

Abstract
Activation of insulin-like growth factor-1 (IGF-1) receptor (IGF1R) signaling induces keratinocyte migration, but little is known about its regulation, including in diabetic wounds. GM3, a lipid raft ganglioside synthesized by GM3 synthase (GM3S), regulates receptor signaling. In diabetic mice, knockout or topically applied nanoconstruct-mediated knockdown of GM3S promotes wound edge IGF1R phosphorylation and re-epithelialization. Through modulating GM3 expression, we explored the role of GM3 in regulating human keratinocyte IGF1R signaling. Increases in GM3 and GM3S expression, including by exposure to high glucose, inhibit keratinocyte migration and IGF-1–induced chemotaxis in association with inhibition of IGF1R phosphorylation, suppression of Rac1 signaling, and activation of RhoA signaling. In contrast, GM3 depletion accelerates cell migration; increases cell velocity, displacement, and persistence; and activates IGF1R-Rac1 signaling. These data implicate GM3 in mediating glucose-induced suppression of IGF1R-Rac1 signaling. Furthermore, our findings provide evidence of a pivotal role for GM3-induced insulin resistance in impairing keratinocyte migration and reinforce the previously published studies in diabetic mice supporting GM3-depleting strategies as an approach for accelerating the healing of human diabetic wounds.
INTRODUCTION
Chronic skin wounds, especially of the plantar surface, occur in approximately 15% of individuals with type 2 diabetes, leading to amputations in 70,000 Americans annually (American Diabetes Association, January 4, 2014). In type 2 diabetes, the insulin receptor (IR)/insulin-like growth factor-1 (IGF-1) receptor (IGF1R) signaling pathway is suppressed (“insulin resistance”), leading to chronic hyperglycemia and accumulation of glycosylated proteins. Poor diabetic control with chronic hyperglycemia has been linked to an increased risk of impaired acral healing (Christman et al., 2011; Markuson et al., 2009).
Normal wound healing requires migration of normal human epidermal keratinocytes (NHEKs) for re-epithelialization, but keratinocyte insulin resistance and its role in diabetic wound healing have received little attention. Activation of the IGF-1/insulin signaling axis leads to actin polymerization and lamellipodia formation, structural reorganization that depends on Rac1 activation (Haase et al., 2003; Stachelscheid et al., 2008) and is critical for wound re-epithelialization in vivo (Tscharntke et al., 2007). Although insulin and IGF-1 stimulate each other’s receptor (insulin more than IGF-1 for IR and IGF-1 more than insulin for IGF1R), IGF-1 stimulates migration at 50- to 500-fold lower concentrations than insulin (Wang et al., 2014) and is also critical for keratinocyte proliferation and morphogenesis (Gunschmann et al., 2013; Sadagurski et al., 2006; Stachelscheid et al., 2008). The regulation of keratinocyte IGF1R activation is poorly understood, but exposure to high concentrations of glucose, commonly used to simulate diabetes in vitro (Ingram et al., 2008; Suzuki et al., 2011), has been shown to slow keratinocyte migration (Lan et al., 2009) and proliferation by inhibiting IGF1R phosphorylation (Spravchikov et al., 2001). The molecular mechanism for glucose-induced keratinocyte slowing and effects on keratinocyte dynamics has not been further explored.
Increasing evidence suggests that ganglioside GM3, a sialylated membrane-based glycosphingolipid and precursor for more complex gangliosides, plays a pivotal role in insulin resistance and diabetes in classical insulin-sensitive cells and tissues. In adipocytes, GM3 suppresses tyrosine phosphorylation of the IR, IGF1R, and IR substrate-1, decreasing cellular glucose uptake (Tagami et al., 2002). Tumor necrosis factor-α increases GM3 expression in adipocytes, myocytes, and hepatocytes (Memon et al., 1999; Tagami et al., 2002), and glucosylceramide synthase inhibitors (GCSIs), which deplete GM3, reverse tumor necrosis factor-α–induced suppression of IR substrate-1 activation in adipocytes (Aerts et al., 2007; Tagami et al., 2002). GM3 synthase (GM3S) promotes GM3 synthesis. GM3S-knockout mice (Yamashita et al., 2003) are obese on a high-fat diet but often show improved glucose tolerance. Furthermore, treating diabetic mice or rats with GCSIs improves insulin sensitivity (Zhao et al., 2009; Zhao et al., 2007), ameliorates hepatic steatosis (Zhao et al., 2009), and prevents the development of diabetic renal hypertrophy (Zador et al., 1993).
We have found that GM3 and GM3S are increased in diet-induced obese and db/db (leptin receptor deficient) diabetic mouse skin (Wang et al., 2014). Despite their high-fat diet–induced obesity and diabetes, GM3S-knockout mice (Wang et al., 2014) and mice with cutaneous GM3S knockdown from topical application of GM3S gene-suppressing nanoconstructs (Randeria et al., 2015) have normal wound healing. However, humans lack a murine salvage pathway for neoganglioside expression with GM3S knockout (Wang et al., 2013); therefore, it was necessary to test the effect of GM3 depletion in human cells before advancing this treatment approach. Furthermore, exploring the effects of GM3 modulation could elucidate how high glucose regulates cutaneous IGF1R signaling. Using genetic and biochemical means to modulate GM3 levels, we addressed the crucial role of GM3 as a regulator of IGF1 R/Rac1 signaling in NHEKs, including in mediating glucose-induced IGF1R suppression.
RESULTS
Human diabetic foot epidermis expresses increased ganglioside GM3
To establish the potential relevance of our mouse studies for human diabetes, we first explored the expression of GM3S (Wang et al., 2014) and ganglioside GM3 in human plantar epidermis from diabetics versus age-matched healthy control subjects. Expression of GM3S and GM3 in human diabetic skin is greater than 3-fold (Wang et al., 2014) and greater than 2-fold higher (Figure 1a), respectively, than in control samples (p < .001).
Figure 1. Manipulation of GM3 levels.
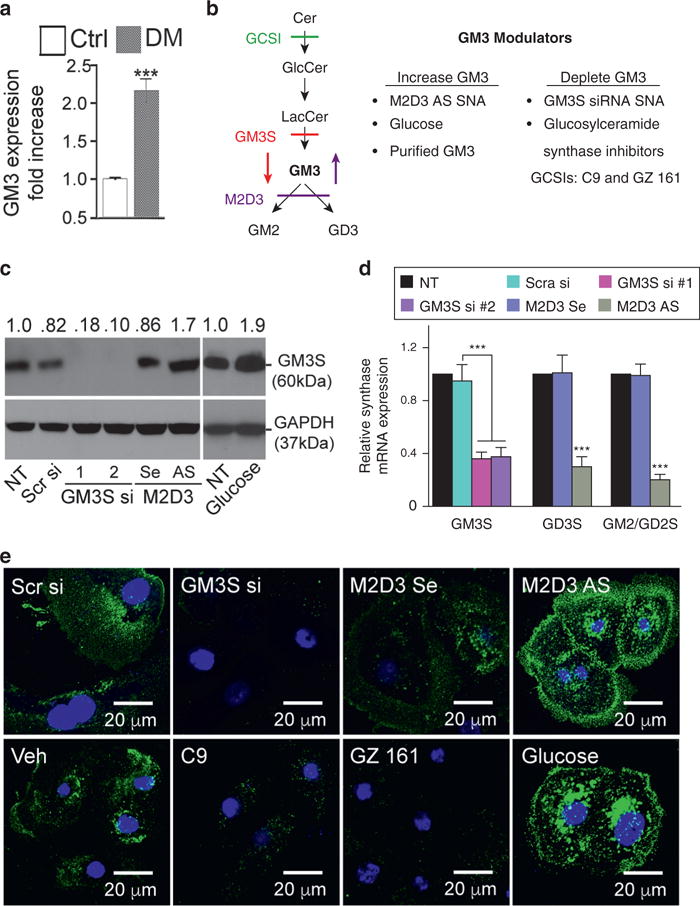
(a) GM3 quantification by ELISA from human diabetic (n = 7, DM) and age- and sex-matched normal (n = 5, Ctrl) plantar epidermis. (b) Left: pathways for GM3 synthesis and metabolism. Right: GM3 manipulation effects. (c) After 72 hours, treatment with two different GM3S siRNA SNAs (GM3S si), bispecific M2D3 AS SNA, supplemental glucose, or controls, GM3S protein was detected by immunoblotting with anti-GM3S antibody. (d) After 72 hours treatment with GM3S siRNA or M2D3 AS SNA, GM3S, GD3S, and GM2/GD2S mRNA expression was detected by quantitative real-time reverse transcriptase–PCR. (e) NHEKs were treated with SNAs, supplemental glucose, glucosylceramide synthase inhibitors (GCSIs, GZ 161, and C9), or controls. GM3 expression was detected by confocal microscopy with anti-GM3 antibody. Scale bar = 20 μm. ***P < 0.001. AS, antisense DNA; Cer, ceramides; Ctrl, control; DM, diabetic; GlcCer, glucosylceramide; LacCer, lactosylceramide; NT, no treatment; Scr, scrambled; Se, sense; si, small interfering RNA; SNA, spherical nucleic acid; Veh, vehicle.
Modulation of GM3 in human keratinocytes
To test the specificity of GM3 in mediating insulin resistance, we designed biochemical and genetic techniques to increase or decrease GM3 while variably altering sphingolipid precursors and metabolites (Figure 1b). GM3 was increased by (i) supplemental GM3 (Wang et al., 2001, 2002), (ii) high glucose (Wang et al., 2014), and (iii) a bispecific antisense oligonucleotide (AS) spherical nucleic acid (SNA) that blocks GM3 metabolism. GM3 was decreased by (i) GM3S small interfering RNA (siRNA) SNAs (Randeria et al., 2015) or (ii) two structurally different, new-generation small molecule GCSIs (Figure 1b) that deplete GM3 and its LacCer precursor: C9 (Genz-123346) (Karman et al., 2010; McEachern et al., 2007; Zhao et al., 2007, 2009) and GZ 161 (Genz-667161) (Cabrera-Salazar et al., 2012; Dodge et al., 2015) (Figure 1 c–e, and see Supplementary Figure S1 online). Although the first-generation GCSI, 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP), increases ceramides, C9 and GZ 161 reduce ceramides by shunting toward sphingosine (Cabrera-Salazar et al., 2012; McEachern et al., 2007). For AS- and siRNA-induced gene modulation, we introduced gold-cored SNAs, spherically arrayed siRNA or DNA antisense oligomers that (i) have excellent uptake in NHEKs without ancillary agents (such as cationic lipids) (Zheng et al., 2012), (ii) cause minimal off-target or immune effects (Zheng et al., 2012), and (iii) potently knock down target genes in vitro and, after topical application, ex vivo and in vivo (Randeria et al., 2015; Zheng et al., 2012) (see Supplementary Table S1 online). Concurrent AS knockdown of GM2/GD2 synthase and GD3 synthase (Nagata et al., 1992; Sasaki et al., 1994) using the bispecific “M2D3” AS SNA leads to accumulation of GM3 and upstream precursors but decreases downstream gangliosides. Glucose treatment also increases GM3S and GM3 expression in NHEK (Figures 1c and e).
GM3 inhibits cell migration, whereas depletion of GM3 accelerates migration
NHEK migration was assessed by scratch assays in complete medium with standard (6 mmol/L; Figure 2a and c) and high (18 mmol/L) (Figure 2b and d) glucose concentrations. In standard medium, GM3 depletion (GM3S siRNA SNA, GZ 161, C9) increased NHEK migration versus controls at 6–18 hours after scratching (Figure 2a and c). In contrast, GM3 accumulation (M2D3 AS SNA) or adding purified GM3 progressively inhibited NHEK migration during the 18 hours (Figure 2a and c). Exposure to GM3 inhibited migration (Figure 2a and c) and repletion with GM3 fully reversed the increased migration in GM3-depleted NHEKs (Figure 2c). GM3 depletion blocked the 2.4-fold slowing induced by excess glucose in untreated NHEKs and even accelerated migration (P < 0.001 from 6 to 18 hours, Figure 2b and d). Repletion of GM3 in GM3-depleted cells also inhibited their migration at all time points in high-glucose medium (Figure 2b and d). These data suggest a role for GM3 expression in regulating NHEK migration. The concordance of effects from GM3 modulation, despite techniques that differentially affect expression of other sphingolipids, and the reversal of accelerated migration with GM3 itself point specifically to GM3 as the key glycosphingolipid intermediate rather than a more complex ganglioside metabolite or GM3 precursor. These data also suggest that excess glucose (simulating hyperglycemia) inhibits migration through eliciting an increase in GM3.
Figure 2. GM3 levels regulate NHEK migration.
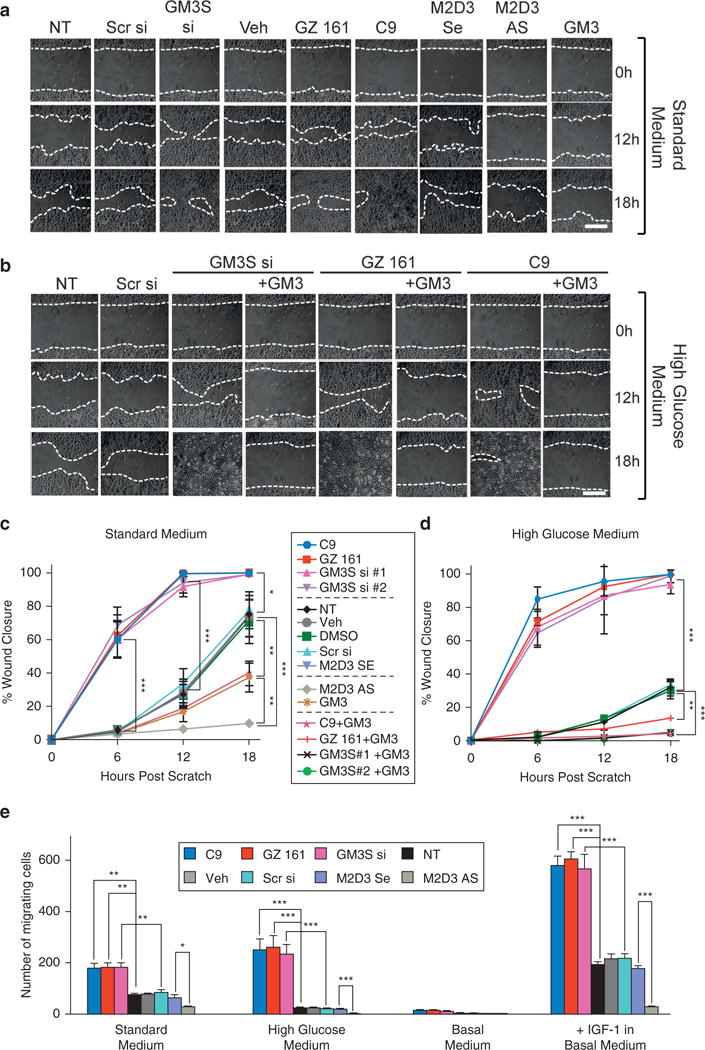
Representative scratch assays after GM3 modulation in the presence of (a, c) standard medium and (b, d) medium supplemented with 12 mmol/L glucose (total = 18 mmol/L). (a, b) Dotted lines highlight the wound edge. Graphs showing wound closure in (c) standard medium and (d) high glucose; the central key identifies lines in both graphs. (e) Boyden chemotaxis assay. Treated NHEKs were grown in standard complete (left) or high glucose (right) medium or were starved overnight (basal medium) and stimulated with IGF1 (100 ng/ml) for 18 hours of migration. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar in a and b = 500 μm. AS, antisense DNA; h, hours; NHEK, normal human epidermal keratinocyte; NT, no treatment; Scr, scrambled; SE, sense; si, small interfering RNA; Veh, vehicle.
To evaluate the impact of GM3 modulation on IGF-1 –induced chemotaxis, we tested NHEK migration using a Boyden chamber (Figure 2e). In 18-hour assays with basal medium in the upper chamber and complete medium in the lower chamber, GM3-depleted cells had greater chemotaxis and M2D3 SNA-treated NHEKs reduced chemotaxis. When complete medium with 18 mmol/L glucose was placed into the lower chamber, migration of control NHEKs stopped, but migration of GM3-depleted cells increased compared with standard medium (P < 0.05). Adding 100 ng/ml IGF-1 in basal medium to the bottom chamber after overnight starvation stimulated NHEK chemotaxis, and GM3-depleted NHEKs showed much greater chemotaxis than controls (P < 0.001). M2D3 SNA treatment prevented response to IGF-1 (P < .05) (Figure 2e, right). These data, showing parallel results in scratch and chemotaxis assay, reinforce the predominant role of the insulin/IGF-1 signaling axis in GM3-induced regulation of migration.
GM3 depletion prevents glucose-induced suppression of single-cell directional movement
To investigate the relative roles of changes in velocity and intrinsic directionality, single NHEKs with and without GM3 modulation were tracked by phase contrast live cell imaging for 2 hours (representative vector diagrams, Figure 3a). NHEKs with M2D3 or high glucose treatment showed virtually no movement (P < 0.001 vs. control samples with standard medium). GM3-depleted cells, however, moved more than control-treated cells in standard medium and even more so in high-glucose medium (both P < 0.0001 vs. control samples). GM3-depleted cells traveled at higher velocities and larger net displacements versus control samples in standard medium (Figure 3b and c). Excess glucose did not alter persistence in GM3-depleted cells, despite its further increase of velocity and displacement (Figure 3b and d). However, glucose supplementation further reduced persistence in control cells (which largely were stationary; all P < 0.001).
Figure 3. GM3 depletion increases NHEK velocity and directional migration.
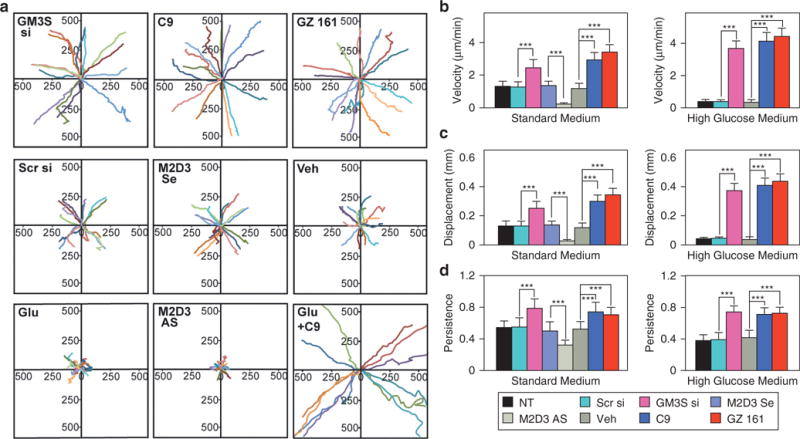
Single-cell movement was monitored in 4-minute intervals for 100–140 minutes using live cell imaging and computerized analysis. (a) Vector maps (rose diagrams) of IGF-1 stimulated cell migration from start (0) to end as distance in μm. shown are 15 representative cells of the total 60–100 NHEKs analyzed per condition. (b) Velocity, (c) net displacement, and (d) persistence coefficient (also called processivity; net displacement/total distance with 1 representing a linear path) of modulated NHEKs in standard complete (left) and high glucose (right) medium. Mean ± standard deviation. ***P< 0.001. AS, antisense DNA; Glu, glucose; IGF-1, insulin-like growth factor-1; min, minutes; NHEK, normal human epidermal keratinocyte; NT, no treatment; Scr, scrambled; Se, sense; si, small interfering RNA; Veh, vehicle.
Adhesion to substrates may also affect velocity. Neither GM3 depletion (from GM3S siRNA SNA or C9) nor increases in GM3 (from M2D3 AS SNA or glucose) affected adhesion on our collagen I substrate, poly-L-lysine, or plastic (see Supplementary Figure S2 online). These data suggest that GM3 and excess glucose cause an intrinsic motility defect with both cell slowing and dysfunctional directional migration.
GM3S depletion activates Rac1 signaling via activated IGF1R signaling but suppresses RhoA signaling
Increased lamellipodial area and cell dynamics with GM3 depletion indicates active actin remodeling. We predicted that GM3 depletion would activate IGF1R and Rac1, leading to cofilin activation and actin polymerization (Ghosh et al., 2004). Increases in GM3 inhibit IGF1R phosphorylation, whereas GM3 depletion activates IGF1R signaling (Figure 4a). GM3S siRNA SNA and GSCIs reduce cofilin Ser3-phosphorylation (p-cofilin; inactive) without affecting cofilin expression (Figure 4b). In addition, M2D3 AS SNA and high glucose increase p-cofilin but not total cofilin expression (Figure 4b), indicating that GM3 increases inactive, filamentous actin-severing p-cofilin.
Figure 4. GM3 expression regulates Rac1 versus RhoA activation.
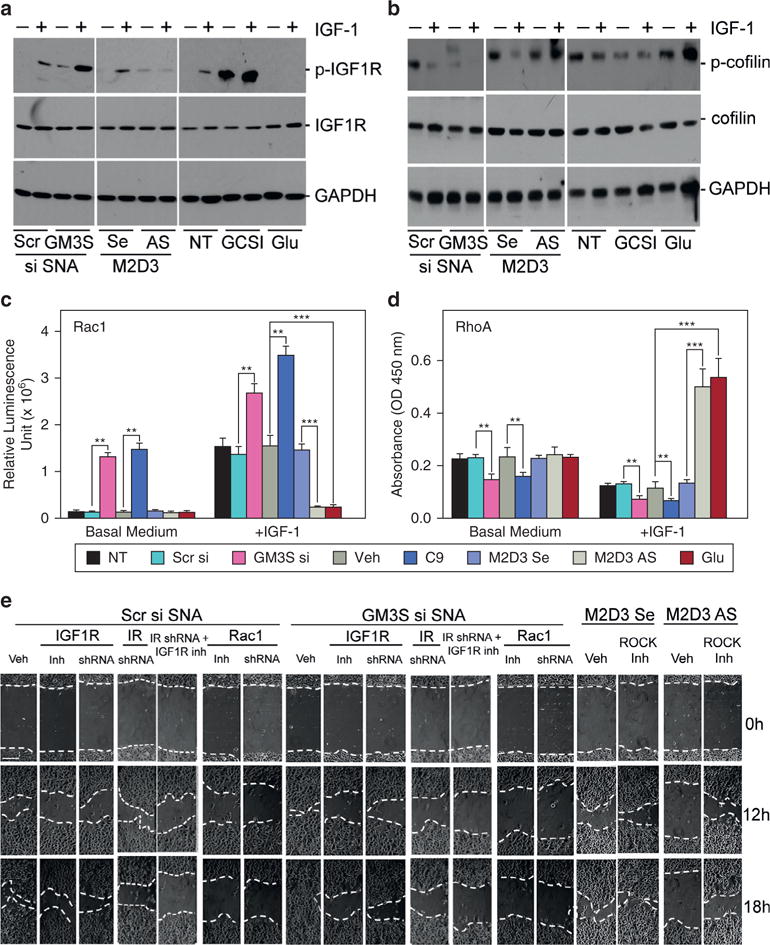
(a–d) NHEKs with and without GM3 modulation were starved overnight and treated for 2 minutes with or without IGF-1. Phosphorylation and expression of (a) IGF1R and (b) cofilin were examined by Western blotting. Activation of (c) Rac1 or (d) RhoA was determined by G-LISA. (e) IGF1R, Rac1, and Rho-associated protein kinase were inhibited with small molecule inhibitors (Inh) or shRNA (see Supplementary Figure S5). Cell migration was accessed by scratch assay. Scale bar (upper left image) = 100 μm. **P < 0.01, ***P < 0.001. AS, antisense DNA; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; G-LISA, small G-protein activation ELISA; Glu, glucose; h, hours; IGF-1, insulin-like growth factor-1; IGF1R, insulin-like growth factor-1 receptor; Inh, inhibitor; IR, insulin receptor; NHEK, normal human epidermal keratinocyte; NT, no treatment; Scr, scrambled; SE, sense; shRNA, short hairpin RNA; si, small interfering RNA; SNA, spherical nucleic acid; Veh, vehicle.
When stimulated with IGF-1, GM3-depleted NHEKs showed a 2.5- to 3.5-fold Rac1 increase by Rac G-LISA, but NHEKs with increased GM3 had basal unstimulated levels of Rad activation. Even starved and without IGF-1 stimulation, GM3-depleted NHEKs had greater p-IGF1R (Figure 4a), less p-cofilin (Figure 4b), and more Rac1 activation (Figure 4c) than starved controls, although starved GM3-depleted cells were not migratory in chemotaxis assays (Figure 2e). RhoA activation, as measured by RhoA small G-protein activation ELISA (enzyme-linked immunosorbent assay), was reduced by GM3 depletion in both basal medium and after IGF-1 stimulation. Increases in GM3 from treatment with M2D3 AS SNA or high glucose increased RhoA activity by 4-fold (Figure 4d).
To further assess the requirement for IGF1R-Rac1 activation in the accelerated wound healing induced by GM3 depletion, we modulated GM3 expression and examined the impact of IGF1R-Rac1 pathway inhibition (Figures 4e and 5, and see Supplementary Figure S3 online). Specific inhibition of IGF1R activation with AG538 (see Supplementary Figure S3a) or short hairpin RNA (shRNA)-induced knockdown of IGF1R (see Supplementary Figure S3b) reduced NHEK migration, particularly the accelerated migration after GM3 depletion (by 78% with AG538 and by 82% with shRNA at 12 hours) (Figures 4e and 5). Rac1 activity inhibition with NSC23766 or shRNA knockdown (see Supplementary Figure S3d) prevented GM3-depletion acceleration of migration to a slightly greater extent than IGF1R inhibition (by 84% at 12 hours, P < 0.05) but not more than the concurrent knockdown of IR (see Supplementary Figure S3c) and inhibition of IGF-1R activation (Figures 4e and 5). Inhibition of Rho-associated protein kinase by Y27632, which blocks the effect of RhoA, reversed the reduction in NHEK migration by M2D3 AS SNA treatment (Figures 4e and 5). These data further implicate GM3 as the mediator of glucose-induced suppression of NHEK migration through suppression of the IGF1R-Rac1 signaling axis.
Figure 5. Quantification of percentage of wound closure of GM3-modulated NHEKs in the presence of inhibitors.
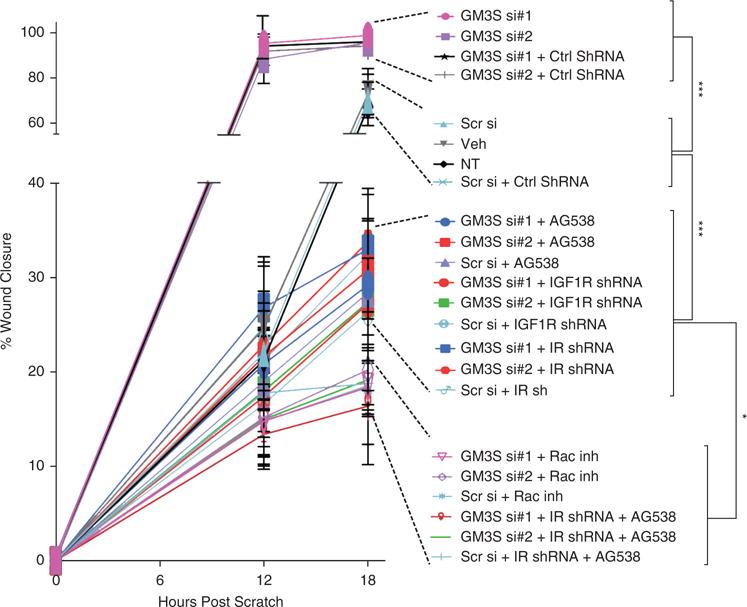
Each condition in the key is shown in descending order of the percentage wound closure at 18 hours, expressed as mean ± standard deviation. ** P < 0.01; ***P < 0.001. Ctrl, control; IGF1R, insulin-like growth factor-1 receptor; inh, inhibitor; IR, insulin receptor; NHEK, normal human epidermal keratinocyte; NT, untreated control; Scr, scrambled control; ShRNA, short hairpin RNA; si, small interfering RNA; Veh, phosphate buffered saline vehicle control.
DISCUSSION
We have previously shown overexpression of GM3 in the keratinocytes and skin of diabetic mice and have reversed their wound healing impairment in vivo by GM3S knockout (Wang et al., 2014) and topical application of GM3S-targeting gene regulation (Randeria et al., 2015; Wang et al., 2014). In these models, GM3S reduction reversed the inhibition of IGF1R phosphorylation at the wound edge (Randeria et al., 2015), suggesting that GM3 depletion activates the insulin/IGF-1 signaling pathway. We now provide evidence that GM3 levels are elevated in human diabetic plantar epidermis and that GM3 mediates the development of insulin/IGF-1 resistance in human keratinocytes, including in response to excessive glucose, simulating diabetic hyperglycemia. Furthermore, we show that GM3 depletion activates the IGF1R/Rac1 pathway that is known to promote keratinocyte migration (Figure 6). These data support the potential translation of our previous mouse studies to the therapy of human diabetic wounds.
Figure 6. Schematic of proposed impact of GM3 modulation on IGF1R and Rac1 signaling.
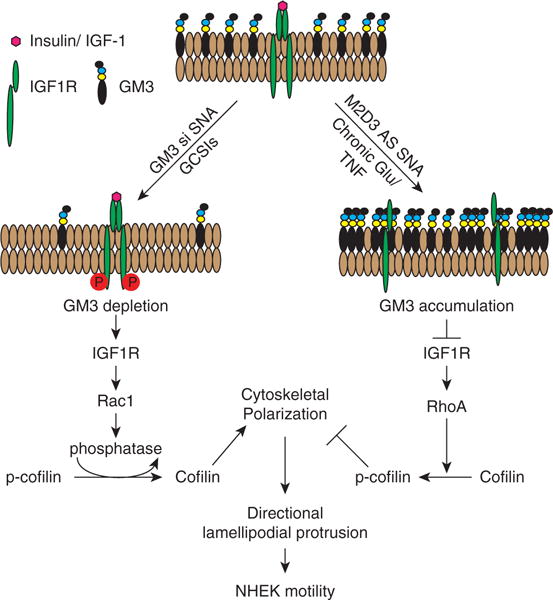
GM3 depletion promotes IGF1R activation, triggering a cascade of phosphorylations that leads to Rac1 activation, cofilin dephosphorylation to polymerize actin, and directional lamellipodial protrusion and increases in both cell velocity and displacement. In contrast, increases in GM3 content suppress IGF1R phosphorylation, leading to RhoA activation, cofilin phosphorylation, and inhibition of cytoskeletal polarization and cell motility. AS, antisense DNA; GCSI, glucosylceramide synthase inhibitor; IGF-1, insulin-like growth factor-1; IGF1R, insulin-like growth factor-1 receptor; NHEK, normal human epidermal keratinocyte; si, small interfering RNA; SNA, spherical nucleic acid; TNF, tumor necrosis factor.
In our previous studies in diabetic mice and mouse keratinocytes, we reduced GM3S and GM3 using a GM3S siRNA nanoconstruct. The dense spherical configuration of siRNA in these nanoconstructs promotes penetration through human epidermis, cell uptake, and RNA stability compared with free siRNA (Randeria et al., 2015; Zheng et al., 2012). The siRNA oligomer used in these mouse studies (our GM3S si #1), and a second GM3S-suppressing SNA (GM3S si #2), recognize 100% concordant stretches of mouse and human GM3S mRNA, knock down GM3S efficiently in both species, and accelerate human keratinocyte migration through IGF1R-Rac1 activation. These observations further suggest that the same GM3S siRNA SNA used in mouse studies could be translated to human wound intervention trials. As an alternative means to deplete GM3, we treated NHEKs with C9 and GZ 161 GCSIs. C9 is closely related to eliglustat, a commercially available small molecule inhibitor for Gaucher disease (Cox et al., 2015), which reduces both GM3 and upstream glucosylceramide. C9 and GZ 161, a newer GCSI used effectively in a Gaucher disease mouse model (Cabrera-Salazar et al., 2012), have been administered systemically to mice without a deleterious impact on skin. Similarly, eliglustat caused no xerosis or epidermal barrier alteration during at least 2 years of evaluation in human trials (Cox et al., 2015). The small molecular weights of C9, eliglustat, and GZ 161 (494, 382, and 405 Da, respectively), raise the possibility of epidermal penetration at the wound border to directly activate epidermal keratinocyte IR/IGF1R signaling in a species-independent manner. Whether through gene regulation of GM3S or pharmacological inhibition of GCS, depletion of GM3 and resulting IGF1R-Rac1 pathway activation is a promising new approach for diabetic wounds.
Previous studies of GM3 as a regulator of insulin resistance have focused on GM3’s regulatory role in IR signaling in canonical insulin-responsive tissues (adipose tissue, muscle, and liver). In epidermis, IGF-1–induced activation of IGF1R is thought to play a more crucial role than insulin and IR activation in insulin/IGF-1 receptor signaling (Ando and Jensen, 1993; Blakytny et al., 2000; Stachelscheid et al., 2008). Keratinocyte-specific overexpression of IGF-1 in mouse epidermis promotes keratinocyte proliferation, migration, and wound re-epithelialization (Semenova et al., 2008), and epidermis-specific IGF1R knockout limits epidermal stratification in morphogenesis (Gunschmann et al., 2013). Indeed, IGF-1 is deficient in streptozotocin-induced diabetic rat wound fluid (Bitar and Labbad, 1996) and in the basal keratinocytes of the wound margin and dermal fibroblasts (the source of skin IGF-1) in human diabetic wounds (Blakytny et al., 2000). Exogenous liposomal IGF-1 gene transfer to diabetic wounds increases re-epithelialization, collagen deposition, and angiogenesis in wounds (Jeschke et al., 2004). Our studies imply that preventing the glucose- or tumor necrosis factor-induced elevation of GM3 can sensitize keratinocytes to the effects of IGF1R ligands and improve wound healing.
Haase et al. (2003) first recognized that IGF-1 induces changes in the actin cytoskeleton and directional migration in keratinocytes. Indeed, activating IGF1R through GM3 depletion not only increases cell velocity, but also stimulates directional membrane protrusion through Rac1 activation and directional migration. In contrast, tripling the ambient glucose concentration to simulate diabetic hyperglycemia or accumulating GM3 through M2D3 AS SNA-induced gene regulation promotes RhoA activation and suppresses Rac1 signaling, limiting keratinocyte migration. Prevention of glucose-induced movement arrest when GM3 is depleted strongly suggests a role for GM3 as a regulator as well of glucose uptake in keratinocytes, as has been suggested in adipocytes (van Eijk et al., 2009). Indeed, we have found in early studies that GM3 depletion increases membrane translocation of GLUT1 and glucose uptake (Shehu et al., 2013), providing a carbon source for increased velocity. How GM3 regulates IGF1R-integrin crosstalk, the relative roles of GM3-promoted activation of RhoA versus suppression of Rac1 activation, and the mechanism by which increasing glucose promotes migration in GM3-deficient cells deserve investigation.
In summary, our studies begin to elucidate the impact of GM3 expression on human keratinocytes and epidermal cell migration in wounds. However, wound closure is affected by a variety of cells and, in diabetic wounds, ischemia and neuropathy are known to be important deterrents to healing (Falanga, 2005). Our GM3-depleted diabetic mouse models have increased numbers of wound bed CD31+ cells, a marker of wound vascularity (Randeria et al., 2015), and have normal cutaneous innervation and pain responses (Menichella et al., 2016), providing evidence that GM3 depletion may also have beneficial effects on wound ischemia and innervation, respectively. Better understanding of the mechanism for these various effects of GM3 modulation and their role in diabetic pathology is a major goal of our ongoing research.
MATERIALS AND METHODS
All in vitro studies were performed three or more times in triplicate. See Supplementary Materials online for details.
Detection and measurement of GM3S and ganglioside GM3 in NHEKs and human skin
Diabetic and control plantar skin were obtained after written and informed consent, approved by the Institutional Review Board of Northwestern University. Epidermis was separated by cold, RNase-free dispase treatment. Total RNA or protein was extracted and purified from treated NHEKs and analyzed by reverse transcriptase–PCR and Western blotting (Wang et al., 2014). GM3 expression in NHEKs was assessed by immunofluorescent staining, flow cytometry, and ganglioside ELISA in human skin using anti-GM3 antibody (Ambios, Santa Cruz, CA).
Cells and GM3 modulation
Foreskin NHEKs were maintained in calcium-free with 75 micromol/L calcium plus growth supplement medium and 75 umol/L Calcium. GSCIs at 1 μmol/L C9 or 200 nmol/L GZ 161, or 2 nmol/L GM3S siRNA SNA, was added for 72 hours to decrease GM3 expression. 5 nmol/L M2D3 antisense SNA, 100 μM purified GM3 in DMSO, or 12 mmol/L extra glucose (total 18 mmol/L) was added for 96 hours to increase GM3 expression. Controls included untreated, DMSO vehicle, scrambled siRNA SNA and sense SNA.
SNA synthesis and characterization
Gold nanoparticles (13 nm) were synthesized by reducing Au(III) salt in citric buffer. Thiolated oligonucleotides were attached through gold-thiol bonds using salt-aging methodology (Randeria et al., 2015). The size distribution and polydispersity of the SNAs were determined using dynamic light scattering.
Immunoblotting
40 mg total protein from each treatment set was separated on a precast gradient TGX gel (4–15%; Bio-Rad, Hercules, CA) and transferred to 0.2-μm nitrocellulose membrane. IGF1R, phospho-IGF1R, IR, GM3S, Rac-1 and, to normalize expression, glyceraldehyde-3-phosphate dehydrogenase were detected with antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Anti-cofilin and anti-phospho-cofilin antibodies were from Cell Signaling (Danvers, MA).
Migration and chemotaxis assays
For scratch assays, treated NHEKs were densely plated in complete serum-free medium, treated with mitomycin C to prevent proliferation, and scratched. Wound closure was serially imaged by Bio-Station (Nikon, Melville, NY). Chemotaxis was assessed using Boyden chambers with complete medium with or without extra glucose and/or IGF-1 in the bottom chamber. Motility was analyzed by ImageJ software (National Institutes of Health, Bethesda, MD).
Cell transduction
Lentiviral IR, IGF1R, or Rac1 shRNA were transduced into NHEKs overnight. Transduced cells were selected with puromycin for 3 days.
G-LISA
Treated cells were seeded at 60–75% confluence, starved overnight, stimulated by 400 ng/ml IGF-1 for 2 minutes before lysis, and analyzed using the G-LISA Kit (Cytoskeleton Inc., Denver, CO).
Statistical analysis
Student t testing was used for all analyses. P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We acknowledge Chad Mirkin for developing the concept of SNAs and both Chad Mirkin and Pratik Randeria for generating the original SNAs and teaching how to make the GM3S siRNA SNA and M2D3 AS SNA, Seng Cheng and Genzyme for providing the C9 and GZ 161, Joshua Rappaport and Constadina Arvanitis from Northwestern’s Center for Advanced Microscopy (NCI, P30CA060553) for their counsel, and Neil Patel for his help with live cell imaging. We thank Jonathan Jones, Spiro Getsios, and Robert Lavker for their careful review of the manuscript. This work was supported by the National Institutes of Health grants R01AR44619, R21AR062898, and R01AR068375 (AP), Postgraduate Training in Cutaneous Biology T32 AR060710 (DD), the Foglia Family Endowment, and the Astellas Research Endowment (XW). This research also used Core resources provided by the Northwestern University Skin Disease Research Center (NIAMS, P30AR057216).
Abbreviations
- AS
antisense oligonucleotide
- GCS
glucosylceramide synthase
- GCSI
glucosylceramide synthase inhibitor
- GM3S
ganglioside GM3 synthase
- IGF-1
insulin-like growth factor-1
- IGF1R
insulin-like growth factor-1 receptor
- IR
insulin receptor
- M2D3
GM2/GD2 and GD3 synthases
- NHEK
normal human epidermal keratinocyte
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- SNA
spherical nucleic acid
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at http://dx.doi.org/10.1016/j.jid.2016.09.028.
References
- Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, et al. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes. 2007;56:1341–9. doi: 10.2337/db06-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association. Statistics about diabetes. 2014 http://www.diabetes.org/diabetes-basics/statistics. (accessed 3 September 2016)
- Ando Y, Jensen PJ. Epidermal growth factor and insulin-like growth factor I enhance keratinocyte migration. J Invest Dermatol. 1993;100:633–9. doi: 10.1111/1523-1747.ep12472297. [DOI] [PubMed] [Google Scholar]
- Bitar MS, Labbad ZN. Transforming growth factor-beta and insulin-like growth factor-I in relation to diabetes-induced impairment of wound healing. J Surg Res. 1996;61:113–9. doi: 10.1006/jsre.1996.0090. [DOI] [PubMed] [Google Scholar]
- Blakytny R, Jude EB, Martin Gibson J, Boulton AJ, Ferguson MW. Lack of insulin-like growth factor 1 (IGF1) in the basal keratinocyte layer of diabetic skin and diabetic foot ulcers. J Pathol. 2000;190:589–94. doi: 10.1002/(SICI)1096-9896(200004)190:5<589::AID-PATH553>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Cabrera-Salazar MA, Deriso M, Bercury SD, Li L, Lydon JT, Weber W, et al. Systemic delivery of a glucosylceramide synthase inhibitor reduces CNS substrates and increases lifespan in a mouse model of type 2 Gaucher disease. PLoS One. 2012;7:e43310. doi: 10.1371/journal.pone.0043310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman AL, Selvin E, Margolis DJ, Lazarus GS, Garza LA. Hemoglobin A1c predicts healing rate in diabetic wounds. J Invest Dermatol. 2011;131:2121–7. doi: 10.1038/jid.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TM, Drelichman G, Cravo R, Balwani M, Burrow TA, Martins AM, et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet. 2015;385:2355–62. doi: 10.1016/S0140-6736(14)61841-9. [DOI] [PubMed] [Google Scholar]
- Dodge JC, Treleaven CM, Pacheco J, Cooper S, Bao C, Abraham M, et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2015;112:8100–5. doi: 10.1073/pnas.1508767112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–43. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–6. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- Gunschmann C, Stachelscheid H, Akyuz MD, Schmitz A, Missero C, Bruning JC, et al. Insulin/IGF-1 controls epidermal morphogenesis via regulation of FoxO-mediated p63 inhibition. Dev Cell. 2013;26:176–87. doi: 10.1016/j.devcel.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase I, Evans R, Pofahl R, Watt FM. Regulation of keratinocyte shape, migration and wound epithelialization by IGF-1- and EGF-dependent signalling pathways. J Cell Sci. 2003;116:3227–38. doi: 10.1242/jcs.00610. [DOI] [PubMed] [Google Scholar]
- Ingram DA, Lien IZ, Mead LE, Estes M, Prater DN, Derr-Yellin E, et al. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes. 2008;57:724–31. doi: 10.2337/db07-1507. [DOI] [PubMed] [Google Scholar]
- Jeschke MG, Schubert T, Klein D. Exogenous liposomal IGF-I cDNA gene transfer leads to endogenous cellular and physiological responses in an acute wound. Am J Physiol Regul Integr Comp Physiol. 2004;286:R958–66. doi: 10.1152/ajpregu.00541.2003. [DOI] [PubMed] [Google Scholar]
- Karman J, Tedstone JL, Gumlaw NK, Zhu Y, Yew N, Siegel C, et al. Reducing glycosphingolipid biosynthesis in airway cells partially ameliorates disease manifestations in a mouse model of asthma. Int Immunol. 2010;22:593–603. doi: 10.1093/intimm/dxq044. [DOI] [PubMed] [Google Scholar]
- Lan CC, Wu CS, Kuo HY, Huang SM, Chen GS. Hyperglycaemic conditions hamper keratinocyte locomotion via sequential inhibition of distinct pathways: new insights on poor wound closure in patients with diabetes. Br J Dermatol. 2009;160:1206–14. doi: 10.1111/j.1365-2133.2009.09089.x. [DOI] [PubMed] [Google Scholar]
- Markuson M, Hanson D, Anderson J, Langemo D, Hunter S, Thompson P, et al. The relationship between hemoglobin A(1c) values and healing time for lower extremity ulcers in individuals with diabetes. Adv Skin Wound Care. 2009;22:365–72. doi: 10.1097/01.ASW.0000358639.45784.cd. [DOI] [PubMed] [Google Scholar]
- McEachern KA, Fung J, Komarnitsky S, Siegel CS, Chuang WL, Hutto E, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucherdisease. Mol Genet Metab. 2007;91:259–67. doi: 10.1016/j.ymgme.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Memon RA, Holleran WM, Uchida Y, Moser AH, Ichikawa S, Hirabayashi Y, et al. Regulation of glycosphingolipid metabolism in liver during the acute phase response. J Biol Chem. 1999;274:19707–13. doi: 10.1074/jbc.274.28.19707. [DOI] [PubMed] [Google Scholar]
- Menichella DM, Jayaraj ND, Wilson HM, Ren D, Food K, Wang XQ, et al. Ganglioside GM3 synthase depletion reverses neuropathic pain and small fiber neuropathy in diet-induced diabetic mice. Mol Pain. 2016;12 doi: 10.1177/1744806916666284. http://dx.doi.org/10.1177/1744806916666284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata Y, Yamashiro S, Yodoi J, Lloyd KO, Shiku H, Furukawa K. Expression cloning of beta-1,4 N-acetylgalactosaminyltransferase CDNAs that determine the expression of GM2 and GD2 Gangliosides. J Biol Chem. 1992;267:12082–9. [PubMed] [Google Scholar]
- Randeria PS, Seeger MA, Wang XQ, Wilson H, Shipp D, Mirkin CA, et al. siRNA-based spherical nucleic acids reverse impaired wound healing in diabetic mice by ganglioside GM3 synthase knockdown. Proc Natl Acad Sci USA. 2015;112:5573–8. doi: 10.1073/pnas.1505951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagurski M, Yakar S, Weingarten G, Holzenberger M, Rhodes CJ, Breitkreutz D, et al. Insulin-like growth factor 1 receptor signaling regulates skin development and inhibits skin keratinocyte differentiation. Mol Cell Biol. 2006;26:2675–87. doi: 10.1128/MCB.26.7.2675-2687.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Kurata K, Kojima N, Kurosawa N, Ohta S, Hanai N, et al. Expression cloning of a GM3-specific alpha-2,8-sialyltransferase (GD3 synthase) J Biol Chem. 1994;269:15950–6. [PubMed] [Google Scholar]
- Semenova E, Koegel H, Hasse S, Klatte JE, Slonimsky E, Bilbao D, et al. Overexpression of mIGF-1 in keratinocytes improves wound healing and accelerates hair follicle formation and cycling in mice. Am J Pathol. 2008;173:1295–310. doi: 10.2353/ajpath.2008.071177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu A, Lu JY, Wilson H, Bach D, Shipp D, Randeria PS, et al. Cytoplasmic sequestion of keratinocyte GLUT1 by ganglioside GM3 mediates impaired diabetic wound healing. J Invest Dermatol. 2013;133:S249. [Google Scholar]
- Spravchikov N, Sizyakov G, Gartsbein M, Accili D, Tennenbaum T, Wertheimer E. Glucose effects on skin keratinocytes: implications for diabetes skin complications. Diabetes. 2001;50:1627–35. doi: 10.2337/diabetes.50.7.1627. [DOI] [PubMed] [Google Scholar]
- Stachelscheid H, Ibrahim H, Koch L, Schmitz A, Tscharntke M, Wunderlich FT, et al. Epidermal insulin/IGF-1 signalling control interfollicular morphogenesis and proliferative potential through Rac activation. EMBO J. 2008;27:2091–101. doi: 10.1038/emboj.2008.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero D, et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci USA. 2011;108:13829–34. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami S, Inokuchi Ji J, Kabayama K, Yoshimura H, Kitamura F, Uemura S, et al. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J Biol Chem. 2002;277:3085–92. doi: 10.1074/jbc.M103705200. [DOI] [PubMed] [Google Scholar]
- Tscharntke M, Pofahl R, Chrostek-Grashoff A, Smyth N, Niessen C, Niemann C, et al. Impaired epidermal wound healing in vivo upon inhibition or deletion of Rac1. J Cell Sci. 2007;120:1480–90. doi: 10.1242/jcs.03426. [DOI] [PubMed] [Google Scholar]
- van Eijk M, Aten J, Bijl N, Ottenhoff R, van Roomen CP, Dubbelhuis PF, et al. Reducing glycosphingolipid content in adipose tissue of obese mice restores insulin sensitivity, adipogenesis and reduces inflammation. PLoS One. 2009;4:e4723. doi: 10.1371/journal.pone.0004723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bright A, Xin B, Bockoven JR, Paller AS. Cutaneous dyspigmentation in patients with ganglioside GM3 synthase deficiency. Am J Med Genet A. 2013;161A:875–9. doi: 10.1002/ajmg.a.35826. [DOI] [PubMed] [Google Scholar]
- Wang X, Rahman Z, Sun P, Meuillet E, George D, Bremer EG, et al. Ganglioside modulates ligand binding to the epidermal growth factor receptor. J Invest Dermatol. 2001;116:69–76. doi: 10.1046/j.1523-1747.2001.00222.x. [DOI] [PubMed] [Google Scholar]
- Wang XQ, Lee S, Wilson H, Seeger M, Iordanov H, Gatla N, et al. Ganglioside GM3 depletion reverses impaired wound healing in diabetic mice by activating IGF-1 and insulin receptors. J Invest Dermatol. 2014;134:1446–55. doi: 10.1038/jid.2013.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Sun P, Paller AS. Ganglioside induces caveolin-1 redistribution and interaction with the epidermal growth factor receptor. J Biol Chem. 2002;277:47028–34. doi: 10.1074/jbc.M208257200. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Hashiramoto A, Haluzik M, Mizukami H, Beck S, Norton A, et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci USA. 2003;100:3445–9. doi: 10.1073/pnas.0635898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zador IZ, Deshmukh GD, Kunkel R, Johnson K, Radin NS, Shayman JA. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J Clin Invest. 1993;91:797–803. doi: 10.1172/JCI116299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Przybylska M, Wu IH, Zhang J, Maniatis P, Pacheco J, et al. Inhibiting glycosphingolipid synthesis ameliorates hepatic steatosis in obese mice. Hepatology. 2009;50:85–93. doi: 10.1002/hep.22970. [DOI] [PubMed] [Google Scholar]
- Zhao H, Przybylska M, Wu IH, Zhang J, Siegel C, Komarnitsky S, et al. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes. 2007;56:1210–8. doi: 10.2337/db06-0719. [DOI] [PubMed] [Google Scholar]
- Zheng D, Giljohann DA, Chen DL, Massich MD, Wang XQ, Iordanov H, et al. Topical delivery of siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation. Proc Natl Acad Sci USA. 2012;109:11975–80. doi: 10.1073/pnas.1118425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.