


Abstract
DNA-single strand annealing proteins (SSAPs) are recombinases frequently encoded in the genome of many bacteriophages. As SSAPs can promote homologous recombination among DNA substrates with an important degree of divergence, these enzymes are involved both in DNA repair and in the generation of phage mosaicisms. Here, analysing Sak and Sak4 as representatives of two different families of SSAPs present in phages infecting the clinically relevant bacterium Staphylococcus aureus, we demonstrate for the first time that these enzymes are absolutely required for phage reproduction. Deletion of the genes encoding these enzymes significantly reduced phage replication and the generation of infectious particles. Complementation studies revealed that these enzymes are required both in the donor (after prophage induction) and in the recipient strain (for infection). Moreover, our results indicated that to perform their function SSAPs require the activity of their cognate single strand binding (Ssb) proteins. Mutational studies demonstrated that the Ssb proteins are also required for phage replication, both in the donor and recipient strain. In summary, our results expand the functions attributed to the Sak and Sak4 proteins, and demonstrate that both SSAPs and Ssb proteins are essential for the life cycle of temperate staphylococcal phages.
INTRODUCTION
Bacteriophages (phages) are recognised as the most abundant and diverse microorganisms on earth (1). The relevant and versatile roles that phages play in nature is demonstrated in the pathogenic bacterium Staphylococcus aureus. In this species, Siphoviridae phages are responsible for different human diseases by encoding important toxins, such as exfoliatin A (responsible for the scalded skin syndrome), enterotoxin A (food poisoning) or the Panton-Valentine leukocidin (PVL) (necrotising pneumonia) (2). Phages are also relevant as a major source of genetic variation when S. aureus strains from different origins are compared (3). Moreover, horizontal gene transfer in S. aureus occurs mainly by phage-mediated transduction (4). A remarkable example of the role of staphylococcal phages in spreading virulence genes is observed with a family of pathogenicity islands, the S. aureus pathogenicity islands (SaPIs), which hijack staphylococcal phage machinery to ensure their promiscuous intra- and inter-generic transfer (recently reviewed in (4)). Since these elements encode factors involved in virulence and host adaptation, phages are considered to be the major evolutionary driving force in this species (5). In spite of their relevance, the biology of staphylococcal phages remains poorly understood.
One group of proteins widespread in bacteriophages are the single strand annealing proteins (SSAPs), a subtype of recombinases which has gained attention in recent years not only because of its use in recombineering systems (6) but also because of its impact in the remodeling of bacterial genomes (7). For simplicity we will refer hereafter to SSAP as SSAP or just recombinases (not to be confused with site-specific recombinases coded by temperate bacteriophages that catalyze integration/excision). Different recombinase families have been identified in prophage genomes from both Gram-positive and Gram-negative bacteria. These include Sak, Redβ, Erf, Sak4 and Gp2.5 (8). Sak, Redβ and Erf belong to the Rad52-like superfamily of recombinases, Sak4 belongs to the unrelated Rad51/RecA family, while Gp2.5 defines the third superfamily of these recombinase proteins. In spite of their promiscuous presence in phage genomes, very few members of the different recombinase families have been characterized in detail. Biochemical studies have shown that all the phage recombinases studied so far are SSAPs that can promote genetic recombination under more permissive conditions than RecA (6,9,10). However, their roles in vivo have not been studied to any great extent. It has long been assumed that these enzymes play a pivotal role in promoting gene shuffling among temperate phages, accelerating phage evolution (10). One study using Bacillus subtilis phage SPP1 suggested that the recombinase G35P (Redβ family) could be involved in the generation of the concatemeric DNA during phage replication (11), whereas another study implicated the phage P22-encoded Erf recombinase in the circularization of the phage genome after infection (12). However, for most members of these families their role in the phage cycle remains to be determined.
Remarkably, staphylococcal temperate phages always encode one of these five aforementioned families of SSAPs. Interestingly, although not related in sequence, the genes encoding the SSAP proteins always share the same location in the phage genomes (Supplementary Figure S1) (8,13), suggesting that all have the same role in the phage cycle. Further analysis indicates the constant presence of a gene coding for a predicted protein, homologous to a single strand DNA binding protein (Ssb), adjacent to these recombinase genes (Supplementary Figure S1). Similar to the SSAP proteins, the role of the Ssb proteins in the staphylococcal phage cycle remains to be established. With this in mind, we have explored here the role that different families of SSAPs and their cognate Ssb proteins play in the life cycle of the staphylococcal Siphoviridae temperate phages. Our results clearly demonstrate that both staphylococcal phage encoded proteins, SSAP and Ssb, play an essential role in phage replication.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The bacterial strains used in this study are listed in Supplementary Table S1. The procedures for preparation and analysis of phage lysates, in addition to transduction and transformation of S. aureus, were performed as previously described (14,15).
DNA methods
General DNA manipulations were performed using standard procedures. The plasmids and oligonucleotides used in this study are listed in Supplementary Tables S2 and S3, respectively. The labeling of probes and DNA hybridizations were performed according to the protocol supplied with the PCR-DIG DNA-labeling and Chemiluminescent Detection Kit (Roche). To produce the phage mutations, we used plasmid pMAD (16), as previously described (17).
Complementation of the mutants
The different phage genes under study were PCR amplified using oligonucleotides listed in Supplementary Table S3. PCR products were cloned into pCN51 (18) and the resulting plasmids (Supplementary Table S2) were introduced into the appropriate recipient strains (Supplementary Table S1).
Analysis of phage DNA replication by pulsed field gel electrophoresis (PFGE)
Bacteria were grown in TSB to OD540 = 0.3, then the wild-type or prophage mutants under study were induced by Mitomycin C (MC; 2 μg/ml). Cultures were grown at 32°C with slow shaking (80 rpm). Samples (1 ml) were removed at various time-points after phage induction and cells were harvested for 5 min at 12 000 rpm, 4°C. Cells were washed with 1 ml of ice cold washing buffer (Tris 20 mM pH 8.5, 100 mM EDTA, 100 mM NaCl), pelleted and stored at –20°C. Cells were resuspended in 50 μl of buffer P1 (50 mM Tris pH 8.0 10 mM EDTA, 0.1 mg/ml RNAse A) and lysed with lysostaphin (0.2 mg/ml) and lysozyme (0.5 mg/ml) for 30 min at 22°C. Then 50 μl of a solution containing proteinase K (1 mg/ml) plus 2% sodium dodecyl sulphate (SDS) was added and samples were further incubated for 30 min at 55°C. 10 μl aliquots were analysed by agarose gel (0.8%) electrophoresis in Tris-acetate buffer or by PFGE followed by Southern blot.
PFGE was performed with a Bio-Rad CHEF-DR II apparatus. Running conditions were 5 V/cm, 0.5× TBE, 0.5–10 switch time for 20 h at 12°C. The molecular weight marker used was the LW range PFG marker (New England Biolabs). The probe used for Southern blot development was a PCR product of 500-bp complementary to the cI (80α) and Orf15 (ϕ11) gene regions. Southern blots were performed with Hybond-N+ membranes as instructed by the manufacturer (GE Healthcare) and detection was performed with the AlkPhos Direct Labeling kit (GE Healthcare).
Real-time quantitative PCR
Whole DNA of S. aureus strains RN4220, RN10359, JP6001, JP6016, JP6017, JP1361, JP4001, JP4012 and JP4013 was extracted using the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich) according to the manufacturer's instructions. Real-time quantitative PCR was performed in triplicate using 1 μl DNA as template and Fast SYBR Green Master Mix (Thermofisher Scientific) according to the manufacturer's instructions. Samples were normalized using the levels of gyrB and the levels of reconstituted empty attachment sites were calculated relative to the RN4220 control. Primers used for quantitative PCR are listed in Supplementary Table S3. To monitor specificity, the final PCR products were analysed by melting curves and electrophoresis. The relative DNA levels within distinct experiments were determined by using the 2−ΔΔCT method (19). The results show the average ± SEM of at least three independent experiments.
Protein expression and purification
The 80α sak gene was PCR amplified and cloned into NcoI and BamHI cleaved pET-15b plasmid to generate plasmid pET-sak. The correct insertion of sak into plasmid pET-sak was confirmed by sequencing. Escherichia coli BL21 (DE3) pLysS cells were transformed with pET-sak and protein expression was induced by addition of 0.2 mM IPTG to cells growing in LB at 18°C at an optical density of 0.2 at 560 nm. These were then further incubated for 16 h at 18°C. Cells were collected by centrifugation and stored at –20°C.
Ten grams of wet cells were resuspended in 50 ml of buffer A (50 mM Tris–HCl pH 7.5, 15% glycerol, 1 mM EDTA, 1 mM DTT) containing 300 mM NaCl and lysed by sonication. Cell debris and insoluble proteins were then removed by centrifugation at 18 000 rpm for 45 min at 4°C. Sak protein was precipitated from the soluble fraction by 50% ammonium sulfate saturation, followed by centrifugation at 18 000 rpm for 30 min at 4°C. The pellet was washed once with 50% ammonium sulfate saturation and centrifuged again. The ammonium sulfate pellet containing Sak was resuspended in buffer A containing 500 mM NaCl and was loaded into a 5 ml hydroxyapatite column (Bio-Rad). The column was washed stepwise with increasing phosphate concentrations in buffer A containing 500 mM NaCl. Pure Sak protein was eluted from this column at 200 mM phosphate. Fractions containing pure protein were pooled and precipitated with 70% ammonium sulfate to concentrate the protein. The pellet was solubilized in buffer A containing 500 mM NaCl and 50% glycerol and purified protein was stored at –20°C.
DNA substrates
Oligo 100-nt-up with the sequence: 5΄- GGGCGAATTGGGCCCGACGTCGCATGCTCCTCTAGACTCGAGGAATTCGGTACCCCGGGTTCGAAATCGATAAGCTTACAGTCTCCATTTAAAGGACAAG-3΄ and its complementary, oligo 100-nt-down, were purchased from SIGMA and purified by PAGE as described before (20).
Oligonucleotide 100-nt-up was radiolabeled at the 5΄-end by polynucleotide kinase and γ32P-ATP and was the source of single strand DNA (ssDNA). Its double strand (dsDNA) control was obtained by heating to 95°C for 5 min an equimolar amount of each oligonucleotide in annealing buffer (100 mM sodium phosphate buffer pH 7.5), and then cooling the sample to room temperature over 2 h. A pUC18 HindIII–NdeI 216 bp dsDNA fragment was the source of dsDNA. It was radiolabeled using α32P-dATP incorporated using Klenow enzyme.
Single-strand annealing and binding assays
Binding of Sak to DNA was analysed through electrophoretic mobility-shift assays (EMSA), using ssDNA and dsDNA substrates (0.5 nM). Reactions were performed in buffer B (25 mM Tris–HCl pH 7.5, 0.50 mg/ml BSA, 100 mM NaCl, 5% glycerol, 2 mM DTT) with 1 mM MgCl2 for 15 min at 37°C. Complexes were separated by PAGE in 6% polyacrylamide gels run in TBE, and the gels were dried before autoradiography.
The single-strand annealing activity of the Sak protein was analyzed using the two complementary synthetic oligonucleotides (oligo 100-nt-up and oligo 100-nt-down) of which oligo 100-nt-up was radiolabeled. In time-course experiments, a reaction mix was prepared containing 40 nM Sak in buffer B containing 5 mM magnesium acetate and 0.5 nM oligo 100-nt-up. Reactions were initiated by the addition of 0.5 nM cold 100-nt-down DNA. Tubes were placed at 30°C and 10 μl aliquots were removed at times 2, 4, 8, 6, 8, 10 and 12 min. Samples were deproteinized by the addition of one volume of stop buffer (40 mM Tris–HCl pH 7.5, 0.2% SDS, 100 mM EDTA, 0.4 mg/ml proteinase K) followed by a 15 min incubation at 37°C. A control reaction was performed in parallel without protein to observe the spontaneous annealing of the two oligonucleotides under the conditions used. DNA products were analysed by 8% PAGE, run in TBE and autoradiography.
RESULTS
The 80α encoded Sak recombinase is required for phage replication
Phage 80α ORF16 (accession number ABF71587) is well conserved among many S. aureus phages. Interestingly, the primary sequence of the ORF16 encoded protein shows 42% identity in a stretch of 150 amino acids to the lactococcal phage p2 Sak3 protein (accession number AAR14300; Supplementary Figure S2), and therefore we renamed ORF16 as Sak. Lactococcal Sak3 has been characterised as a single strand-annealing protein (SSAP) belonging to the Sak family of recombinases (8,9). Although the biochemical characterization of Sak3 clearly demonstrated that this protein promotes homologous recombination (9), its role in the phage cycle remains unclear.
We initially analyzed whether the 80α-encoded Sak protein had a similar ability to that of the lactococcal Sak recombinases in its ability to preferentially bind to ssDNA and anneal two complementary ssDNA sequences. We purified the protein and first performed DNA binding studies. As shown in Figure 1A, experiments performed increasing the Sak concentration and keeping the ssDNA concentration constant confirmed that Sak forms stable complexes with ssDNA. The affinity (Kapp) for this interaction was ∼200 nM, which is similar to that previously reported for lactococcal Sak3 (9). Similar experiments were performed with dsDNA, but the amount of dsDNA retarded was reduced and the Kapp was >4 μM (Figure 1B). Next, since the 80α-encoded Sak does not contain any Walker motif in its sequence (Supplementary Figure S2), we tested whether Sak can anneal complementary 100-mer oligonucleotides in the absence of ATP. This was the case, and a time-course experiment revealed that Sak annealed complementary oligonucleotides with similar kinetics to Sak3 (Figure 1C) (9). Taken together, these results confirm that the 80α-encoded Sak recombinase is a bona fide SSAP.
Figure 1.

ssDNA binding and annealing activities of S. aureus phage 80α Sak protein. (A) Electrophoretic mobility shift assay showing the binding of Sak protein to ssDNA. Radiolabelled oligo 100-nt-up (0.5 nM) was incubated with increasing amounts of Sak protein (from 30 nM to 1 μM) in buffer B containing 1 mM MgCl2 for 15 min at 37°C. The two types of complexes formed are denoted by I and II. (B) Binding to dsDNA. Radiolabelled pUC18 HindIII–NdeI 216 bp dsDNA fragment (0.5 nM) was incubated with increasing amounts of Sak protein (from 60 nM to 4 μM) in buffer B containing 1 mM MgCl2 for 15 min at 37°C. The three types of complexes formed are denoted by I, II and III. (C) Sak promotes single-strand annealing. Radiolabelled oligo 100-nt-up and its complementary (0.5 nM) were incubated with a fixed concentration of Sak (40 nM) in the presence of 5 mM magnesium acetate. At the given time points, aliquots were removed and deproteinized. The reaction products were analyzed by 8% PAGE and autoradiopgraphy. Lane 1: control annealed 100 bp ds DNA; lane 2: radiolabelled oligo 100-nt-up; lanes 3–8: control reaction without protein (2–12 min); lanes 9–14: reaction with Sak of phage 80α (40 nM, 2–12 min).
Next, we introduced an in-frame deletion in this gene in an 80α lysogen and evaluated how this mutation influenced the phage cycle. Surprisingly, this deletion severely affected phage reproduction and phage-mediated bacterial lysis. Thus, cellular lysis after mitomycin C (MC) induction of the 80α sak mutant prophage occurred overnight, in clear contrast to the cells carrying the wt 80α prophage which lysed 4 h after MC induction. Moreover, the analysis of the different phage lysates showed that the 80α mutant did not generate plaques in the recipient strain. Complementation of the recipient strain with a pCN51 derivative plasmid expressing the sak gene under the control of the Pcad promoter showed that the 80α mutant was able to generate a few functional phage particles in the donor strain in the absence of the sak gene (Table 1). Complementation of the donor but not the recipient strain showed that sak is also absolutely required in the recipient strain to form plaques (Table 1). In summary, our complementation studies demonstrated that the effect observed in the 80α mutant depends on the sak gene, with this gene being required both in the donor and the recipient strains (Table 1).
Table 1. Effect of mutations on phage titera.
| Recipient strain | |||
|---|---|---|---|
| Donor lysogen | RN4220 | RN4220 pCN51-sak | RN4220 pCN51-ssb80α |
| 80α | 1.3 × 1010 | NDb | ND |
| 80α Δsak | <10 | 5.2 × 104 | ND |
| 80α Δsak pCN51-sak | <10 | 4.3 × 108 | ND |
| 80α Δssb | 1.2 × 108, c | ND | 1.3 × 108, d |
| 80α Δssb pCN51-ssb80α | 3.2 × 109, c | ND | 7.2 × 109, d |
| Donor lysogen | RN4220 | RN4220 pCN51-sak4 | RN4220 pCN51-ssb ϕ11 |
| ϕ11 | 6.7 × 108 | ND | ND |
| ϕ11 Δsak4 | <10 | 3.2 × 103 | ND |
| ϕ11Δsak4 pCN51-sak4 | <10 | 8.2 × 108 | ND |
| ϕ11Δssb | 1.2 × 106, c | ND | 1.3 × 106, d |
| ϕ11Δssb pCN51-ssbϕ11 | 1.2 × 108, c | ND | 5.2 × 108, d |
aPfu/ml of induced culture.
bND: Not determined.
cSmall plaques.
dNormal sized plaques.
To get insights into the mechanism by which Sak influences the phage cycle, we analyzed its role in phage replication. To do this, both the 80α wt and the sak mutant prophages were induced by MC addition and Southern blot analyses with samples taken at different time points were performed. As shown in Figure 2A, in accordance with the results shown in Table 1, replication of the sak mutant was significantly reduced.
Figure 2.

Replication of the 80α sak and ssb mutants. (A) Time course of bacteriophage DNA replication after MC induction. Samples of 80α wt, Δsak and Δssb mutants were taken at the indicated times after MC induction and separated on agarose. An ethidium bromide stained gel and its Southern blot with a phage-specific probe are shown. (B) The 80α Sak and Ssb proteins are not involved in prophage induction. Excision of the 80α wt, Δsak, Δssb and Δint mutant prophages was investigated at different times (0, 30 or 60 min) after MC induction of the phage lytic cycle. Samples were normalized using the levels of gyrB (housekeeping gene). Levels of reconstituted empty attC sites were calculated relative to the non-lysogenic strain RN4220 used as a control. Error bars represent SEM. A two-way ANOVA with Holm–Sidak multiple comparisons test was performed to compare mean differences between strains. Differences among the phages were not significant, except for the 80α Δint mutant (*P < 0.0001).
To rule out the possibility that the observed defects in phage replication were due to ineffective prophage induction, we measured by qPCR the excision activities of the prophages 80α and 80α Δsak, using primers hybridising to the regions flanking the region of phage integration. After 80α induction, the phage is excised and the chromosomal attC site is reconstituted. As a control for the experiment, we also included the 80α prophage mutant in the integrase gene (int), which does not excise (21). As shown in Figure 2B, excision of the 80α Δsak mutant was similar to wt, while no excision was observed in the 80α Δint mutant, confirming the role of Sak in phage replication but not in excision.
The fact that the Sak recombinase was required for phage replication in S. aureus was surprising since in the prototypical phage λ the Redβ recombinase is not required for λ phage replication (22). It has been proposed that phage 80α encodes a replication module of the initiator-helicase loader type, similar to that present in phage λ (23). However, this has not been experimentally demonstrated and the sequence homology of ORF20 and ORF21 with λ proteins O and P is very limited (Supplementary Figures S3A and S4A). A Blast analysis showed that ORF20 has 26% sequence identity (Supplementary Figure S3B) with SPP1 G38P, a protein that is essential for DNA replication in phage SPP1, and that has two activities: origin-binding (24), and a PriA-like activity (25). ORF21 showed 27% sequence identity with the E. coli DnaC protein (Supplementary Figure S4B, (26)), which acts as a loader of the DnaB replicative helicase (27). Therefore, both proteins are expected to be essential for the initiation of DNA replication in phage 80α. To analyze this, since the precise boundaries of Staphylococcal siphovirus DNA replication modules are unknown, we individually deleted in the lysogenic strain containing the 80α prophage the ORFs from ORF9 to ORF37. This region comprises the early transcript of phage 80α (28). Note that although ORFs 7 and 8 also belong to the early transcript of phage 80α, these encode the Cro-like (ORF7) and antirepressor (ORF8) proteins, which are not involved in replication, and consequently, are outside the scope of this study. Analogously, ORFs 22 (Sri) and 38 (RinA), which also belong to this region, have been previously analyzed (14,21,28) and since they do not have a role in replication they will not be further characterized here.
Each strain was SOS induced using MC. Total DNA, containing both bacterial and phage DNA, was prepared from each strain after 60 min, separated on agarose, stained and photographed before Southern blotting with a phage-specific probe. Table 2 summarizes the results obtained with the 80α mutants. Interestingly, deletion of ORFs 20 or 21 completely abolished phage replication (Table 2). In accordance with this, these mutant strains did not lyse, and no phage particles were obtained in the supernatant of the induced strains (Table 2). Moreover, while the 80α ΔORF21 could be complemented in trans, the 80α ORF20 could not (JP14567 and JP15037 respectively, Table 2), suggesting that the 80α replication origin (ori) is embedded in this gene. These results showed that just ORF20 and ORF21 are strictly required to initiate DNA replication. However, quantification of the amount of DNA synthesized after 60 min MC induction showed that DNA replication was highly affected in the Δsak (ORF16), as previously reported, and in the Δssb (ORF17) mutant (Table 2 and Figure 2).
Table 2. Effect of phage mutations on 80α titera.
| 80α | Donor lysogen | Lysisb | Replicationb | Complementationc | Phage titerd |
|---|---|---|---|---|---|
| wt | RN451 | +++ | +++ | 2.8 × 1010 | |
| Δ9 | JP5262 | +++ | +++ | 2.1 × 1010 | |
| Δ10 | JP5224 | +++ | +++ | 1.7 × 1010 | |
| Δ11 | JP5412 | +++ | +++ | 2.1 × 1010 | |
| Δ12-13 | MS1531 | +++ | +++ | 1.1 × 1010 | |
| Δ14 | JP6014 | +++ | +++ | 2.1 × 1010 | |
| Δ15 | JP6015 | +++ | +++ | 2.8 × 1010 | |
| Δ16 (sak) | JP6016 | + | + | See Table 1 | <10 |
| Δ17 (ssb) | JP6017 | +++ | ++ | See Table 1 | 4.5 × 108 |
| Δ18 | JP6018 | +++ | +++ | 1.7 × 1010 | |
| Δ19 | JP6019 | +++ | +++ | 1.7 × 1010 | |
| Δ20 | JP6020 | - | 0 | - | < 10 |
| JP15037 | - | 0 | + | < 10 | |
| Δ21 | JP6021 | - | 0 | - | < 10 |
| JP14567 | +++ | ++ | + | 1.7 × 107 | |
| *Δ22 | JP6022 | +++ | +++ | 9.8 × 109 | |
| Δ23 | JP6023 | +++ | +++ | 1.8 × 1010 | |
| Δ24 | JP6024 | +++ | +++ | 1.5 × 1010 | |
| Δ25 | JP6025 | +++ | +++ | 8.2 × 109 | |
| Δ26 | JP6026 | +++ | +++ | 1.0 × 1010 | |
| Δ27 | JP6027 | +++ | +++ | 1.7 × 1010 | |
| Δ28 | JP6028 | +++ | +++ | 1.4 × 1010 | |
| Δ29 | JP6029 | +++ | +++ | 2.0 × 109 | |
| Δ30 | JP6030 | +++ | +++ | 3.0 × 1010 | |
| Δ31 | JP6031 | +++ | +++ | 1.0 × 1010 | |
| Δ32 | JP4480 | +++ | +++ | 2.5 × 1010 | |
| Δ33 | JP6033 | +++ | +++ | 1.8 × 1010 | |
| Δ34 | JP5416 | +++ | +++ | 2.2 × 1010 | |
| Δ35 | JP5417 | +++ | +++ | 1.6 × 1010 | |
| Δ36 | JP6036 | +++ | +++ | 2.2 × 1010 | |
| Δ37 | JP6037 | +++ | +++ | 2.7 × 1010 |
aThe means of results from three independent experiments are shown. Variation was within ±5% in all cases.
bPhage replication. +++: not affected; ++: reduced; +: severely reduced; 0: abolished.
cIn this experiment, only those phage mutants that had affected their life cycle were included. For complementation, the empty pCN51 (–) or the pCN51 derivative plasmid expressing the gene under study (+) was introduced both in the donor and recipient strains, and the phage titer was measured in the recipient strain.
dPFU/ml induced culture, using RN4220 as recipient strain.
*Previously analyzed in (14).
To gain further insight into the role of these proteins in DNA replication we analysed whether they were sufficient to support replication of a suicide plasmid. Thus, the 80α genes encoding ORFs 20 and 21 were cloned into a suicide plasmid capable of replicating in E. coli but not in S. aureus, under the control of a Cd-inducible promoter, generating plasmid pJP1900. This plasmid was successfully transferred to S. aureus strain RN4220, where it replicated autonomously. The ability of the transformed strain to form colonies also indicated that the plasmid could segregate. As controls for the experiment two derivatives of the aforementioned plasmid carrying mutations in the 80α ORF20 or ORF21 were generated. Neither plasmid generated colonies in S. aureus strain RN4220, suggesting both proteins are necessary and sufficient for 80α replication. To rule out that these suicide plasmids, carrying mutations in the ORFs 20 or 21, did not generate colonies in RN4220 because of a defect in the transformation process, we generated the same constructs but using the thermosensitive (ts) plasmid vector pCN50, which replicates very poorly at 43°C. We would expect that all three plasmids (wt carrying the ORF20-21 insert; ORF20 mutant or ORF21 mutant) replicate at 30°C because of the activity of the pCN50-encoded ts replicon. However, we would expect that at 43°C only the wt plasmid expressing the ORFs 20 and 21 would support autonomous replication of the pCN50 plasmid. This was the case, and following plating at 43°C, only the strain carrying the wt plasmid expressing the complete 80α replicating module generated colonies. In summary, on the basis of these results we conclude that the 80α ORFs 20–21 complex can drive autonomous replication and segregation of a plasmid in S. aureus.
The single-stranded binding protein Ssb is also required for phage replication
In addition to the aforementioned ORFs, 16 (Sak), 20 and 21, deletion of ORF17 affected DNA replication and phage titre, although to a minor extent (Figure 2 and Table 2). As occurred with the sak mutant, the 80α ORF17 mutant did not show any defect in excision after prophage induction (Figure 2B). As previously mentioned, ORF17 encodes a single-strand binding protein (Ssb, Supplementary Figure S5), which is located next to the sak gene in all the analyzed phages (Supplementary Figure S1). The fact that the deletion of the phage coded ssb gene affects phage reproduction is surprising since 80α Ssb protein shows a high degree of identity with one of the host coded Ssb proteins, the SsbA protein (29). The sequence identity between these two proteins is 79% (Supplementary Figure S5). In SPP1 phage, the phage coded Ssb (G36P protein) can be replaced by host SsbA since both proteins have a high degree of sequence identity (25). This putative replacement could partially explain why although most of the plaques generated by this mutant were reduced in size, some plaques were normal (Supplementary Figure S6). To further explore this, we performed complementation studies that demonstrated that expression of the ssb gene in the donor strain partially restored the phage titre. The ssb gene is required in the recipient strain to fully recover the phage titre and to generate normal sized plaques (Table 1). Thus, the ssb gene, as the sak gene, is required both in the donor and the recipient strains (Table 1).
To analyze how Ssb affects phage DNA replication, the ssb mutant prophage was SOS induced and Southern blot analysis with samples taken at different time points was performed. Similar DNA replication kinetics were observed in the sak and ssb mutants (Figure 2 and Table 1), confirming that both proteins, Sak and Ssb, are required for 80α replication.
Sak and Ssb are involved in concatemer formation
There are two modes of bacteriophage λ DNA replication during its lytic development in E. coli cells. The circle-to-circle (theta; θ) replication predominates at early stages of the phage growth, and depends on the aforementioned O and P proteins (23,30). The rolling-circle (sigma; σ) replication occurs late after infection to produce long concatemers that serve as substrates for packaging of unit-length λ DNA into phage proheads (31). Similarly, in bacteriophage SPP1 DNA replication starts by binding of G38P to one of the origins of replication and proceeds by a θ mechanism, before it is shifted later to σ replication (32). The σ mode of DNA replication is essential for concatemer production, which are packaged into the empty proheads by the headful mechanism (33). Phage 80α also packages its concatemeric DNA by a headful mechanism. It has been proposed that in bacteriophage SPP1 the G35P recombinase (Redβ family) could be essential for the shift from θ to σ replication (11). Based on the fact that the phage titres observed for both the sak and the ssb mutants were severely affected compared with phage replication, our hypothesis was that the Sak recombinase and the Ssb protein may be required for the transition from θ to σ replication or for concatemer production in phage 80α. To test this, we analyzed the presence of the concatemeric form in the 80α wt and the sak and ssb mutant phages. To facilitate this study, we deleted the small terminase gene (terS) both in the 80α wt and the sak and ssb mutant. Note that TerS is absolutely required for phage DNA packaging (31,34). Consequently, the terS mutant is unable to use the concatemeric form as a substrate for packaging, facilitating the analyses of the presence of this replicating form in the wt and mutant phages. As expected, phage replication was affected in the sak ΔterS and ssb ΔterS double mutants (Figure 3). However, it was still possible to observe the concatemeric form in these mutants, although to a lesser extent. A possible explanation for this result involving the cellular RecA protein is discussed below.
Figure 3.

PFGE analysis of 80α replication intermediates. Southern blot showing the time course of bacteriophage DNA replication after MC induction of Δsak, ΔterS, and Δsak ΔterS mutants (A) or after MC induction of Δssb, ΔterS, and Δssb ΔterS mutants (B). M is the Low Range PFG Marker. The size of the bands is stated in kilobases.
As a complementary strategy, we cloned the PCR product containing ORFs 16–21 under the control of a Cd-inducible promoter, into the aforementioned suicide plasmid. This generated plasmid pJP1901, which expressed Sak (ORF16), Ssb (ORF17) the G38P- and DnaC-like proteins (ORFs 20 and 21) plus ORFs 18 and ORF19. As controls we generated a set of derivative suicide plasmids carrying single mutations in each of the aforementioned ORFs. Since expression of ORFs 20 and 21 are sufficient to support θ replication, and since our hypothesis is that both Sak and Ssb can be involved in σ replication, the rationale of the experiment was that DNA replication, or at least the accumulation of high molecular weight DNA (HMW), would be increased in the wt plasmid compared with that present in the sak and ssb mutants. As expected, plasmid mutants in ORFs 20 and 21 were incapable of replicating autonomously in S. aureus. Interestingly, plasmids that could be maintained in S. aureus generated different replicating species, some of them HMW, compatible with σ replication, the exception being the plasmid bearing the mutation in the sak gene, in which replication was clearly reduced (clearly involving this protein in phage replication). With this plasmid predominantly supercoiled monomeric forms were formed (Figure 4), which are compatible with θ replication. We were unable to see any effect for the ssb mutation in these analyses, compatible with its less relevant role in phage replication.
Figure 4.
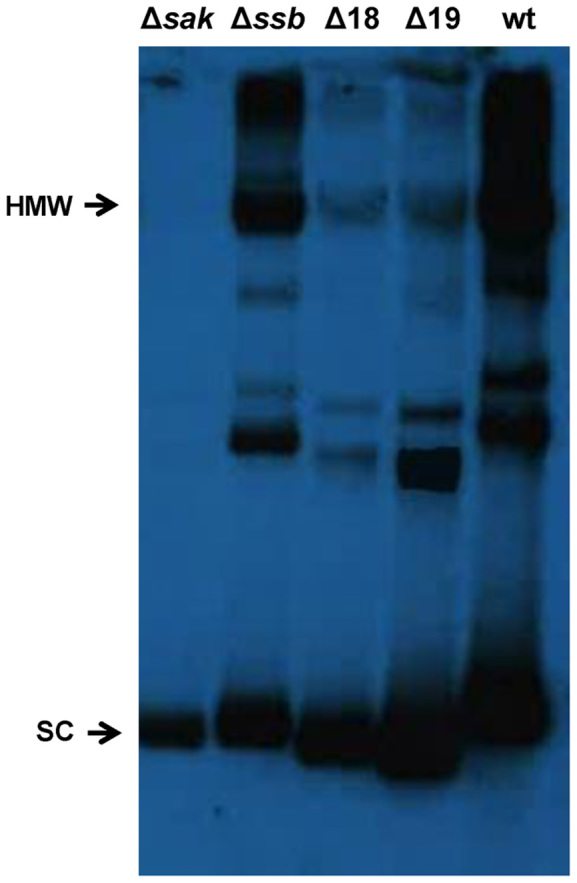
Replication products of 80α subclones. Total DNA from bacterial cultures carrying plasmid pJP1901 (lane 5, designated as wt), or derivatives mutant in the different Orfs (Δsak to Δ19) was extracted and separated by agarose gel electrophoresis, then Southern blotted using a plasmid-specific probe. SC: supercoiled monomers; HMW: high-molecular weight DNA (multimers).
The unrelated Sak4 recombinase is also required for phage replication
As previously indicated, at least five different recombinase families can be found in the staphylococcal temperate phages: Sak, Erf, Redβ, Sak4 and Gp2.5 families (8). The Sak4 family is less well studied than other families of recombinases. To date, only two reports exist in the literature about Sak4. The first Sak4 protein described was discovered in a genetic screen that showed that mutations in Sak4 of phage 31 conferred the ability to infect a phage-resistant strain of Lactococcus lactis expressing the AbiK protein, as it occurs with mutations in the Sak3 proteins (35). A preliminary study of the Sak4 protein encoded by phage PA73 of Pseudomonas aeruginosa showed that this protein has a moderate activity in a single strand recombineering assay in E. coli (8). As is the case with the Sak family, the role of Sak4 recombinases in the staphylococcal phage cycles remains unsolved, although this family is widely represented in staphylococcal phages (8). To solve this, we analysed the role of the unrelated recombinase Sak4 (NP_803265; Supplementary Figure S7) in the life cycle of ϕ11. An in-frame mutant of the sak4 (ORF12) gene was obtained in the ϕ11 prophage. This mutant and the wt prophage were SOS induced, and the phage cycle analysed. As reported for the Sak protein, the Sak4 recombinase was essential for phage replication and transfer (Figure 5 and Table 1), but not for excision of the induced prophage (Figure 5). Complementation studies also confirmed that Sak4 was required both in the donor and recipient cells (Table 1).
Figure 5.

PFGE analysis of DNA replication in ϕ11 mutants. Ethidium bromide gel (A) and Southern blot (B) showing the time course of bacteriophage DNA replication after MC induction of phage ϕ11 Δsak4, ΔterS, and Δsak4 ΔterS mutants. In (C), and (D), an ethidium bromide gel and Southern blot showing the time course of bacteriophage DNA replication after MC induction of Δssb, ΔterS and Δssb ΔterS mutants. M, is the Low Range PFG Marker. The size of the bands is stated in kilobases. (E) The ϕ11 Sak4 and Ssb proteins are not involved in prophage induction. Excision of the ϕ11 wt, Δsak, Δssb and Δint mutant prophages was investigated at different times (0, 30 or 60 min) after MC induction of the phage lytic cycle. Samples were normalized using the levels of gyrB (house keeping gene). Levels of reconstituted empty attC sites were calculated relative to the non-lysogenic strain RN4220 used as a control. Error bars represent SEM. A two-way ANOVA with Holm–Sidak multiple comparisons test was performed to compare mean differences between strains. Differences among the phages were not significant, except for the 80α Δint mutant (*P < 0.0001).
The recombinase and Ssb proteins form a functional cluster involved in phage replication
As previously indicated, in the staphylococcal prophage genomes the sak and ssb genes cluster together, and this also occurred in prophages with sak4 instead of sak (Supplementary Figure S1). Since the recombinase-Ssb pair was present in all of the phages analyzed, we investigated whether they comprise a functional unit. First we analysed whether the different recombinases cluster with cognate Ssb proteins, or instead, if there is no association among these proteins. To do this, we randomly selected 14 phages, 7 encoding Sak and 7 encoding Sak4 proteins (see Supplementary Table S4 for details). Supplementary Figure S8A and B shows a comparison among the different phage coded SSAP proteins. Next, we analysed the Ssb proteins encoded by these phages (Supplementary Table S4). As shown in Supplementary Figure S9A, there were two different variants of the Ssb protein: one was present in all the phages encoding Sak (Supplementary Figure S9B), and the other present in phages encoding Sak4 (Supplementary Figure S9C). Thus, the analyses of the different recombinase genes showed that each one is genetically linked with a specific cognate ssb gene, suggesting co-ordinated evolution.
To test whether the cognate recombinase-Ssb pairs work co-ordinately, we analysed whether the expression (both in the donor and recipient strains) of the ϕ11 sak4 gene could complement the 80α sak mutant. Similarly, we investigated whether the expression of the 80α sak gene could complement the ϕ11 sak4 mutant. Note, as is shown in Supplementary Figure S7, the 80α Sak and the ϕ11 Sak4 proteins are unrelated in sequence. As shown in Table 3, while the ϕ11 Sak4 did not restore the functionality of the phage 80α sak mutant, expression of the 80α sak gene complemented the ϕ11 sak4 mutant. In order to explain this result we hypothesised that the 80α Ssb protein is unable to interact with the ϕ11 Sak4 protein, while the ϕ11 Ssb can interact somehow with the 80α Sak protein. If our hypothesis was true, we would expect that: i) the ϕ11 ssb gene has a role in phage replication, as previously reported for the 80α ssb gene; ii) the 80α ssb mutant could be complemented with the ϕ11 ssb but not vice versa; and iii) complementation of the 80α sak mutant with a plasmid expressing both the ϕ11 Sak4 and Ssb proteins will restore the phage titre.
Table 3. Cross-complementation of the sak and sak4 mutantsa.
| Recipient strain | |||
|---|---|---|---|
| Donor lysogen | RN4220 | RN4220 pCN51-sak | RN4220 pCN51-sak4 |
| 80α | 8.5 × 109 | —b | — |
| 80α Δsak | <10 | — | — |
| 80α Δsak pCN51-sak | — | 2.1 × 109 | — |
| 80α Δsak pCN51-sak4 | — | — | <10 |
| ϕ11 | 6.4 × 108 | — | — |
| ϕ11 Δsak4 | <10 | — | — |
| ϕ11Δsak4 pCN51-sak | — | 2.3 × 108 | — |
| ϕ11Δsak4 pCN51-sak4 | — | — | 1.1 × 108 |
aPfu/ml of induced culture.
b—: Not determined.
To solve these questions, we deleted the ssb gene (ORF13) in the ϕ11 prophage. As occurred with the 80α Ssb, the ϕ11 Ssb is required for phage replication (Figure 5). Moreover, in this mutant both the ϕ11 phage titre and the size of the plaques were significantly affected (Table 1, Supplementary Figure S6). In support of our hypothesis, while expression of the ϕ11ssb gene complemented both the 80α and ϕ11 ssb mutants, expression of the 80α ssb only complemented the cognate 80α ssb mutant (Table 4). Finally, while the 80α sak mutant cannot be complemented with the ϕ11 sak4 gene, expression of both the ϕ11 Sak4 and Ssb proteins in the 80α sak mutant restored the phage titre (Table 5). These results confirmed the idea that both the phage coded recombinase and Ssb proteins are required for phage replication, working coordinately. In order to know whether the production of concatemeric DNA is affected in ϕ11 sak4 and ssb mutants we combined these mutations with a mutation in the terminase (terS). As occurred in 80α, the formation of concatemeric DNA was reduced, but could still be observed in these mutants (Figure 5).
Table 4. Cross-complementation of the ssb mutantsa.
| Recipient strain | |||
|---|---|---|---|
| Donor lysogen | RN4220 | RN4220 pCN51- ssb80α | RN4220 pCN51- ssbϕ11 |
| 80α | 1.3 × 1010 | —b | — |
| 80α Δssb | 4.0 × 107 | — | — |
| 80α Δssb pCN51-ssb80α | — | 5.1 × 109 | — |
| 80α Δssb pCN51-ssbϕ11 | — | — | 1.0 × 109 |
| ϕ11 | 6.1 × 108 | — | — |
| ϕ11 Δssb | 1.1 × 106 | — | — |
| ϕ11Δssb pCN51-ssb80α | — | 1.7 × 106 | — |
| ϕ11Δssb pCN51-ssbϕ11 | — | — | 2.3 × 107 |
aPfu/ml of induced culture.
b—: Not determined.
Table 5. Complementation of the 80α sak mutant with the sak4-ssb pair from ϕ11a.
| Recipient strain | |||
|---|---|---|---|
| Donor lysogen | RN4220 | RN4220 pCN51-sak4 | RN4220 pCN51-sak4-ssbϕ11 |
| 80α | 1.3 × 1010 | —b | — |
| 80α Δsak | <10 | — | — |
| 80α Δsak pCN51-sak4 | — | <10 | — |
| 80α Δssb pCN51-sak4-ssbϕ11 | — | — | 1.8 × 108 |
aPfu/ml of induced culture.
b—: Not determined.
DISCUSSION
In this work, we have used biochemical and genetic approaches to decipher the roles in DNA replication of genes present in the putative replication operon of staphylococcal phage 80α. Our results show that ORFs 20 and 21 are essential for initiation of DNA replication. ORF20 may be the replisome organizer, bearing in its gene sequence the origin of replication, as it occurs with its homolog the phage SPP1 G38P protein (Supplementary Figure S3) (24,36), or with the unrelated lambda O protein (37). In the λ phage, the P protein facilitates replication initiation through recruitment of the E. coli replicative helicase DnaB at the oriλ by its interaction with the O protein (38,39). Similarly, in phage SPP1, G38P binds specifically to the origin of replication (oriL) embedded in gene 38 (24,36). In this phage, the helicase and helicase loader are coded by the phage (40) and protein–protein interactions between the phage replicative helicase and the host DnaG primase and the PolIII holoenzyme are essential for recruitment and assembly of a replisome at phage SPP1 oriL site (41,42). Since phage 80α does not encode a DNA helicase, and 80α ORF20 and ORF21 are necessary and sufficient to drive autonomous replication in S. aureus, the product of the 80α ORF21, a protein homologous to the E. coli DnaC-helicase loader (DnaI helicase loader in Gram positives), may be involved in the initiation of DNA replication by recruitment of the host replicative helicase (the DnaC replicative helicase in Gram positives) at the viral origin by protein-protein interaction (43).
We propose that similarly to the λ and SPP1 phages, in phage 80α DNA replication is initiated by the θ mode, and shifts to σ replication, which produces concatemeric DNA. Although extensively studied, the exact mechanism regulating the switch from θ to σ replication in these two phages has not been completely deciphered. In phage λ, host DnaA is considered the main factor involved in the control of the switch from θ- to σ-mode of DNA replication (44–48). In phage SPP1, it is believed that G38P tightly bound to the other origin of replication may cause replication stalling and formation of a double strand break. This substrate could be used by the G35P recombinase to re-start DNA replication by the σ mechanism (11). Our mutational analyses showed that mutations in the 80α genes 16 and 17, coding for a recombinase (Sak family) and for an Ssb respectively, affected DNA replication and concatemeric DNA formation. The involvement of recombinases and Ssb proteins in viral DNA replication was further explored in another staphylococcal phage, ϕ11. This phage also codes for a recombinase-Ssb pair although the recombinase belongs to a different protein family, the Sak4 family, and its Ssb has limited homology with the 80α Ssb. The same defect in DNA replication was observed with the ϕ11 mutants. To our knowledge, this study is pioneering in the analyses of the roles of the Sak and Sak4 families of recombinases in DNA replication. We also show for the first time that these proteins work in concert with their cognate Ssb.
Previous work has characterized some recombinases of the Redβ family, and different results were obtained regarding their role in the phage cycle. G35P (Redβ family) from phage SPP1 is an essential protein (11). The Redβ recombinase from phage λ is not essential for λ growth in wild-type hosts (49), although λ red mutants grow less well than the wild-type phages (50), and both Redα (a 5΄ to 3΄ exonuclease) and Redβ become essential in recA mutant strains (51). Similarly, Salmonella phage P22 erf gene is only essential in recA and recJ mutant hosts (52,53), and again the P22 erf mutant grows less well than wild-type P22 following infection of wild-type host cells (52).
Our results show that not only Sak and Sak4 recombinases, but also their cognate Ssb proteins are required for DNA replication. Interestingly in the lactococcal phage p2, in addition to a Sak recombinase there is a Ssb protein that, as occurs with RecA, interacts with the Sak protein (9), although its role in recombination or in vivo has not been analysed. In some coliphages like T4 and T7, phage-encoded Ssbs are essential for DNA replication and cannot be replaced by their host counterparts with which they share no homology (54,55). In contrast, B. subtilis phage SPP1 encodes a protein highly homologous to the bacterial-host Ssb (56), and experiments in vitro and in vivo showed that the bacterial SsbA protein can replace the viral SSB (G36P) at the SPP1 replisome (25). Remarkably, our experiments showed that none of the Ssb proteins encoded in the S. aureus genome could complement the phages mutant in the ssb genes. The genome of S. aureus codes for two single-stranded DNA binding proteins called SsbA and SsbB (29), as occurs in other Gram positive bacteria (57) (58). SsbA shows the typical features of the essential bacterial Ssbs, which are homotetramers consisting of four short polypeptides (160–180 aa) with an amphipathic C-terminus involved in the interaction with numerous proteins involved in DNA replication, repair and recombination (59,60). SsbB proteins are non-essential, shorter (130–110 aa), and share identity with the N-terminal DNA-binding domain of SsbA, but lack the characteristic C-terminal region that mediates protein interactions in SsbA (57). The Ssbs encoded by the 80α and ϕ11 phages are very similar to the SsbA protein, and remarkably the C-terminal region is almost identical (Supplementary Figure S5). This homology is even higher between the 80α-Ssb and host SsbA (Supplementary Figure S5). Although the ssb mutants are not affected as the sak or sak4 mutants, the 80α ssb mutant shows a 100-fold reduction in the phage titer, while the ϕ11 ssb mutant shows a 500-fold reduction in the phage titer. These results suggest that despite its sequence identity, the host SsbA can only partially replace the viral Ssb proteins, which are required for phage replication, at least in the two staphylococcal phages we tested (80α and ϕ11). The fact that the Ssb proteins interact with their cognate SSAPs also suggests that both families of proteins work co-ordinately. Thus, as proposed for the SSAPs proteins, our current hypothesis is that the Ssb proteins could be involved in the transition between the θ and σ replication. This is currently under study.
Although deletion of the sak and sak4 genes generated a strong defect in phage replication, it was still possible to see in the PFGE analysis some concatemeric forms in the mutant phages. We hypothesize that in these phages, RecA-dependent and RecA-independent pathways of recombination could occur as reported for phage λ (22). In the RecA-independent pathway Sak and Sak4 recombinases may be the main actors in the generation of a concatemer by recombination. In the RecA-dependent pathway, Sak and Sak4 recombinases may act as mediators of RecA, so the concatemeric DNA observed in these mutants may be due to a poorly efficient recombination reaction performed by the host RecA protein in the absence of the SSAPs mediators.
The 80α Sak protein does not have the ATP binding (Walker A) and hydrolysis (Walker B) motifs that are present in many but not all lactococcal Sak proteins (Supplementary Figure S2). Our biochemical analysis showed that 80α Sak protein is able to bind and anneal ssDNA in the absence of ATP. In contrast, the phage p2 Sak3 protein was shown to possess an ATPase activity that is required both for RecA stimulation and DNA binding. None of the staphylococcal Sak proteins that we analyzed possess the ATP binding and hydrolysis motifs (Supplementary Figures S2 and S8). Interestingly, the ATPase domain is widely conserved in the Sak4 superfamily (8). However, mutation of the Walker A motif in the Sak4 recombinase from Pseudomonas aeruginosa phage PA73 did not affect its recombination activity, raising questions as to why this domain is required in some but not in all Sak proteins.
What are the roles in vivo for the staphylococcal recombinases? Staphylococcal phages have extensive genome mosaicism (61), which, as occurs in other systems, could be attributed to the presence of different recombinases in the phages’ genomes (7). Furthermore, a recent report has shown that different prophage proteins of unknown function activate the host Stk2 kinase, blocking phage infection (62). All the activator proteins described in that report as of unknown functions show a high degree of homology with either the Sak recombinase or the Sak4 protein described in this work, suggesting a novel role for the recombinases. We show in this report that all the recombinases are essential for phage reproduction. Here two modes of recombination, a single-stranded DNA-annealing pathway and a RecA-assisted pathway might take place in the viral replication cycle for the formation of the viral concatemer.
Supplementary Material
Footnotes
Present address: Ignacio Mir-Sanchis, Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, IL, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Medical Research Council (UK) [MR/M003876/1]; Biotechnology and Biological Sciences Research Council (BBSRC, UK) [BB/N002873/1]; from European Union [ERC-ADG-2014 Proposal n° 670932 Dut-signal to J.R.P.]; MINECO (Spain) [BFU2012-39879-C02-02, BFU2015-67065-P to S.A.]; MINECO (Spain) [BIO2013-42619-P]; Valencian Government [Prometeo II/2014/029 to A.M.]. Funding for open access charge: Medical Research Council (UK).
Conflict of interest statement. None declared.
REFERENCES
- 1. Salmond G.P.C., Fineran P.C.. A century of the phage: past, present and future. Nat. Rev. Microbiol. 2015; 13:777–786. [DOI] [PubMed] [Google Scholar]
- 2. Brüssow H., Canchaya C., Hardt W.-D.. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004; 68:560–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Viana D., Comos M., McAdam P.R., Ward M.J., Selva L., Guinane C.M., González-Muñoz B.M., Tristan A., Foster S.J., Fitzgerald J.R. et al. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat. Genet. 2015; 47:361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Penadés J.R., Christie G.E.. The phage-inducible chromosomal islands: a family of highly evolved molecular parasites. Annu. Rev. Virol. 2015; 2:181–201. [DOI] [PubMed] [Google Scholar]
- 5. Viana D., Blanco J., Tormo-Más M.Á., Selva L., Guinane C.M., Baselga R., Corpa J.M., Lasa I., Novick R.P., Fitzgerald J.R. et al. Adaptation of Staphylococcus aureus to ruminant and equine hosts involves SaPI-carried variants of von Willebrand factor-binding protein. Mol. Microbiol. 2010; 77:1583–1594. [DOI] [PubMed] [Google Scholar]
- 6. Datta S., Costantino N., Zhou X., Court D.L.. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:1626–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Menouni R., Hutinet G., Petit M.-A., Ansaldi M.. Bacterial genome remodeling through bacteriophage recombination. FEMS Microbiol. Lett. 2015; 362:1–10. [DOI] [PubMed] [Google Scholar]
- 8. Lopes A., Amarir-Bouhram J., Faure G., Petit M.-A., Guerois R.. Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51 and Gp2.5 remote homologs. Nucleic Acids Res. 2010; 38:3952–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scaltriti E., Launay H., Genois M.-M., Bron P., Rivetti C., Grolli S., Ploquin M., Campanacci V., Tegoni M., Cambillau C. et al. Lactococcal phage p2 ORF35-Sak3 is an ATPase involved in DNA recombination and AbiK mechanism. Mol. Microbiol. 2011; 80:102–116. [DOI] [PubMed] [Google Scholar]
- 10. De Paepe M., Hutinet G., Son O., Amarir-Bouhram J., Schbath S., Petit M.-A.. Temperate phages acquire DNA from defective prophages by relaxed homologous recombination: the role of Rad52-like recombinases. PLoS Genet. 2014; 10:e1004181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ayora S., Missich R., Mesa P., Lurz R., Yang S., Egelman E.H., Alonso J.C.. Homologous-pairing activity of the Bacillus subtilis bacteriophage SPP1 replication protein G35P. J. Biol. Chem. 2002; 277:35969–35979. [DOI] [PubMed] [Google Scholar]
- 12. Botstein D., Matz M.J.. A recombination function essential to the growth of bacteriophage P22. J. Mol. Biol. 1970; 54:417–440. [DOI] [PubMed] [Google Scholar]
- 13. Delattre H., Souiai O., Fagoonee K., Guerois R., Petit M.-A.. Phagonaute: A web-based interface for phage synteny browsing and protein function prediction. Virology. 2016; 496:42–50. [DOI] [PubMed] [Google Scholar]
- 14. Tormo-Más M.Á., Mir I., Shrestha A., Tallent S.M., Campoy S., Lasa I., Barbé J., Novick R.P., Christie G.E., Penadés J.R.. Moonlighting bacteriophage proteins derepress staphylococcal pathogenicity islands. Nature. 2010; 465:779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tormo-Más M.Á., Donderis J., García-Caballer M., Alt A., Mir-Sanchis I., Marina A., Penadés J.R.. Phage dUTPases control transfer of virulence genes by a proto-oncogenic G protein-like mechanism. Mol. Cell. 2013; 49:947–958. [DOI] [PubMed] [Google Scholar]
- 16. Arnaud M., Chastanet A., Débarbouillé M.. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 2004; 70:6887–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quiles-Puchalt N., Carpena N., Alonso J.C., Novick R.P., Marina A., Penadés J.R.. Staphylococcal pathogenicity island DNA packaging system involving cos-site packaging and phage-encoded HNH endonucleases. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:6016–6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Charpentier E., Anton A.I., Barry P., Alfonso B., Fang Y., Novick R.P.. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 2004; 70:6076–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 20. Zecchi L., Piano Lo A., Suzuki Y., Cañas C., Takeyasu K., Ayora S.. Characterization of the Holliday junction resolving enzyme encoded by the Bacillus subtilis bacteriophage SPP1. PLoS ONE. 2012; 7:e48440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferrer M.D., Quiles-Puchalt N., Harwich M.D., Tormo-Más M.Á., Campoy S., Barbé J., Lasa I., Novick R.P., Christie G.E., Penadés J.R.. RinA controls phage-mediated packaging and transfer of virulence genes in Gram-positive bacteria. Nucleic Acids Res. 2011; 39:5866–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stahl F.W. Recombination in phage lambda: one geneticist's historical perspective. Gene. 1998; 223:95–102. [DOI] [PubMed] [Google Scholar]
- 23. Weigel C., Seitz H.. Bacteriophage replication modules. FEMS Microbiol. Rev. 2006; 30:321–381. [DOI] [PubMed] [Google Scholar]
- 24. Missich R., Weise F., Chai S., Lurz R., Pedré X., Alonso J.C.. The replisome organizer (G38P) of Bacillus subtilis bacteriophage SPP1 forms specialized nucleoprotein complexes with two discrete distant regions of the SPP1 genome. J. Mol. Biol. 1997; 270:50–64. [DOI] [PubMed] [Google Scholar]
- 25. Seco E.M., Zinder J.C., Manhart C.M., Piano Lo, A., McHenry C.S., Ayora S.. Bacteriophage SPP1 DNA replication strategies promote viral and disable host replication in vitro. Nucleic Acids Res. 2013; 41:1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christie G.E., Matthews A.M., King D.G., Lane K.D., Olivarez N.P., Tallent S.M., Gill S.R., Novick R.P.. The complete genomes of Staphylococcus aureus bacteriophages 80 and 80α–implications for the specificity of SaPI mobilization. Virology. 2010; 407:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Funnell B.E., Baker T.A., Kornberg A.. In vitro assembly of a prepriming complex at the origin of the Escherichia coli chromosome. J. Biol. Chem. 1987; 262:10327–10334. [PubMed] [Google Scholar]
- 28. Quiles-Puchalt N., Tormo-Más M.Á., Campoy S., Toledo-Arana A., Monedero V., Lasa I., Novick R.P., Christie G.E., Penadés J.R.. A super-family of transcriptional activators regulates bacteriophage packaging and lysis in Gram-positive bacteria. Nucleic Acids Res. 2013; 41:7260–7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glanzer J.G., Endres J.L., Byrne B.M., Liu S., Bayles K.W., Oakley G.G.. Identification of inhibitors for single-stranded DNA-binding proteins in eubacteria. J. Antimicrob. Chemother. 2016; 71:3432–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Węgrzyn G., Węgrzyn A.. Genetic switches during bacteriophage lambda development. Prog. Nucleic Acid Res. Mol. Biol. 2005; 79:1–48. [DOI] [PubMed] [Google Scholar]
- 31. Rao V.B., Feiss M.. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 2008; 42:647–681. [DOI] [PubMed] [Google Scholar]
- 32. Piano Lo, A., Martínez-Jiménez M.I., Zecchi L., Ayora S.. Recombination-dependent concatemeric viral DNA replication. Virus Res. 2011; 160:1–14. [DOI] [PubMed] [Google Scholar]
- 33. Oliveira L., Tavares P., Alonso J.C.. Headful DNA packaging: bacteriophage SPP1 as a model system. Virus Res. 2013; 173:247–259. [DOI] [PubMed] [Google Scholar]
- 34. Quiles-Puchalt N., Martínez-Rubio R., Ram G., Lasa I., Penadés J.R.. Unravelling bacteriophage ϕ11 requirements for packaging and transfer of mobile genetic elements in Staphylococcus aureus. Mol. Microbiol. 2014; 91:423–437. [DOI] [PubMed] [Google Scholar]
- 35. Bouchard J.D., Moineau S.. Lactococcal phage genes involved in sensitivity to AbiK and their relation to single-strand annealing proteins. J. Bacteriol. 2004; 186:3649–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pedré X., Weise F., Chai S., Lüder G., Alonso J.C.. Analysis of cis and trans acting elements required for the initiation of DNA replication in the Bacillus subtilis bacteriophage SPP1. J. Mol. Biol. 1994; 236:1324–1340. [DOI] [PubMed] [Google Scholar]
- 37. Denniston-Thompson K., Moore D.D., Kruger K.E., Furth M.E., Blattner F.R.. Physical structure of the replication origin of bacteriophage lambda. Science. 1977; 198:1051–1056. [DOI] [PubMed] [Google Scholar]
- 38. Mallory J.B., Alfano C., McMacken R.. Host virus interactions in the initiation of bacteriophage lambda DNA replication. Recruitment of Escherichia coli DnaB helicase by lambda P replication protein. J. Biol. Chem. 1990; 265:13297–13307. [PubMed] [Google Scholar]
- 39. Alfano C., McMacken R.. Ordered assembly of nucleoprotein structures at the bacteriophage lambda replication origin during the initiation of DNA replication. J. Biol. Chem. 1989; 264:10699–10708. [PubMed] [Google Scholar]
- 40. Ayora S., Stasiak A., Alonso J.C.. The Bacillus subtilis bacteriophage SPP1 G39P delivers and activates the G40P DNA helicase upon interacting with the G38P-bound replication origin. J. Mol. Biol. 1999; 288:71–85. [DOI] [PubMed] [Google Scholar]
- 41. Ayora S., Langer U., Alonso J.C.. Bacillus subtilis DnaG primase stabilises the bacteriophage SPP1 G40P helicase-ssDNA complex. FEBS Lett. 1998; 439:59–62. [DOI] [PubMed] [Google Scholar]
- 42. Martínez-Jiménez M.I., Mesa P., Alonso J.C.. Bacillus subtilis tau subunit of DNA polymerase III interacts with bacteriophage SPP1 replicative DNA helicase G40P. Nucleic Acids Res. 2002; 30:5056–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hood I.V., Berger J.M.. Viral hijacking of a replicative helicase loader and its implications for helicase loading control and phage replication. Elife. 2016; 5:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baranska S., Gabig M., Wegrzyn A., Konopa G., Herman-Antosiewicz A., Hernandez P., Schvartzman J.B., Helinski D.R., Wegrzyn G.. Regulation of the switch from early to late bacteriophage lambda DNA replication. Microbiology (Reading, Engl.). 2001; 147:535–547. [DOI] [PubMed] [Google Scholar]
- 45. Narajczyk M., Barańska S., Węgrzyn A., Węgrzyn G.. Switch from theta to sigma replication of bacteriophage lambda DNA: factors involved in the process and a model for its regulation. Mol. Genet. Genomics. 2007; 278:65–74. [DOI] [PubMed] [Google Scholar]
- 46. Narajczyk M., Barańska S., Szambowska A., Glinkowska M., Węgrzyn A., Węgrzyn G.. Modulation of lambda plasmid and phage DNA replication by Escherichia coli SeqA protein. Microbiology (Reading, Engl.). 2007; 153:1653–1663. [DOI] [PubMed] [Google Scholar]
- 47. Węgrzyn G., Licznerska K., Węgrzyn A.. Phage λ—new insights into regulatory circuits. Adv. Virus Res. 2012; 82:155–178. [DOI] [PubMed] [Google Scholar]
- 48. Wegrzyn G., Wegrzyn A., Konieczny I., Bielawski K., Konopa G., Obuchowski M., Helinski D.R., Taylor K.. Involvement of the host initiator function dnaA in the replication of coliphage lambda. Genetics. 1995; 139:1469–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Manly K.F., Signer E.R., Radding C.M.. Nonessential functions of bacteriophage lambda. Virology. 1969; 37:177–188. [DOI] [PubMed] [Google Scholar]
- 50. Echolas H., Gingery R.. Mutants of bacteriophage lambda defective in vegetative genetic recombination. J. Mol. Biol. 1968; 34:239–249. [DOI] [PubMed] [Google Scholar]
- 51. Enquist L.W., Skalka A.. Replication of bacteriophage lambda DNA dependent on the function of host and viral genes. I. Interaction of red, gam and rec. J. Mol. Biol. 1973; 75:185–212. [DOI] [PubMed] [Google Scholar]
- 52. Fenton A.C., Poteete A.R.. Genetic analysis of the erf region of the bacteriophage P22 chromosome. Virology. 1984; 134:148–160. [DOI] [PubMed] [Google Scholar]
- 53. Mahan M.J., Garzón A., Casadesús J.. Host RecJ is required for growth of P22 erf bacteriophage. J. Bacteriol. 1993; 175:288–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim Y.T., Richardson C.C.. Bacteriophage T7 gene 2.5 protein: an essential protein for DNA replication. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:10173–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alberts B.M., Frey L.. T4 bacteriophage gene 32: a structural protein in the replication and recombination of DNA. Nature. 1970; 227:1313–1318. [DOI] [PubMed] [Google Scholar]
- 56. Alonso J.C., Lüder G., Stiege A.C., Chai S., Weise F., Trautner T.A.. The complete nucleotide sequence and functional organization of Bacillus subtilis bacteriophage SPP1. Gene. 1997; 204:201–212. [DOI] [PubMed] [Google Scholar]
- 57. Yadav T., Carrasco B., Myers A.R., George N.P., Keck J.L., Alonso J.C.. Genetic recombination in Bacillus subtilis: a division of labor between two single-strand DNA-binding proteins. Nucleic Acids Res. 2012; 40:5546–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yadav T., Carrasco B., Hejna J., Suzuki Y., Takeyasu K., Alonso J.C.. Bacillus subtilis DprA recruits RecA onto single-stranded DNA and mediates annealing of complementary strands coated by SsbB and SsbA. J. Biol. Chem. 2013; 288:22437–22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shereda R.D., Kozlov A.G., Lohman T.M., Cox M.M., Keck J.L.. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008; 43:289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Costes A., Lecointe F., McGovern S., Quevillon-Cheruel S., Polard P.. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010; 6:e1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kwan T., Liu J., DuBow M., Gros P., Pelletier J.. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:5174–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Depardieu F., Didier J.-P., Bernheim A., Sherlock A., Molina H., Duclos B., Bikard D.. A eukaryotic-like serine/threonine kinase protects Staphylococci against phages. Cell Host Microbe. 2016; 20:471–481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.