

Abstract
An unprecedented method that makes use of the cooperative interplay between molecular iodine and photoredox catalysis has been developed for dual light‐activated intramolecular benzylic C−H amination. Iodine serves as the catalyst for the formation of a new C−N bond by activating a remote C −H bond (1,5‐HAT process) under visible‐light irradiation while the organic photoredox catalyst TPT effects the reoxidation of the molecular iodine catalyst. To explain the compatibility of the two involved photochemical steps, the key N−I bond activation was elucidated by computational methods. The new cooperative catalysis has important implications for the combination of non‐metallic main‐group catalysis with photocatalysis.
Keywords: 1,5-HAT processes; amination; cooperative catalysis; iodine catalysis; photoredox catalysis
A fundamental goal in developing new catalytic processes is to establish environmentally benign and effective conditions. The activation of small organic molecules by photoredox catalysis under visible‐light irradiation has opened up a whole new field in catalysis, leading to a plethora of effective transformations.1 Here, processes merging transition‐metal and photoredox catalysis are of exceptional value as the utilization of stoichiometric amounts of oxidizing reagents other than molecular oxygen is no longer required.2 Although this strategy has enabled the development of organic transformations, including C−H bond activations with a number of transition‐metal catalysts,3 no strategies for combining photoredox‐catalyzed reoxidation with iodine redox catalysis have been reported to date.4 Devising a practical method for the functionalization of non‐activated C −H bonds still represents a great challenge in modern organic synthesis. Transition‐metal‐catalyzed transformations of C−H bonds into new C−C and C−heteroatom bonds have been established over the past years, but this strategy usually requires functional groups in close proximity to ensure regioselectivity and to overcome activation barriers.5 The modification of remote positions, however, by C−H bond activation is even more challenging. Lately, our group developed an iodine‐catalyzed oxidative Hofmann–Löffler reaction of sulfonamides for the formation of pyrrolidines (Figure 1).6a In this process, an in situ formed N−I bond facilitates the formation of a nitrogen‐centered amidyl radical, which induces a selective 1,5‐hydrogen atom transfer (1,5‐HAT) process,7 activating a remote C −H bond for subsequent C−N bond formation. While the reaction is synthetically efficient, the requirement for stoichiometric amounts of hypervalent iodine reagents indicates a clear disadvantage. Rovis and Knowles independently reported a photoredox‐based concept for the generation of amidyl radicals and subsequent activation of C −H bonds by a 1,5‐HAT process (Figure 1).8, 9 They extended the applicability of these processes by intermolecular radical interception with activated alkenes. Within this new C−C bond‐forming approach, however, an amination reaction is no longer feasible, and the role of the amide is restricted to the generation of the requisite radicals.
Figure 1.
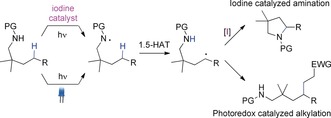
Recent examples for the nitrogen‐promoted application of 1,5‐HAT processes for the functionalization of remote C −H bonds. EWG=electron‐withdrawing group, PG=protecting group.
To further improve the photochemical amidyl‐radical‐promoted C −H bond activation, we envisaged combining a 1,5‐HAT process and a subsequent cyclization reaction to reestablish the original pathway to synthetically valuable pyrrolidines. We surmised that molecular iodine would play an essential role in the cyclization step of the envisaged process.10 In a cooperative catalysis with two individual photoinduced processes, the iodine catalyst would promote pyrrolidine formation through activation of the N−H bond with a subsequent 1,5‐HAT process, while the photoredox catalyst would enable the reoxidation of the iodine that is extruded during the cyclization event. The two catalysts would therefore be working cooperatively, and this process would constitute the first photoredox‐catalyzed reoxidation of a molecular homogeneous iodine catalyst. As we intended to develop a metal‐free process and because of iodine's known ability to quench metal‐to‐ligand charge transfer (MLCT) excited states of metal‐based photoredox catalysts,11 organic dyes were the preferred choice.12
To this end, a solution of the model substrate 1 a, molecular iodine (10 mol %), and a photoredox catalyst (5 mol %) in 1,2‐dichloroethane (DCE) under anhydrous or oxygen‐free conditions was exposed to blue light (λ max=456 nm±12 nm) at room temperature. However, for all reactions with different organic dyes (eosin Y, Fukuzumi's catalyst, and TPT; Figure 2), only trace amounts of the desired pyrrolidine 2 a were detected (Table 1, entry 1).
Figure 2.
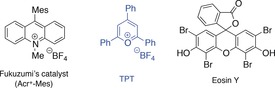
Organic dyes tested as photoredox catalysts. Mes=2,4,6‐trimethylphenyl.
Table 1.
Optimization of the reaction conditions for the iodine/photoredox‐catalyzed process.[a]

| Entry | x | y | Solvent | Yield [%] |
|---|---|---|---|---|
| 1[b,c] | 5 | 10 | DCE | trace |
| 2[c] | 5 | 10 | DCE | 20 |
| 3 | 5 | 10 | HFIP/DCE | 84 |
| 4 | 2 | 10 | HFIP/DCE | 58 |
| 5 | 2 | 5 | HFIP/DCE | 80 |
| 6 | 1 | 5 | HFIP/DCE | 90 |
| 7 | 1 | 2 | HFIP/DCE | 31[d] |
| 8 | 0.5 | 5 | HFIP/DCE | 76[d] |
| 9 | – | 5 | HFIP/DCE | 5[d] |
| 10 | 1 | – | HFIP/DCE | 14[d] |
| 11 | 1 | 5 | HFIP/DCE | –[e] |
| 12 | 1 | 5 | HFIP/DCE | 83[f] |
[a] All reactions were performed with 0.3 mmol of 1 a and stirred for 18 h under blue light irradiation in 3 mL of solvent (1:1) without external heating. Yields refer to isolated material after purification. [b] Reaction performed under anhydrous or oxygen‐free conditions. [c] The use of eosin Y or Fukuzumi's catalyst (Figure 2) led to trace amounts of the product. [d] Yield determined by 1H NMR spectroscopy with 1,3,5‐trimethoxybenzene as the internal standard. [e] Reaction performed in the absence of light. [f] Reaction performed on 2 mmol scale, 22 h.
The first positive result was obtained when the reaction was performed in unpurified solvent with TPT13 as the photoredox catalyst of choice under air (20 %; entry 2). A solvent mixture of DCE and hexafluoropropan‐2‐ol (HFIP)14 dramatically improved the yield of 2 a to 84 % (entry 3). When the TPT loading was reduced to 2 mol %, the yield dropped to 58 % (entry 4). However, a simultaneous reduction in the amount of iodine (5 mol %) led to similar yields (80 %; entry 5), which indicates that iodine causes an unproductive absorption of visible light at high concentrations. With only 5 mol % of iodine, the TPT loading was further reduced to 1 mol %, leading to 90 % of 2 a (entry 6). A further reduction in the amount of iodine (2 mol %) only gave 31 % of the desired product (entry 7), accompanied by large amounts of side products stemming from overoxidation. With 0.5 mol % of TPT, 2 a was still formed in 76 % yield (entry 8). These results indicate the importance of the I2/TPT ratio for the effectiveness of the cooperative catalysis. To probe the necessity of both catalysts, control experiments were conducted. With iodine alone, 2 a was formed in 5 % yield, which corresponds to a stoichiometric reaction of the molecular iodine (entry 9). When TPT was added as the only catalyst, pyrrolidine 2 a was formed in 14 % yield along with large amounts of decomposition products (entry 10). The pyrrolidine was not formed in the absence of light, demonstrating that the reaction is indeed light‐driven (entry 11). At this point, we would like to emphasize that the intensity of the LED light source was found to be crucial.15 To finally demonstrate the robustness of the process, the reaction was carried out on 2 mmol scale, leading to 83 % of 2 a under the standard conditions with a slightly increased reaction time (entry 12).
Mechanistically, we propose a reaction sequence consisting of two individual light‐induced catalytic reactions within several intertwined individual cycles (Figure 3). Raman spectroscopy was employed to identify the active molecular iodine catalyst. Rapid formation of hypoiodite16 through disproportionation of the molecular iodine was observed in the wet reaction medium, which explains the requirement for water traces to initiate the reaction.15 This catalyst state is in agreement with Ishihara's work on related hypoiodite catalysis.17 The concomitantly formed HI plays an essential rule in the final catalyst reoxidation event (cycle A).
Figure 3.
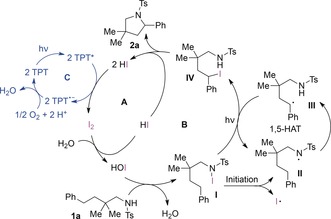
Proposed mechanism for the dual light‐induced cooperative iodine‐ and photoredox‐catalyzed intramolecular amination of sulfonamides. Ts=4‐methylbenzenesulfonyl.
The active hypoiodite catalyst initiates the organic transformation by N‐iodination of substrate 1 a to form the intermediate species I (cycle B).18 Upon irradiation with visible light, the N‐iodinated species I collapses to generate the amidyl radical species II. A subsequent 1,5‐HAT process (III) within a radical chain reaction generates alkyl iodide IV. Isotope‐labeling experiments19 revealed the hydrogen abstraction from II to III to be the rate‐limiting step with a kinetic isotope effect of 2.3.15 The successful incorporation of iodine into the carbon framework precludes the requirement for intermolecular radical quenching.8a,8b It drives the reaction to the C−N bond formation, in which the sulfonamide attacks the intermediary C−I bond in an intramolecular substitution reaction to form pyrrolidine 2 a as the product. The extruded hydrogen iodide is then effectively reoxidized by TPT to molecular iodine in a single‐electron‐transfer process with molecular oxygen (cycle C).20 This discussion characterizes the new iodine photoredox catalysis as an unprecedented iodine (−I/I) manifold, and demonstrates for the first time that the catalytic functionalization of benzylic positions is possible without the involvement of iodine(III) oxidation states.
The conceptually most important feature of this reaction is the compatibility of the two roles of light. First, it participates in the photoredox catalysis that provides the terminal oxidant for the overall process. Second, it is involved in the homolytic cleavage of the N−I bond as the initial step of the C−H bond functionalization. To understand the molecular requirements for the elusive light‐induced N−I bond cleavage at stage I, theory was employed.
The excitation energies for the ten lowest excited states of species I were calculated by time‐dependent density functional theory (TDDFT) with the range‐separated CAMY‐B3LYP functional,21 and environmental effects were included with a continuum solvation model. We accounted for the pronounced conformational freedom by carrying out TDDFT calculations on a closed and an open structure, which were both optimized by DFT with the PBE functional.22 The energies of both structures differ by less than 2.5 kcal mol−1 in favor of the open conformation. With an empirical dispersion correction,23 both energies are almost identical, with the closed conformation being 0.15 kcal mol−1 lower in energy. A Table with the energies and oscillator strengths for all states and the computational setups and more details on the method are included in the Supporting Information.15
The seven lowest excited states correspond to excitations into the N−I antibonding lowest unoccupied molecular orbital (LUMO), and therefore weaken this bond and facilitate bond cleavage (Figure 4). Depending on the computational setup, the transition energies of these excitations cover a range of λ=100–130 nm with the lowest transition energy being approximately λ=380 nm. Several effects shift the individual transition energies. One of them is the conformational freedom of I, which causes a shift of up to Δλ=15 nm for the open and closed structures considered in this study, and other conformers might further extend that range. Another effect is the direct interaction of solvent molecules with I, which leads to a significant broadening of the individual bands owing to a plethora of possible interactions. Furthermore, the error of TDDFT transition energies lies typically in the range of 0.2 eV,24 which corresponds to a wavelength of about λ=30 nm. These effects, and the fact that the photolytic reaction does not need to be highly efficient owing to the long reaction times, explain the discrepancy between the calculated lowest energy transitions and the LED wavelength. While we were mainly interested in identifying the character of the excitations that cause the photolytic reaction, we expect this reaction step to be more efficient for shorter wavelengths. It should be noted that both the inclusion of solvent effects and structural changes in both conformers have a pronounced effect on the oscillator strength.
Figure 4.
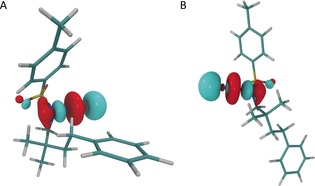
N−I antibonding LUMO of intermediate I in closed (A) and open (B) conformations. The orbitals were calculated by DFT employing the range‐separated CAMY‐B3LYP functional.
We confirmed that the lowest excited states have mainly LUMO character with complete‐active‐space self‐consistent‐field calculations25 employing an active space consisting of 14 electrons in 16 orbitals, where the orbitals were selected with our automated active‐space‐selection protocol.26
With this concise mechanistic explanation of the dual light‐activated iodine‐ and photoredox‐catalyzed C−H amination and the theoretical understanding of the photolytic cleavage of the N−I bond in hand, we investigated the scope of the reaction (Figure 5).
Figure 5.
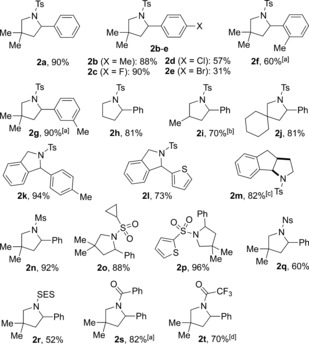
Scope of the intertwined iodine‐ and photoredox‐catalyzed cyclization. Reaction conditions: 1 (0.3 mmol), I2 (5 mol %), TPT (1 mol %), HFIP/DCE (3 mL), RT, blue light, 18 h. All C−N bonds formed in this process are highlighted in blue. Yields of isolated product after purification by column chromatography are generally given. [a] Two rotamers were observed by NMR spectroscopy. [b] Isolated as a 1:1 diastereomeric mixture. [c] Isolated as a single diastereomer. [d] Yield determined by 1H NMR spectroscopy with 1,3,5‐trimethoxybenzene as the internal standard. Ms=methanesulfonyl, Ns= 4‐nitrobenzenesulfonyl, SES=2‐(trimethylsilyl)ethanesulfonyl.
This method constitutes an effective procedure for the synthesis of various 2‐arylpyrrolidines in good to excellent yields. Substituents in the 4‐position of the aryl group are well tolerated (2 a–e; 31–90 %), with only the 4‐chloro‐ and 4‐bromo‐substituted substrates leading to decreased yields because of overoxidation (2 d, e; 57 and 31 %) by the photoredox catalyst. Substrates with 2‐ or 3‐substituted arenes also formed pyrrolidines 2 f and 2 g in 60 and 90 % yield, respectively, demonstrating that steric factors play only a minor role. A possible Thorpe–Ingold effect was shown to have little influence on the reaction as different substitution patterns in the β‐position of the amide were tolerated (2 h–j, m; 70–82 %). Substrates with a phenyl backbone were then subjected to the reaction conditions, effectively yielding previously inaccessible isoindolines (2 k, l; 94 and 73 %). Different sulfonyl groups are also well tolerated in this process, leading to 60–96 % of the desired pyrrolidines 2 n–r. For the first time, benzamide and trifluoroacetamide derivatives are also applicable, leading to the corresponding pyrrolidines 2 s and 2 t in high yields (82 and 70 %).27 In contrast to the previously described iodine(I/III) catalysis for the Hofmann–Löffler reaction, the intermediary benzyl iodide of the iodine(−I/I) approach holds a weaker nucleofuge, requiring additional activation by the benzylic position for internal substitution by the amide nucleophile.
In summary, we have introduced a new concept based on dual light‐activated cooperative iodine and photoredox catalysis and applied it to the intramolecular amination of remote C −H bonds. The iodine acts as the primary catalyst enabling the activation of the C −H bond by light‐induced homolytic cleavage of in situ generated N−I bonds followed by a 1,5‐HAT process. After radical iodine transfer and intramolecular substitution, the molecular iodine catalyst is reoxidized by photoredox catalysis in a second light‐induced process. The key step of the reaction, the cleavage of the intermediary N−I bond, was rationalized by computational methods while the presence of the active iodine species, hypoiodite, was confirmed by Raman spectroscopy.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the Spanish Ministry for Economy and Competitiveness, FEDER (CTQ2014‐56474R grant to K.M. and Severo Ochoa Excellence Accreditation 2014–2018 to ICIQ, SEV‐2013‐0319), and the Schweizerischer Nationalfonds (No. 20020_169120 to M.R.). P.B. and C.J.S. thank the Alexander von Humboldt Foundation for a postdoctoral fellowship and the Fonds der Chemischen Industrie for a Kekulé fellowship, respectively. We are grateful to the CERCA Programme of the Government of Catalonia and to COST Action CA15106 “C−H Activation in Organic Synthesis (CHAOS)”.
P. Becker, T. Duhamel, C. J. Stein, M. Reiher, K. Muñiz, Angew. Chem. Int. Ed. 2017, 56, 8004.
Contributor Information
Prof. Dr. Markus Reiher, Email: markus.reiher@phys.chem.ethz.ch.
Prof. Dr. Kilian Muñiz, Email: kmuniz@iciq.es.
References
- 1.
- 1a. Shaw M. H., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898–6926; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For recent examples, see: Pd:
- 2a. Neufeldt S. R., Sanford M. S., Adv. Synth. Catal. 2012, 354, 3517; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Kalyani D., McMurtrey K. B., Neufeldt S. R., Sanford M. S., J. Am. Chem. Soc. 2011, 133, 18566; Au: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Hopkinson M. N., Sahoo B., Glorius F., Adv. Synth. Catal. 2014, 356, 2794; [Google Scholar]
- 2d. Shu X. Z., Zhang M., He Y., Frei H., Toste F. D., J. Am. Chem. Soc. 2014, 136, 5844; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Tlahuext-Aca A., Hopkinson M. N., Garza-Sanchez R. A., Glorius F., Chem. Eur. J. 2016, 22, 5909; Ni: [DOI] [PubMed] [Google Scholar]
- 2f. Tellis J. C., Primer D. N., Molander G. A., Science 2014, 345, 433; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2g. Noble A., McCarver S. J., MacMillan D. W. C., J. Am. Chem. Soc. 2015, 137, 624; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2h. Chu L., Lipshultz J. M., MacMillan D. W. C., Angew. Chem. Int. Ed. 2015, 54, 7929; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8040; [Google Scholar]
- 2i. Murphy J. J., Melchiorre P., Nature 2015, 524, 297. [DOI] [PubMed] [Google Scholar]
- 3.For reviews on C−H bond functionalization in combination with photoredox catalysis, see:
- 3a. Fabry D. C., Rueping M., Acc. Chem. Res. 2016, 49, 1969; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b.Qin, L. Zhu, S. Luo, Chem. Rev 2017, 117, https://doi.org/10.1021/acs.chemrev.6b00657, and references therein. [DOI] [PubMed]
- 4.For synthetic applications of hypervalent iodine(III) reagents under photoredox catalysis, see:
- 4a. Wang L., Liu J., Eur. J. Org. Chem. 2016, 1813, and references therein; [Google Scholar]
- 4b. Sakamoto R., Inada T., Selmadurai S., Moteki S. A., Maruoka K., Chem. Commun. 2016, 52, 3758. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Yan M., Lo J. C., Edwards J. T., Baran P. S., J. Am. Chem. Soc. 2016, 138, 12692; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Yamaguchi J., Yamaguchi A. D., Itami K., Angew. Chem. Int. Ed. 2012, 51, 8960; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9092. [Google Scholar]
- 6.For recent reports on the Hofmann–Löffler reaction, see:
- 6a. Martínez C., Muñiz K., Angew. Chem. Int. Ed. 2015, 54, 8287; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8405; [Google Scholar]
- 6b. O'Broin C. Q., Fernández P., Martínez C., Muñiz K., Org. Lett. 2016, 18, 436; [DOI] [PubMed] [Google Scholar]
- 6c. Wappes E. A., Fosu S. C., Chopko T. C., Nagib D. A., Angew. Chem. Int. Ed. 2016, 55, 9974; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10128; [Google Scholar]
- 6d. Paz N. R., Rodríguez-Sosa D., Valdés H., Marticorena R., Melián D., Copano M. B., González C. C., Herrera A. J., Org. Lett. 2015, 17, 2370. [DOI] [PubMed] [Google Scholar]
- 7.For references regarding HAT processes, see:
- 7a. Mayer J. M., Acc. Chem. Res. 2011, 44, 36; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Liu W., Huang X., Cheng M.-J., Nielsen R. J., W. A. Goddard III , Groves J. T., Science 2012, 337, 1322; [DOI] [PubMed] [Google Scholar]
- 7c. Jeffrey J. L., Terrett J. A., MacMillan D. W. C., Science 2015, 349, 1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Chu J. C. K., Rovis T., Nature 2016, 539, 272; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Choi G. J., Zhu Q., Miller D. C., Gu C. J., Knowles R. R., Nature 2016, 539, 268; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Hu X.-Q., Chen J.-R., Xiao W.-J., Angew. Chem. Int. Ed. 2017, 56, 1960; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1988. [Google Scholar]
- 9.For alternative metal-mediated methods for the generation of N-centered radicals, see:
- 9a. Xiong T., Zhang Q., Chem. Soc. Rev. 2016, 45, 3069; [DOI] [PubMed] [Google Scholar]
- 9b. Chen J.-R., Hu X.-Q., Hu L.-Q., Lu L.-Q., Xiao W.-J., Chem. Soc. Rev. 2016, 45, 2044; [DOI] [PubMed] [Google Scholar]
- 9c. Nguyen L. Q., Knowles R. R., ACS Catal. 2016, 6, 2894. [Google Scholar]
- 10.For general concepts for iodine-catalyzed C−H/N−H couplings, see:
- 10a. Finkbeiner P., Nachtsheim B., Synthesis 2013, 45, 979; [Google Scholar]
- 10b. Uyanik M., Ishihara K., ChemCatChem 2012, 4, 177; [Google Scholar]
- 10c. Li J., Lear M. J., Kawamoto Y., Umemiya S., Wong A. R., Kwon E., Sato I., Hayashi Y., Angew. Chem. Int. Ed. 2015, 54, 12986; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13178. [Google Scholar]
- 11.
- 11a. Clark C. C., Marton A., Meyer G. J., Inorg. Chem. 2005, 44, 3383; [DOI] [PubMed] [Google Scholar]
- 11b. Gardner J. M., Abrahamsson M., Famum B. H., Meyer G. J., J. Am. Chem. Soc. 2009, 131, 16206. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075; [DOI] [PubMed] [Google Scholar]
- 12b. Hari D. P., König B., Chem. Commun. 2014, 50, 6688; [DOI] [PubMed] [Google Scholar]
- 12c. Fukuzumi S., Ohkubo K., Org. Biomol. Chem. 2014, 12, 6059; [DOI] [PubMed] [Google Scholar]
- 12d. Majek M., von Wangelin A. J., Acc. Chem. Res. 2016, 49, 2316. [DOI] [PubMed] [Google Scholar]
- 13. Miranda M. A., García H., Chem. Rev. 1994, 94, 1063. [Google Scholar]
- 14.The beneficial effect of HFIP might be aiding the protonation of the intermediary hypoiodite species and shifting the disproportionation equilibrium.
- 15.Please see the Supporting Information for more experimental details.
- 16.In wet media, molecular iodine disproportionates into iodide and hypoiodite, among others; see the Supporting Information for details.
- 17. Uyanik M., Hayashi H., Ishihara K., Science 2014, 345, 291. [DOI] [PubMed] [Google Scholar]
- 18.This is in notable agreement with common stoichiometric Hofmann–Löffler reactions using the analogous hypobromite; see:
- 18a. Hofmann A. W., Ber. Dtsch. Chem. Ges. 1883, 16, 558; [Google Scholar]
- 18b. Löffler K., Ber. Dtsch. Chem. Ges. 1909, 42, 3427; [Google Scholar]
- 18c. Wolff M. E., Chem. Rev. 1963, 63, 55. [Google Scholar]
- 19. Corey E. J., Hertler W. R., J. Am. Chem. Soc. 1960, 82, 1657. [Google Scholar]
- 20.We cannot completely rule out the potential participation of the reduced TPT in the N−I activation step. However, this would only permit a stoichiometric radical recombination from III to IV.
- 21.
- 21a. Seth M., Ziegler T., J. Chem. Theory Comput. 2012, 8, 901; [DOI] [PubMed] [Google Scholar]
- 21b. Akinaga Y., Ten-no S., Chem. Phys. Lett. 2008, 462, 348. [Google Scholar]
- 22.
- 22a. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865; [DOI] [PubMed] [Google Scholar]
- 22b. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1997, 78, 1396. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Roos B. O., Taylor P. R., Siegbahn P. E., Chem. Phys. 1980, 48, 157; [Google Scholar]
- 23b. Werner H.-J., Knowles P. J., J. Chem. Phys. 1985, 82, 5053. [Google Scholar]
- 24. Casida M. E., J. Mol. Struct. THEOCHEM 2009, 914, 3. [Google Scholar]
- 25.
- 25a. Grimme S., J. Comput. Chem. 2004, 25, 1463; [DOI] [PubMed] [Google Scholar]
- 25b. Grimme S., J. Comput. Chem. 2006, 27, 1787; [DOI] [PubMed] [Google Scholar]
- 25c. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Stein C. J., Reiher M., J. Chem. Theory Comput. 2016, 12, 1760; [DOI] [PubMed] [Google Scholar]
- 26b. Stein C. J., Reiher M., Chimia 2017, 71, 170. [DOI] [PubMed] [Google Scholar]
- 27.In agreement with our presented calculations, the previous limitation on sulfonamides[6] is therefore not due to the potential role of this group as a photosensitizer. For a discussion of the nature of amides and their amidoyl radicals in Hofmann–Löffler reactions, see: Šakić D., Zipse H., Adv. Synth. Catal. 2016, 358, 3983. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary