transcripts that escaped our detection. In spt6-1004 cells, the very high abundance of these transcripts may saturate the capacity of the Nrd1 pathway so that we may be capturing the ‘escapees’ that terminate at the bidirectional terminators of the upstream gene as a backup mechanism.
Interestingly, our analysis of published RNA-Seq data from spt6-1 S. pombe cells (33) shows that cryptic antisense transcripts in this organism preferentially terminate around promoter regions, rather than terminator regions, suggesting that fission yeast has evolved different mechanisms for coping with the termination of antisense cryptic transcription (data not shown). Why both yeasts use different mechanisms is not clear but differences with regards to transcription termination between these two species have been reported previously (See (42) and references therein).
Our analysis was performed on RNA-Seq data from cells that have functional RNA degradation pathways, which implies that the cryptic transcripts we detected are stable. Future experiments using combinations of Spt6 and exosome mutations, or using techniques such as NET-Seq or GRO-Seq that measure ongoing transcription, may reveal additional cryptic transcripts, perhaps terminated by alternative pathways, in histone chaperone mutants. The use of higher resolution approaches to map the 5΄ and 3΄-ends of these transcripts should also help decipher how cryptic promoters emerge and how cryptic transcription is terminated.
Another important aspect of cryptic transcription is the impact it may have on the expression of bona fide genes. This question is especially important knowing that cryptic transcription occurs in cancer cells. It is increasingly recognized that non-coding transcripts and non-coding transcription can regulate gene expression through multiple mechanisms (38,43). It therefore appears likely that the massive spurious transcription observed in spt6-1004 cells would impact the expression of bona fide genes, notably those that host a cryptic transcript. Unexpectedly, however, we failed to show any significant effect of the presence of a cryptic transcript (should it be sense or antisense) on the expression of the gene hosting it. Indeed, while the expression of protein-coding genes is widely affected in spt6-1004, we found no correlation between these defects and the presence of cryptic transcription (data not shown). This is not to say that cryptic transcription has no impact on gene expression, but simply that our data does not allow us to measure it. We surmise that using mutants with less dramatic effect on chromatin structure (e.g. mutants in the Set2 pathway) may be better suited to address this important question.
AVAILABILITY
yCrypticRNAs is available at https://cran.r-project.org/web/packages/yCrypticRNAs/index.html.
ACCESSION NUMBERS
RNA-Seq data are available at the Gene Expression Omnibus (GEO) database with accession number GSE89601.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Robert lab for their help with the visual validation of the cryptic transcript predictions. We also thank Alexis Blanchette for bioinformatics support and Nicole Francis for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research [MOP-133648 to F.R.]; P.C. held a studentship from Le Fonds de recherche du Québec – Santé. Funding for open access charge: Canadian Institutes of Health Research.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lenhard B., Sandelin A., Carninci P.. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat. Rev. Genet. 2012; 13:233–245. [DOI] [PubMed] [Google Scholar]
- 2. Albert I., Mavrich T.N., Tomsho L.P., Qi J., Zanton S.J., Schuster S.C., Pugh B.F.. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007; 446:572–576. [DOI] [PubMed] [Google Scholar]
- 3. Ernst J., Kellis M.. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010; 28:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kwak H., Lis J.T.. Control of transcriptional elongation. Annu. Rev. Genet. 2013; 47:483–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smolle M., Workman J.L., Venkatesh S.. reSETting chromatin during transcription elongation. Epigenetics. 2013; 8:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smolle M., Workman J.L.. Transcription-associated histone modifications and cryptic transcription. Biochim. Biophys. Acta. 2013; 1829:84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muratani M., Deng N., Ooi W.F., Lin S.J., Xing M., Xu C., Qamra A., Tay S.T., Malik S., Wu J. et al. . Nanoscale chromatin profiling of gastric adenocarcinoma reveals cancer-associated cryptic promoters and somatically acquired regulatory elements. Nat. Commun. 2014; 5:4361. [DOI] [PubMed] [Google Scholar]
- 8. Carvalho S., Raposo A.C., Martins F.B., Grosso A.R., Sridhara S.C., Rino J., Carmo-Fonseca M., de Almeida S.F.. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 2013; 41:2881–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaplan C.D., Laprade L., Winston F.. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003; 301:1096–1099. [DOI] [PubMed] [Google Scholar]
- 10. Cheung V., Chua G., Batada N.N., Landry C.R., Michnick S.W., Hughes T.R., Winston F.. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 2008; 6:e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Bakel H., Tsui K., Gebbia M., Mnaimneh S., Hughes T.R., Nislow C.. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet. 2013; 9:e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carrozza M.J., Li B., Florens L., Suganuma T., Swanson S.K., Lee K.K., Shia W.J., Anderson S., Yates J., Washburn M.P. et al. . Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005; 123:581–592. [DOI] [PubMed] [Google Scholar]
- 13. Silva A.C., Xu X., Kim H.S., Fillingham J., Kislinger T., Mennella T.A., Keogh M.C.. The replication-independent histone H3-H4 chaperones HIR, ASF1, and RTT106 co-operate to maintain promoter fidelity. J. Biol. Chem. 2012; 287:1709–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Imbeault D., Gamar L., Rufiange A., Paquet E., Nourani A.. The Rtt106 histone chaperone is functionally linked to transcription elongation and is involved in the regulation of spurious transcription from cryptic promoters in yeast. J. Biol. Chem. 2008; 283:27350–27354. [DOI] [PubMed] [Google Scholar]
- 15. Joshi A.A., Struhl K.. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol. Cell. 2005; 20:971–978. [DOI] [PubMed] [Google Scholar]
- 16. Du H.N., Briggs S.D.. A nucleosome surface formed by histone H4, H2A, and H3 residues is needed for proper histone H3 Lys36 methylation, histone acetylation, and repression of cryptic transcription. J. Biol. Chem. 2010; 285:11704–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Du H.N., Fingerman I.M., Briggs S.D.. Histone H3 K36 methylation is mediated by a trans-histone methylation pathway involving an interaction between Set2 and histone H4. Genes Dev. 2008; 22:2786–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hainer S.J., Martens J.A.. Identification of histone mutants that are defective for transcription-coupled nucleosome occupancy. Mol. Cell. Biol. 2011; 31:3557–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smolle M., Venkatesh S., Gogol M.M., Li H., Zhang Y., Florens L., Washburn M.P., Workman J.L.. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat. Struct. Mol. Biol. 2012; 19:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Radman-Livaja M., Quan T.K., Valenzuela L., Armstrong J.A., van Welsem T., Kim T., Lee L.J., Buratowski S., van Leeuwen F., Rando O.J. et al. . A key role for Chd1 in histone H3 dynamics at the 3΄ ends of long genes in yeast. PLoS Genet. 2012; 8:e1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li B., Jackson J., Simon M.D., Fleharty B., Gogol M., Seidel C., Workman J.L., Shilatifard A.. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 2009; 284:7970–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chu Y., Simic R., Warner M.H., Arndt K.M., Prelich G.. Regulation of histone modification and cryptic transcription by the Bur1 and Paf1 complexes. EMBO J. 2007; 26:4646–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jeronimo C., Robert F.. Histone chaperones FACT and Spt6 prevent histone variants from turning into histone deviants. Bioessays. 2016; 38:420–426. [DOI] [PubMed] [Google Scholar]
- 24. Jeronimo C., Watanabe S., Kaplan C.D., Peterson C.L., Robert F.. The Histone Chaperones FACT and Spt6 Restrict H2A.Z from Intragenic Locations. Mol. Cell. 2015; 58:1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brunelle M., Coulombe C., Poitras C., Robert M.A., Markovits A.N., Robert F., Jacques P.E.. Aggregate and heatmap representations of genome-wide localization data using VAP, a versatile aggregate profiler. Methods Mol. Biol. 2015; 1334:273–298. [DOI] [PubMed] [Google Scholar]
- 29. Coulombe C., Poitras C., Nordell-Markovits A., Brunelle M., Lavoie M.A., Robert F., Jacques P.E.. VAP: a versatile aggregate profiler for efficient genome-wide data representation and discovery. Nucleic Acids Res. 2014; 42:W485–W493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graber J.H., Cantor C.R., Mohr S.C., Smith T.F.. In silico detection of control signals: mRNA 3΄-end-processing sequences in diverse species. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:14055–14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carroll K.L., Pradhan D.A., Granek J.A., Clarke N.D., Corden J.L.. Identification of cis elements directing termination of yeast nonpolyadenylated snoRNA transcripts. Mol. Cell. Biol. 2004; 24:6241–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li B., Gogol M., Carey M., Pattenden S.G., Seidel C., Workman J.L.. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007; 21:1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DeGennaro C.M., Alver B.H., Marguerat S., Stepanova E., Davis C.P., Bahler J., Park P.J., Winston F.. Spt6 regulates intragenic and antisense transcription, nucleosome positioning, and histone modifications genome-wide in fission yeast. Mol. Cell. Biol. 2013; 33:4779–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lickwar C.R., Rao B., Shabalin A.A., Nobel A.B., Strahl B.D., Lieb J.D.. The Set2/Rpd3S pathway suppresses cryptic transcription without regard to gene length or transcription frequency. PLoS One. 2009; 4:e4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Egli C.M., Braus G.H.. Uncoupling of mRNA 3΄ cleavage and polyadenylation by expression of a hammerhead ribozyme in yeast. J. Biol. Chem. 1994; 269:27378–27383. [PubMed] [Google Scholar]
- 36. Egli C.M., Duvel K., Trabesinger-Ruf N., Irniger S., Braus G.H.. Sequence requirements of the bidirectional yeast TRP4 mRNA 3΄-end formation signal. Nucleic Acids Res. 1997; 25:417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Irniger S., Egli C.M., Braus G.H.. Different classes of polyadenylation sites in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 1991; 11:3060–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mellor J., Woloszczuk R., Howe F.S.. The interleaved genome. Trends Genet. 2016; 32:57–71. [DOI] [PubMed] [Google Scholar]
- 39. Neil H., Malabat C., d’Aubenton-Carafa Y., Xu Z., Steinmetz L.M., Jacquier A.. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009; 457:1038–1042. [DOI] [PubMed] [Google Scholar]
- 40. Schulz D., Schwalb B., Kiesel A., Baejen C., Torkler P., Gagneur J., Soeding J., Cramer P.. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell. 2013; 155:1075–1087. [DOI] [PubMed] [Google Scholar]
- 41. Porrua O., Libri D.. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell. Biol. 2015; 16:190–202. [DOI] [PubMed] [Google Scholar]
- 42. Lemay J.F., Marguerat S., Larochelle M., Liu X., van Nues R., Hunyadkurti J., Hoque M., Tian B., Granneman S., Bahler J. et al. . The Nrd1-like protein Seb1 coordinates cotranscriptional 3΄ end processing and polyadenylation site selection. Genes Dev. 2016; 30:1558–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quinn J.J., Chang H.Y.. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016; 17:47–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.