

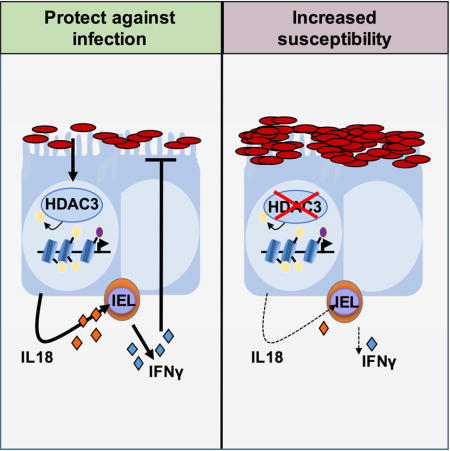
SUMMARY
Mucosal tissues are constantly in direct contact with diverse beneficial and pathogenic microbes, highlighting the need for orchestrating complex microbial signals to sustain effective host defense. Here we show an essential role for intestinal epithelial cell expression of histone deacetylase 3 (HDAC3) in responding to pathogenic microbes and activating protective innate immunity. Mice lacking HDAC3 in intestinal epithelial cells were more susceptible to Citrobacter rodentium when under tonic stimulation by the commensal microbiota. This impaired host defense reflected significantly decreased IFNγ production by intraepithelial CD8+ T cells early during infection. Further, HDAC3 was necessary for infection-induced epithelial expression of the IFNγ-inducing factor IL-18 and administration of IL-18 restored IFNγ activity to resident CD8+ T cells and reduced infection. Thus, HDAC3 mediates communication between intestinal epithelial cells and resident lymphocytes, revealing that epithelial priming by an epigenetic modifier may direct mucosal regulation of host defense against pathogenic microbes.
Keywords: Infection, Intestine, Microbiota, HDAC3, Epigenetic, Mucosal Immunology, Epithelial, Defense, Innate
Graphical abstract
INTRODUCTION
Enteric infections are a major cause of morbidity and mortality globally. Bacterial enteritis alone causes four to six million deaths per year with pathogenic strains of Escherichia coli (E. coli) accounting for an estimated 280 million diarrheal episodes (Black, 1993; DuPont, 2009, 2014). In addition to the impact of infection-induced enteritis, the intestinal mucosa is a major site for invasion and replication of pathogens, such as Listeria, Salmonella, and HIV, that cause disseminated infection and systemic disease (Cossart and Sansonetti, 2004). Therefore, deciphering the innate mechanisms that drive local immunity and host defense against enteric pathogens is critical for developing new approaches for treating and preventing infection.
Enteric pathogens commonly associate with intestinal epithelial cells (IECs) that line the lumen of the intestine. Although non-hematopoietic in origin, critically located IECs are equipped to sense pathogens, produce antimicrobial peptides, and secrete cytokines that regulate the immune system (Gallo and Hooper, 2012; Giacomin et al., 2015; Peterson and Artis, 2014; Ramanan and Cadwell, 2016). Despite continuous exposure to microbes, little is known about the mechanisms regulating how IECs instruct local protective responses to enteric pathogens. Recent studies have highlighted that intraepithelial lymphocytes (IELs) that are interspersed between IECs constitute a critical population of lymphocytes that produce effector cytokines (Cheroutre et al., 2011), however how these resident lymphocytes are instructed by surrounding IECs to defend against pathogens is not well understood.
The constant exposure to diverse microbial signals in the intestine, along with the need to limit pathogenic inflammation, suggests the existence of tightly regulated protective immune regulatory pathways in the intestine. Histone deacetylases (HDACs) are enzymes that can respond to environmental signals and regulate gene expression through the removal of acetyl groups from histones and transcription factors(Chang et al., 2014; Chen et al., 2012; Haberland et al., 2009). In previous work, we identified that the class I HDAC, HDAC3, regulates histone acetylation in IECs and enables integration of microbiota-derived signals to maintain IEC homeostasis (Alenghat et al., 2013), however whether HDAC-mediated regulation directs protective immune response against infection is unknown.
Citrobacter rodentium is a murine bacterial pathogen with similar pathogenesis to enteropathogenic E. coli infection in humans. These Gram-negative extracellular bacteria attach to IECs and initial susceptibility to infection is critically dependent on host innate immune responses (Mundy et al., 2005). Here, we discovered that enteric infection with C. rodentium increased HDAC enzymatic activty in IECs. Further, IEC-intrinsic expression of HDAC3 was necessary to direct microbiota-sensitive innate immune responses that controlled pathogen levels in the intestine. Interestingly, resident CD8+ IELs from mice with IEC-specific deletion of HDAC3 failed to produce significant IFNγ following C. rodentium infection, suggesting that IEC-intrinsic regulation by HDAC3 is necessary to activate effective IEL responses. C. rodentium-triggered IEC expression of the IFNγ-inducing cytokine, IL-18, was impaired in the colon of HDAC3ΔIEC mice. By employing an ex vivo enteric infection system, we found that IL-18 restored IFNγ effector function in tissue-resident IELs without signals from circulating immune cells. Further, administration of IL-18 rescued pathogen control in HDAC3ΔIEC mice. Collectively, these data identify a previously unknown role for epithelial expression of a HDAC in directing protective antibacterial immunity through activation of local resident lymphocytes.
RESULTS
Epithelial cell HDAC3 protects against enteric bacterial infection
Citrobacter rodentium infects the murine large intestine with similar pathology to clinically important attaching and effacing Escherichia coli human pathogens (Koroleva et al., 2015). C. rodentium histologic changes and peak infection occur at 12 days following exposure to the pathogen. Thus, to assess HDAC activity in the intestinal epithelium, IECs were harvested from the colon of naive mice and mice infected with C. rodentium for 12 days. To directly test HDAC activity in IECs without contribution from leukocytes in IEC preparations, contaminating CD45+ leukocytes were depleted prior to assessing HDAC activity (Supplemental Figure 1). Interestingly, quantification of HDAC enzymatic activity revealed significantly elevated HDAC activty in colonic IECs following infection (Figure 1A), indicating that host IECs increase HDAC function in response to the pathogen. We previously determined that HDAC3 is uniquely responsive in IECs to commensal microbial signals (Alenghat et al., 2013). Therefore, to test whether IEC expression of HDAC3 impacts susceptibility to an intestinal infection, we employed C57BL/6 control floxed HDAC3 mice (HDAC3FF) and floxed HDAC3 mice expressing Cre-recombinase under the control of the IEC-specific villin promoter (HDAC3ΔIEC). Following oral infection with C. rodentium for 12 days, significantly higher bacterial burdens were quantified in HDAC3ΔIEC mice compared to floxed HDAC3FF littermate controls (Figure 1B). To specifically examine C. rodentium at the site of infection at this time, mice were infected with GFP-expressing C. rodentium, revealing that HDAC3ΔIEC mice exhibited markedly elevated C. rodentium within the colon adjacent to IECs (Figure 1C).
Figure 1. Epithelial cell HDAC3 expression protects against intestinal bacterial infection.
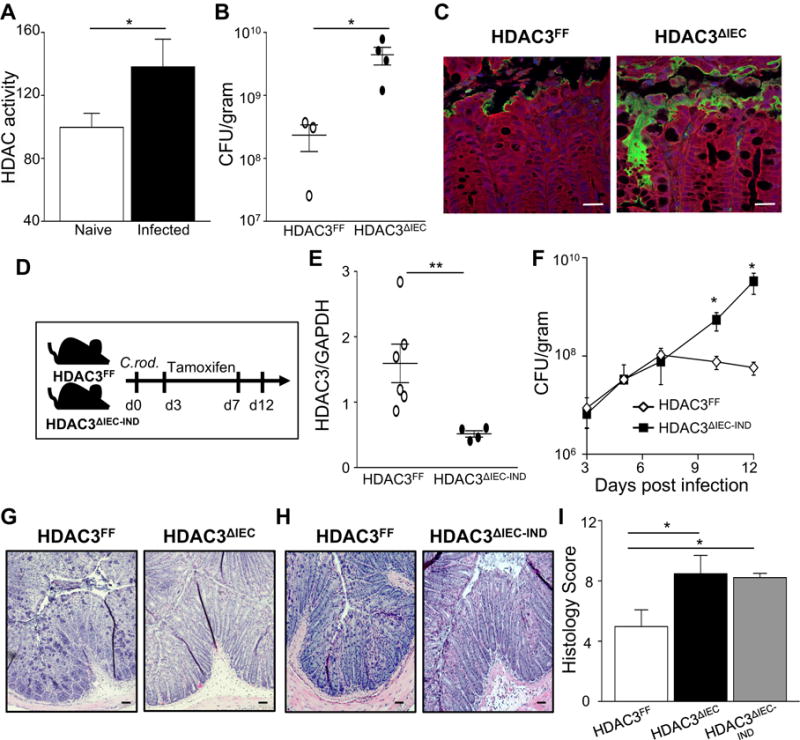
(A) HDAC activity in IECs of naïve and C. rodentium infected C57BL/6 mice. Infection was assessed at 12 days. (B) C. rodentium colony forming units (CFU) in stool of HDAC3FF and HDAC3ΔIEC mice. (C) Immunofluorescent staining of GFP-expressing C. rodentium (green), intestinal cells (red) and nuclei (DAPI, blue) in the colon, scale bar 50 μm. (D) Experimental plan for induced deletion of HDAC3 from IECs after C. rodentium infection. Tamoxifen was administered to HDAC3FF and HDAC3ΔIEC-IND mice. (E) HDAC3 mRNA in IECs from colon. (F) C. rodentium CFU in stool of HDAC3FF and HDAC3ΔIEC-IND mice infected with C. rodentium pre tamoxifen (day 3) and post tamoxifen (days 6–12). (G, H) H&E-stained sections from colon of (G) HDAC3FF and HDAC3ΔIEC mice and (H) tamoxifen-treated HDAC3FF and HDAC3ΔIEC-IND mice, scale bar 50 μm. (I) Histologic scores for C. rodentium-induced intestinal pathology. Scores reflect severity of the following C. rodentium-associated histologic parameters: (a) mural edema (1–5), (b) epithelial lesions (1–5), and (c) leukocyte infiltration (1–5). Data are representative of at least 3 independent experiments containing 3–6 mice per group at day 12 post infection. CFUs are normalized to fecal weight. Results are mean ± s.e.m. *p<0.05, **p<0.01.
Constitutive deletion of HDAC3 has the potential to alter the development of cells in the intestine. Therefore, to directly test whether IEC-intrinsic HDAC3 expression actively regulates host defense against an enteric pathogen, we employed an inducible IEC-specific HDAC3 knockout mouse model (HDAC3ΔIEC-IND). HDAC3ΔIEC-IND mice and littermate control HDAC3FF mice were equally infected with C. rodentium. Tamoxifen administration was initiated in both genotypes at day 3 when the pathogen colonizes the distal colon (Figure 1D) and reduction in HDAC3 expression was confirmed (Figure 1E). As expected, similar pathogen loads were quantified in HDAC3FF and HDAC3ΔIEC-IND mice prior to tamoxifen-induced deletion of HDAC3 (Figure 1F). However, targeted deletion of HDAC3 in IECs following infection led to increased C. rodentium levels (Figure 1F). Collectively, these data indicate that mice lacking epithelial HDAC3 expression are more susceptible to the pathogen and that IEC-intrinsic HDAC3 actively regulates the infection in adult mice.
Consistent with significantly increased pathogen exposure, mice with deletion of HDAC3 in IECs exhibited more severe colonic pathology associated with C. rodentium infection (Figures 1G and 1H). Histologic features were characterized by exaggerated submucosal edema, leukocyte expansion of the lamina propria, and mucosal hyperplasia (Figures 1G and 1H) and scoring the severity of these C. rodentium-associated histologic parameters resulted in a significantly higher histologic score in mice lacking HDAC3 relative to control mice (Figure 1I). Therefore, these data demonstrate a critical role for IEC-intrinsic expression of HDAC3 in controlling host response to an intestinal bacterial pathogen.
Epithelial HDAC3 expression promotes induction of a protective IFNγ response to infection
Several pathogens initially contact and/or adhere to epithelial cells lining mucosal surfaces and their clearance is critically dependent on effective expression of the type 1 cytokine IFNγ (Fuchs et al., 2013; Shiomi et al., 2010; Simmons et al., 2002). Previous work has highlighted a critical role for IFNγ in regulation of susceptibility to C. rodentium, in part through stimulation of goblet cells in the intestine (Chan et al., 2013). We found that IFNγ is increased in the colon 12 days after C. rodentium infection and that HDAC3ΔIEC mice exhibited defective induction of IFNγ at this stage (Figure 2A). Therefore, despite a markedly increased amount of an IFNγ-inducing pathogen in HDAC3ΔIEC mice, these mice were unable to initiate an appropriate IFNγ effector cytokine response. Levels of other C. rodentium-induced cytokines, IL-17 and IL-22, were less altered in the colon by day 12 post infection in HDAC3ΔIEC mice (Supplemental Figures 2A and 2B). Previous work demonstrated that mice lacking IFNγ exhibited significantly elevated bacterial loads (Simmons et al., 2002). Consistently, we found that IFNγ−/− mice infected with C. rodentium displayed elevated bacterial burdens (Figures 2B and 2C). Further, loss of HDAC3 within IECs resulted in increased infection (Figure 2B) and more severe intestinal pathology that were remarkably similar to the differences observed in infected IFNγ−/− mice (Figures 2D and 2E).
Figure 2. HDAC3 in IECs induces IFNγ-mediated protection in the intestine during infection.
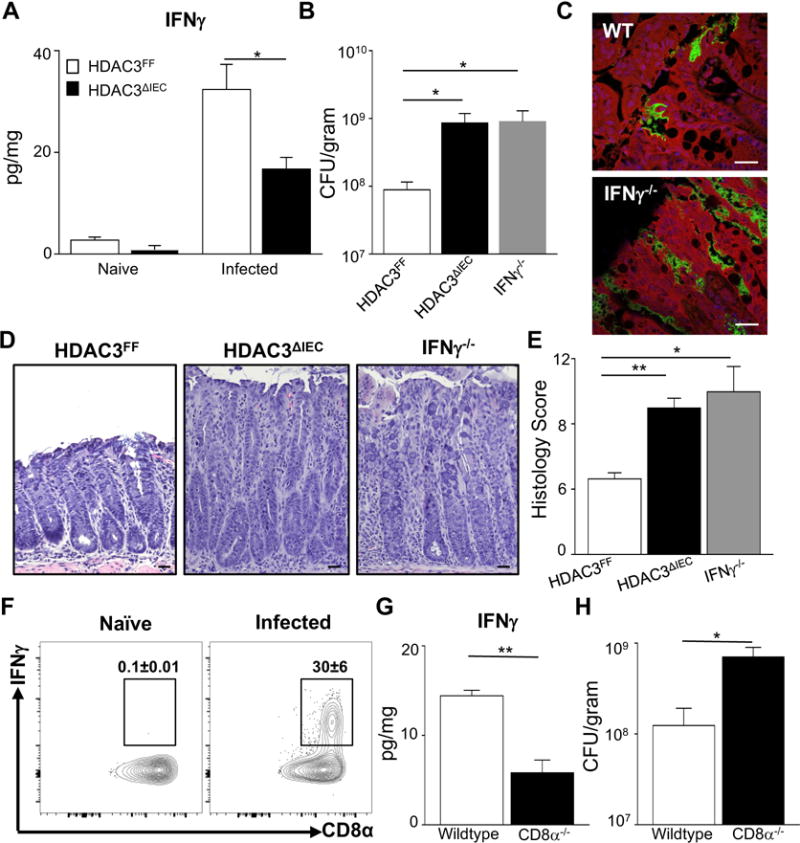
(A) IFNγ in colonic tissue measured by ELISA. (B) C. rodentium CFU in stool of HDAC3 FF, HDAC3ΔIEC and IFNγ−/− mice. (C) Immunofluorescent staining of GFP-expressing C. rodentium (green), intestinal cells (red) and nuclei (DAPI, blue) in the colon, scale bar 50 μm. (D) H&E-stained sections from colon of C. rodentium infected mice, scale bar 50μm. (E) Histologic scores for C. rodentium-induced intestinal pathology. Scores reflect severity of the following C. rodentium-associated histologic parameters: (a) mural edema (1–5), (b) epithelial lesions (1–5), and (c) leukocyte infiltration (1–5). (F) Representative flow cytometry plots of IFNγ-producing CD45+ IELs from wildtype (WT) mice, naïve and 12 days post C. rodentium. (G) IFNγ levels in the colon of WT and CD8α−/− mice, measured by ELISA. (H) C. rodentium CFU in stool of infected WT and CD8α−/− mice. Data are representative of at least 3 independent experiments containing 3 to 6 mice per group at day 12 post infection. CFUs are normalized to fecal weight. Results are mean ± s.e.m. *p<0.05.
Consistent with previous work, CD45+ cells in the intestine produced significant levels of IFNγ relative to naïve mice. CD8+ T cells comprised a large proportion of IFNγ-producers during infection (Figure 2F) and mice lacking CD8+ T cells exhibited decreased IFNγ within the colon (Figure 2G) and increased pathogen load at day 12 post infection (Figure 2H), suggesting a functional role for CD8+ cells in IFNγ-mediated defense against C. rodentium. Staining of glycoproteins demonstrated that wildtype mice exhibited goblet cell depletion during infection, whereas HDAC3ΔIEC mice displayed reduced goblet cell depletion and maintained their mucous layer (Supplemental Figure 2C). These data are consistent with findings that mucosal-adherent pathogens are cleared in part through IFNγ-triggered depletion of goblet cells (Chan et al., 2013). To test whether the microbiota composition of HDAC3ΔIEC mice alone can increase C. rodentium burden, germ-free wildtype mice were colonized with microbiota from either HDAC3FF mice or HDAC3ΔIEC mice then infected with C. rodentium. Unlike the impaired control of C. rodentium observed in conventionally-housed HDAC3ΔIEC mice, germ-free mice colonized with HDAC3ΔIEC microbiota did not exhibit increased C. rodentium burden (Supplemental Figures 2D and 2E) or altered colonic IFNγ at day 12 (Supplemental Figure 2F). These data suggest that the microbiota composition of HDAC3ΔIEC mice is not sufficient to transfer altered host defense against C. rodentium infection.
HDAC3-dependent regulation of C. rodentium and intestinal IFNγ occurs early during infection in a microbiota-dependent manner
To identify the initial time point of altered pathogen dynamics in HDAC3ΔIEC mice, the level of infection was closely monitored following original exposure. Interestingly, significant impairment of pathogen control (Figure 3A) and induction of colonic IFNγ (Figure 3B) was already evident by day 6 post infection in mice lacking IEC-intrinsic HDAC3 expression, prior to significant histologic alterations. In the complimentary model, induced deletion of HDAC3 in IECs of infected HDAC3ΔIEC-IND adult mice also prevented induction of IFNγ in the colon within 5 days after tamoxifen (Supplemental Figure 3A). To test whether impaired early IFNγ induction in the colon may contribute to increased susceptibility of HDAC3ΔIEC mice to C. rodentium infection, a Th1-inducing immunostimulatory oligonucleotide (ISS-ODN) (Ciorba et al., 2010) was administered intraperitoneally at day 3 and 5 post infection. ISS-ODN partially restored IFNγ levels in the colon of C. rodentium-infected HDAC3ΔIEC mice (Figure 3C). Further, ISS-ODN treatment of mice lacking IEC-intrinsic expression of HDAC3, constitutively or inducibly, resulted in improved regulation of C. rodentium infection compared to vehicle-treated HDAC3ΔIEC mice (Figure 3D and Supplemental Figure 3B). IFNγ is essential for the protective benefits of ISS-ODN on pathogen load since the reduction in bacterial burden was not observed following administration of ISS-ODN to IFNγ−/− mice (Figure 3D). Taken together, these data indicate that epithelial expression of HDAC3 is necessary to mount an effective IFNγ immune response to an enteric bacterial infection.
Figure 3. HDAC3-dependent regulation of C. rodentium and intestinal IFNγ occurs early during infection in a microbiota-dependent manner.
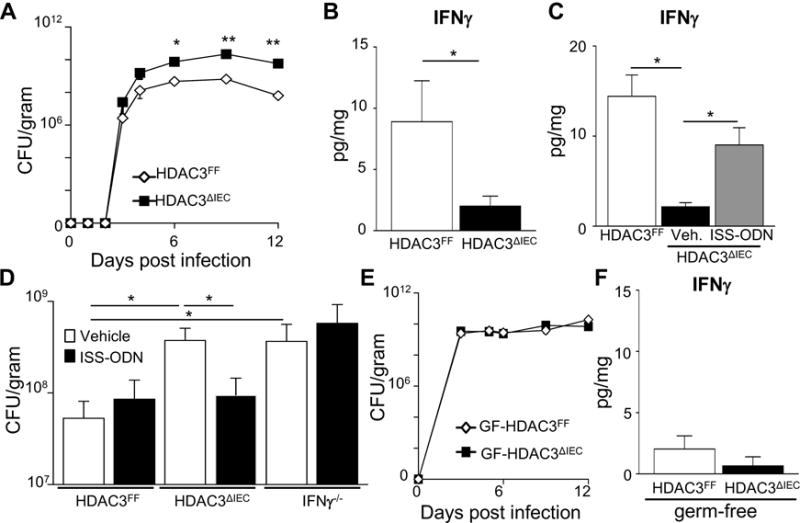
(A) C. rodentium colony forming units (CFU) in stool of infected HDAC3FF and HDAC3ΔIEC mice. (B) IFNγ in colonic tissue measured by ELISA 6 days post infection. (C) IFNγ in colonic tissue 6 days post infection following treatment with PBS or Th1-inducing immunostimulatory oligonucleotide (ISS-ODN). (D) C. rodentium CFU in stool mice treated with Vehicle (PBS) or ISS-ODN 6 days post infection. (E) C. rodentium CFU in stool of infected germ-free HDAC3FF and germ-free HDAC3ΔIEC mice. (B) IFNγ in colonic tissue of germ-free mice measured by ELISA 6 days post infection. Data are representative of at least 3 independent experiments containing 3 to 6 mice per group. CFUs are normalized to fecal weight. Results are mean ± s.e.m. *p<0.05.
To interrogate the role of microbiota-derived signals in HDAC3-dependent regulation of C. rodentium, the response to infection was compared in germ-free (GF) wildtype mice (GF-HDAC3FF) and GF-HDAC3ΔIEC mice. Consistent with previous work (Kamada et al., 2012), GF wildtype mice (GF-HDAC3FF) exhibited increased C. rodentium loads in the intestine relative to microbiota-replete conventionally-housed wildtype mice. Remarkably, in the absence of the microbiota, epithelial-intrinsic expression of HDAC3 was dispensable for resistance against C. rodentium (Fig. 3E). Unlike conventionally-housed microbiota-replete HDAC3ΔIEC mice, GF-HDAC3ΔIEC mice did not exhibit increased luminal C. rodentium burden (Fig. 3E) or decreased colonic IFNγ levels (Fig. 3F) relative to GF control mice. Although potential differences in pathogen localization can not be ruled out, these data suggest that HDAC3-dependent integration of microbiota-derived signals permits effective control of enteric bacterial infection.
Early infection-induced IFNγ production by intraepithelial CD8+ lymphocytes is impaired in HDAC3ΔIEC mice
HDAC3ΔIEC mice exhibited impaired control of pathogen loads and failed to induce effective IFNγ production in the colon early in the course of infection (Figures 3A and 3B), suggesting that resident immune cells in HDAC3ΔIEC mice may not be primed to effectively activate protective local IFNγ responses during infection. Intraepithelial lymphocytes (IELs) constitute a heterogeneous population of immune cells that are in close proximity to IECs and produce IFNγ in response to mucosal pathogens (Cheroutre et al., 2011). Remarkably, we found that CD45+ IELs harvested from the colon 6 days post infection already exhibited significant activation of IFNγ relative to naïve mice (Figure 4A). The majority of infection-induced IFNγ-producing IELs represented T cells and CD8α+ TCRß+ lymphocytes, not CD4+ cells, γδ T cells, or NK cells, were the major initial source of IFNγ in the IEL compartment of C. rodentium-infected wildtype mice (Figures 4B and 4C, Supplemental Figure 4A). More in depth characterization identified the predominant IFNγ-producing CD8α+ TCRß+ IELs during C. rodentium infection as resident CD8αα+, CD103+ IELs, whereas a smaller portion were CD8αß+IELs (Figures 4D and 4E).
Figure 4. Intraepithelial lymphocytes activate a rapid IFNγ response following C. rodentium infection.
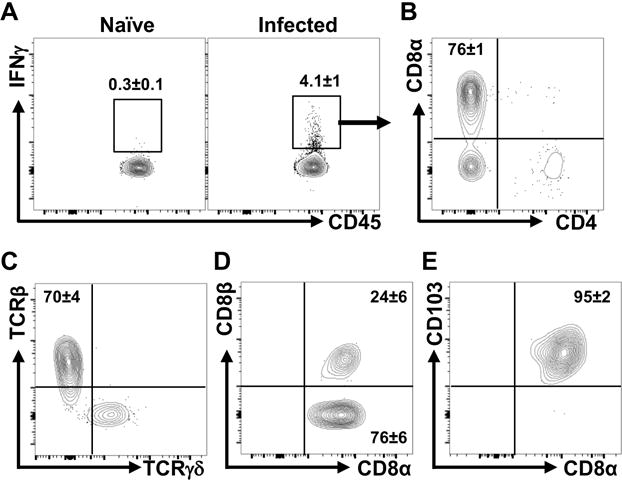
(A) Representative flow cytometry plots of IFNγ-producing CD45+ IELs from wildtype (WT) mice, naïve and 6 days post C. rodentium. (B) Representative plots of IFNγ-producing CD8α+ and CD4+ cells from infected mice. Gated on CD45+, IFNγ-producing cells in (A). (C) Representative plots of TCR staining. Gated on CD8α+ cells in (B). (D, E) Characterization of CD8+ cells in (B) for (D) CD8αα versus CD8αβ and (E) CD103 expression. Data are representative of at least 3 independent experiments containing 3 mice per group at day 6 post infection.
To test whether HDAC3 expression in IECs is necessary to activate initial IFNγ-producing IELs, these cells were compared in HDAC3ΔIEC mice. Similar to wildtype mice, CD8α+TCRß+ cells represented the predominant IEL population within the colon of C. rodentium infected HDAC3ΔIEC mice (Figure 5A). Interestingly, however, induction of IFNγ-producing CD8α+TCRß+ IELs was significantly impaired in the colons of HDAC3ΔIEC mice relative to control HDAC3FF mice post-infection (Figures 5B and 5C). These data indicate that epithelial HDAC3 is required to activate early cytokine production by the main IFNγ-producing IEL population during enteric infection. Moreover, IFNγ-producing γδ IELs, which were not a major source of IFNγ following C. rodentium infection, were also reduced within the colon of HDAC3ΔIEC mice (Supplemental Figures 4B and 4C), whereas IL-17 and IL-22 IEL responses were less affected at day 6 post infection (Supplemental Figures 4D and 4E). Collectively, these data demonstrate that expression of HDAC3 in IECs is required for early activation of local IFNγ-producing IELs during an enteric infection.
Figure 5. Intraepithelial lymphocytes in HDAC3ΔIEC mice exhibit impaired IFNγ response to an enteric bacterial infection.
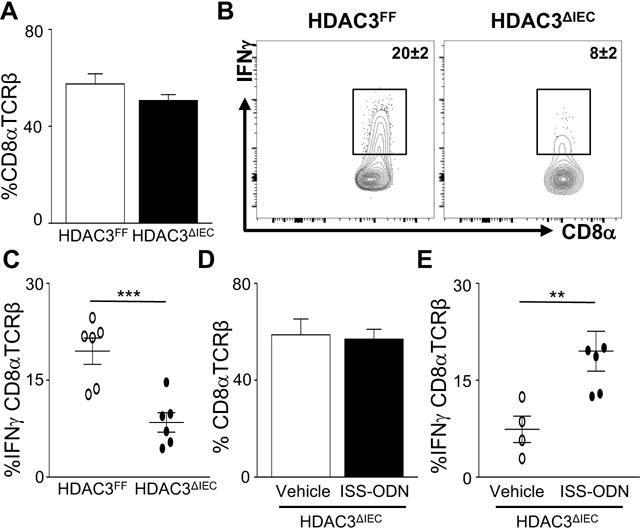
(A) %CD8α+TCRβ+ IELs in colon of HDAC3FF and HDAC3ΔIEC mice. (B) Representative flow cytometry plots of IFNγ-producing CD8α+TCRβ+ IELs. (C) %IFNγ-producing CD8α+TCRβ+ IELs in colons from infected mice. (D) %CD8α+TCRβ+ IELs in infected HDAC3ΔIEC mice following ISS-ODN treatment. (E) %IFNγ-producing CD8α+TCRβ+ IELs in colons from HDAC3ΔIEC mice treated with Vehicle (PBS) or ISS-ODN. Data are representative of at least 3 independent experiments containing 3–6 mice per group at day 6 post infection. Results are mean ± s.e.m. *p<0.05, **p<0.01.
To decipher whether CD8α+TCRß+ IELs from mice lacking IEC-intrinsic HDAC3 were developmentally blocked from producing IFNγ, a Th1-inducing immunostimulatory oligonucleotide (ISS-ODN) was administered to HDAC3ΔIEC mice. ISS-ODN treatment did not alter the frequency of CD8α+TCRß+ IELs (Figure 5D), but increased IFNγ-producing CD8α+TCRß+ IELs (Figure 5E) in HDAC3ΔIEC mice. Thus, IELs from mice lacking epithelial HDAC3 remain capable of producing IFNγ, suggesting that IEL-extrinsic activating signals are impaired in HDAC3ΔIEC mice during infection.
Epithelial HDAC3 regulates IEL activation independent of circulating signals
While CD8αα+ TCRαß+ IELs are thought to localize within the intestinal epithelium early in development, protection against C. rodentium infection reflects both local and systemic immune response. Therefore, to distinguish whether regulation of IELs by epithelial HDAC3 represents local intestinal immunity or requires input from non-colonic or circulating cells, an ex vivo infection system was established in which naïve and infected colon tissues from the same mouse were compared (Figure 6A). Infection of colon explants resulted in association of the pathogen with IECs (Figure 6B) along with a significant increase in local colonic IFNγ production (Figure 6C), confirming that tissue-resident cells can independently induce IFNγ expression during infection. Similar to C. rodentium infection in vivo, CD8α+TCRß+ IELs within colon explants were the main producers of IFNγ following C. rodentium infection (Figures 6D and 6E). Further, HDAC3 expression in IECs was necessary to induce IFNγ expression by CD8α+TCRß+ IELs following ex vivo infection (Figure 6F). Therefore, activation of these tissue-resident IELs during infection largely reflects intestine-intrinsic regulation rather than signals received from circulating factors or recruited immune cells.
Figure 6. Epithelial HDAC3 regulates intestinal lymphocyte activation independent of circulating signals.
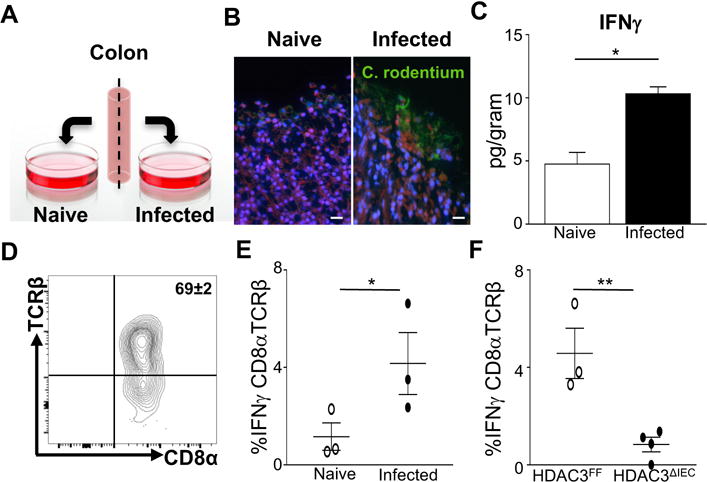
(A) Ex vivo colonic infection. (B) Immunofluorescent staining of ex vivo cultured colon from the same mouse, naïve or infected with GFP-expressing C. rodentium (green) as in (A). Mammalian cell membrane and nuclei are stained with CellMask (red) and DAPI (blue), respectively, scale bar 50 μm. (C) IFNγ in colon samples from (B) measured by ELISA. (D) Identification of CD8α+TCRβ+ IELs in colon explant by flow cytometry. (E) %IFNγ-producing CD8α+TCRβ+ IELs in colon samples from (B). (F) %IFNγ-producing CD8α+TCRβ+ IELs of C. rodentium infected colon explants from HDAC3FF and HDAC3ΔIEC mice. Data are representative of at least 3 independent experiments containing 3–6 mice per group 24h post ex vivo infection. Results are mean ± s.e.m. *p<0.05.
IEC-intrinsic HDAC3 coordinates local host protection against enteric infection through epithelial IL-18 expression
To investigate potential molecular mediators linking IEC regulation by HDAC3 to activation of local IFNγ-producing IELs, the IFNγ-inducing cytokine IL-18 was examined. Following infection, C. rodentium induced IL-18 expression initially in colonic IECs (Figure 7A). Interestingly, IECs from HDAC3ΔIEC mice were unable to induce significant IL-18 in response to the pathogen (Figures 7B and 7C), indicating that HDAC3 expression is necessary for activation of IL-18 production by IECs during infection. Other cytokines expressed by epithelium, such as IL-1β, IL-15, and IL-7, were minimally altered in HDAC3ΔIEC mice (Supplemental Figure 5A), indicating that IL-18 represents a cytokine that is more specifically regulated by HDAC3 during C. rodentium infection. Further, IECs from infected HDAC3ΔIEC mice exhibited decreased expression of multiple genes that can induce IL-18 transcription and post-translational processing (Supplemental Figure 5B), suggesting that networks of pathways mediate how HDAC3 regulates IL-18 expression and activation in IECs.
Figure 7. Regulation of epithelial IL-18 by HDAC3 directs local innate IFNγ-mediated antibacterial immunity.
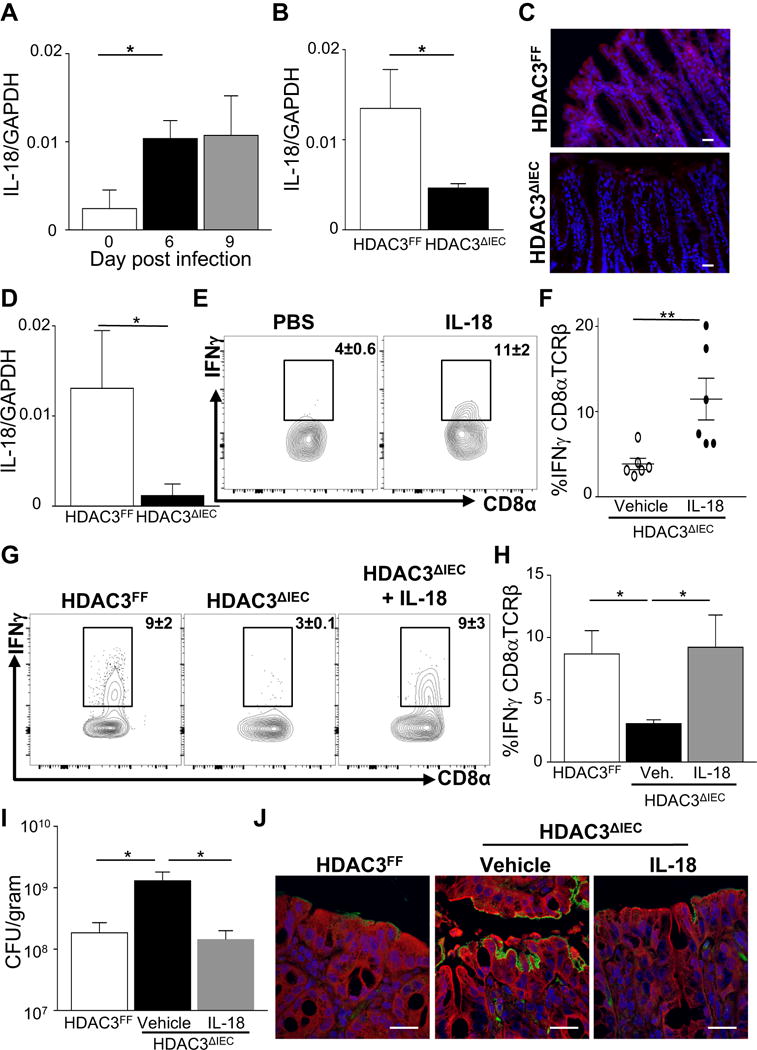
(A) IL-18 mRNA expression in IECs from the colon of wildtype mice during infection. (B) IL-18 mRNA expression in IECs at day 6 post C. rodentium infection. (C) Immunofluorescent staining of IL-18 (Red) and nuclei (DAPI, blue) in the colon post infection, scale bar 50 μm. (D) IL-18 mRNA expression in IECs following 24h ex vivo infection with C. rodentium. (E) Representative flow cytometry plots of IFNγ-producing CD8α+TCRβ+ IELs following vehicle (PBS) or IL-18 treatment of ex vivo infected colon from the same HDAC3ΔIEC mouse. (F) %IFNγ-producing CD8α+TCRβ+ IELs in colon samples from (E). (G) Representative plots of IFNγ-producing CD8α+TCRβ+ IELs following in vivo vehicle (PBS) or IL-18 treatment 6 days post infection. (H) %IFNγ-producing CD8α+TCRβ+ IELs in colon from (G). (I) C. rodentium CFU in stool at day 6 post infection following vehicle (PBS) or IL-18 treatment. (J) Immunofluorescent staining of GFP-expressing C. rodentium (green), intestinal cells (red) and nuclei (DAPI, blue), scale bar 50 μm. Data are representative of at least 3 independent experiments containing 4–6 mice per group. CFUs are normalized to fecal weight. Results are mean ± s.e.m. *p<0.05.
To test whether HDAC3-dependent regulation of IL-18 in IECs triggered resident IELs to produce IFNγ independently of circulating factors, we employed the ex vivo colon infection model (Figure 6). Consistent with alterations observed in vivo, infection of explanted colons lacking IEC-intrinsic HDAC3 failed to induce significant epithelial IL-18 expression (Figure 7D). Interestingly, direct administration of IL-18 to infected colon tissue from HDAC3ΔIEC mice significantly increased the frequency of IFNγ-producing CD8α+TCRß+ IELs (Figures 7E and 7F). Collectively, these data identify that intestine-intrinsic regulation of IL-18 by HDAC3 is essential for activating local type 1 effector function by IELs in response to an enteric infection.
In the intestine, recent evidence suggests that the in vivo effect of IL-18 production can vary depending on the immune context, microbial signals, and cellular source (Nowarski et al., 2015; Siegmund, 2010). To test whether regulation of IL-18 by HDAC3 mediates protection to an enteric infection, IL-18 was administered intraperitoneally during infection to HDAC3ΔIEC mice. Similar to ex vivo explant analyses, IL-18 increased the frequency of IFNγ-producing CD8α+TCRß+ IELs in HDAC3ΔIEC mice at day 6 post infection (Figures 7G and 7H). Interestingly, IL-18-treated HDAC3ΔIEC mice also exhibited significantly improved control of the pathogen relative to vehicle-treated HDAC3ΔIEC mice (Figures 7I and 7J). Taken together, these findings reveal that regulation of IL-18 expression by HDAC3 in IECs is critical for innate defense in the intestine and tissue-intrinsic activation of resident lymphocytes.
DISCUSSION
These findings highlight a previously unrecognized level of intestine-intrinsic regulation between IECs and resident lymphocytes in immunity against a mucosal pathogen. Specifically, epithelial regulation by a microbiota-responsive HDAC is necessary to control enteric infection and induce adjacent lymphocytes to initiate defense against the pathogen (Supplemental Figure 6). Similar to other cells in which bactieral-derived fatty acids can modulate HDACs, metabolites from C. rodentium may be sensed by HDAC3 in the epithelium. Alternatively, or in addition to, HDAC3 activation may function downstream of pattern recognition signaling or nuclear hormone receptor-dependent regulation. Although excess production of pro-inflammatory cytokines such as IL-18 are associated with autoimmune disease, these findings indicate that epithelial priming of CD8αα+ intraepithelial lymphocytes by IL-18 maintains optimal protective immunity. Previous studies indicate that CD8αα+TCRαβ+ IELs possess an antigen-experienced phenotype (Cheroutre et al., 2011) and we have identified that these cells are a dominant source of intestinal IFNγ initially following C. rodentium infection. While our studies highlight the significance of crosstalk between IECs and IELs, we cannot rule out that myeloid cells or other innate populations in the intestine play a contributory role in mediating IEC-initiated communication with IELs. Along this line, myeloid IL-12 production or IL-22 from innate lymphoid cells may synergize with IL-18 to maintain optimal IFNγ responses (Freeman et al., 2012; Simmons et al., 2002; Zheng et al., 2008). The frequency of IFNγ-producing γδ cells was also reduced in infected HDAC3ΔIEC mice. Thus, our data provoke the intriguing possibility that epithelial HDAC3 is required to broadly activate local lymphocytes early during an enteric infection.
Innate pathways differentially protect against C. rodentium infection. For instance, mice lacking innate lymphoid cells or IL-23/IL-22 signaling quickly succumb to C. rodentium infection due to impaired barrier function and translocation of the pathogen (Cella et al., 2009; Satoh-Takayama et al., 2008; Satpathy et al., 2013; Sonnenberg et al., 2011; Zheng et al., 2008). IL-22-induced production of antimicrobial peptides from IECs appears to be a critical downstream pathway that mediates containment of the bacterial pathogen (Zheng et al., 2008). Mast cells have also been shown to prevent dissemination of the pathogen (Wei et al., 2005). Similar to HDAC3 in IECs, other innate pathways regulate the severity of C. rodentium infection itself, more so than intestinal containment and survival. Depletion of CD32−NK1.1⁺ cells results in decreased colonic IFNγ, TNFα and IL-12 followed by increased bacterial load (Reid-Yu et al., 2013). In addition, mice lacking IL-17 exhibit increased bacterial burden, prolonged infection and more severe C. rodentium induced pathology (Ishigame et al., 2009; Zheng et al., 2008). Further, while expression IFNγ often associates with more severe colitis, IFNγ production during C. rodentium infection reduced the severity of the infection and improved clearance from the intestine (Simmons et al., 2002). Our results indicate that this protective role for IFNγ reflects HDAC3-dependent intestine-intrinsic regulation.
Recent studies have demonstrated seemingly opposing effects of IL-18 in the intestine, suggesting that IL-18 is central to the balance between healthy immune homeostasis and pathologic inflammation in the intestine (Elinav et al., 2011; Harrison et al., 2015; Levy et al., 2015; Nowarski et al., 2015; Siegmund et al., 2001; Wlodarska et al., 2014). Increased IL-18 expression from IECs, macrophages and dendritic cells has been found in samples from Crohn’s disease patients (Pizarro et al., 1999). In line with this data, blocking IL-18 signaling protects mice from DSS and TNBS damage-inducing models of colitis (Siegmund et al., 2001; Sivakumar et al., 2002). Further, deletion of IL-18/IL-18R in IECs reduced DSS-induced colitis whereas loss of IL-18 binding protein, a negative regulator of IL-18, increased the severity of colitis by inhibiting goblet cell maturation and development (Nowarski et al., 2015). In contrast, IL-18/IL-18R deficient mice, as well as Casp1 and Nlrp6 deficient mice that cannot activate IL-18, exhibit a more severe DSS-induced colitis and decreased survival during C. rodentium infection (Elinav et al., 2011; Takagi et al., 2003; Wlodarska et al., 2014).
Taken together, these findings suggest that IL-18 equilibrium needs to be tightly regulated in the intestine to protect against infection while limiting the risk of intestinal or systemic pathology. Our work identifies the HDAC3 enzyme as a necessary regulator in calibrating production of IL-18 locally in the intestine during enteric bacterial infection. The most well characterized function of the HDAC3 enzymatic complex is to repress transcription through histone deacetylation of direct targets. Because IL-18 is decreased in HDAC3ΔIEC mice, it is likely that HDAC3 directly regulates IL-18 repressive pathways. The altered response to Citrobacter infection of mice with IEC specific HDAC3 deletion is unlikely due to abnormalities in a single downstream pathway. Instead, our data suggest that the changes observed in HDAC3ΔIEC mice reflect an integrated effect of multiple transcriptional networks that result from impaired HDAC3 activity. Further, we cannot exclude that HDAC3-dependent regulation of inflammasomes, deacetylation of non-histone proteins, such as those involved in NF-κB signaling, or enzyme-independent processes represent additional mechanisms for how HDAC3 regulates IL-18 transcription and activation in IECs.
Recent studies have highlighted that commensal microbes can greatly impact how the mammalian host immune system responds to infection (Abt et al., 2012; Benson et al., 2009; Ganal et al., 2012; Ichinohe et al., 2011). However, little is known about host mechanisms that regulate how mammalian cells integrate beneficial microbial signals and simultaneously instruct clearance of intestinal pathogens. The work described here suggests that microbiota-dependent epigenetic mechanisms prime mammalian cells and render them more responsive to pathogens in the context of the diverse microbial signals. Because HDAC3 in IECs integrates signals from the microbiota to regulate transcription, this epigenetic enzyme reflects a microbiota-sensitive target that enables optimal host responsiveness to enteric pathogens. Therefore, HDAC3-triggered epithelial-IEL crosstalk represents a pathway that can be exploited to improve how the microbiota directs innate immunity to mucosal infection.
EXPERIMENTAL PROCEDURES
Mice
HDAC3FF mice (Mullican et al., 2011) were bred to C57BL/6 mice expressing Cre-recombinase(Madison et al., 2002) or tamoxifen-dependent Cre recombinase(el Marjou et al., 2004) under the control of the villin promoter to generate HDAC3ΔIEC and HDAC3ΔIEC-IND mice, respectively(Alenghat et al., 2013). Germ-free (GF) mice were maintained in isolator units in the CCHMC Gnotobiotic Mouse Facility, fed autoclaved feed and water, and routinely monitored to ensure absence of microbial contamination. Deletion of HDAC3 in HDAC3ΔIEC-IND mice was induced by intraperitoneal injection of 1 mg of tamoxifen (Sigma) once per day for 5 days starting day 3 post infection. For immunostimulatory DNA (ISS-ODN), mice were injected with 10 μg ISS-ODN (5′-TGACTGTGAACGTTCGAGATGA-3′) at day 3 and 5 post infection. Colonization of GF mice was performed with intestinal contents from either HDAC3FF or HDAC3ΔIEC mice. Mice were used at 7–12 weeks old, and age- and gender-matched mice were used for all experiments. Animals were housed up to 4 per cage in a ventilated cage with 12 h light/dark cycle and free access to water and chow. Animals were provided with appropriate care by a licensed veterinarian. All experiments were done according to guidelines of the Institutional Animal Care and Use Committee.
C. rodentium infection
Mice were infected orally with 1×109 GFP-C. rodentium (Bergstrom et al., 2010). Stool was collected in phosphobuffered saline (PBS) and homogenized in a Tissue Lyser II at 30 Hz. Homogenates were serially diluted and plated on MacConkey. CFUs were counted and normalized to stool weight after 24h. For IL-18 treatment, 0.5 μg/mouse rIL-18 (MBL) was injected at day 2 and 4 post infection. For explants, the whole colon was removed and divided longitudinally, washed and placed in complete media (DMEM, 10% FBS, 1% glutamine, 1% HEPEs and 0.1% 2-Mercaptoethanol). The colon explant was incubated with 1×108 GFP-C. rodentium for 24 h. Longitudinal sections from the same mouse were treated with PBS or 100ng/ml rIL-18.
IEC harvest, real-time PCR, ELISA
IECs were isolated from murine samples as described (Alenghat et al., 2013) by shaking intestinal tissue in 1 mM EDTA/1 mM DTT and 5% FCS at 37°C for 10 minutes. RNA was isolated from cells using RNeasy Kit (Qiagen) and subjected to reverse transcription (Verso, Thermoscientific). Real-time PCR was performed using SYBR (Applied Biosystems) and custom primers (Invitrogen, Supplemental Table 1). Reactions were run on a real-time PCR machine (Applied Biosystems) and analyzed with a threshold in the linear range of amplification and based on a standard of serial ten-fold dilutions. Samples were normalized to an endogenous control gene. For tissue ELISA, 1 cm colon sections were rinsed with PBS and homogenized in RIPA. Samples were centrifuged and assayed by ELISA; IFNγ (BD), IL-17 (eBio), IL-22 (BioLegend). Concentrations were normalized to colon weight.
HDAC activity
IECs were isolated followed by CD45+ depletion using Mojosort mouse CD45 nanobeads and magnet (Biolegend). IECs were lysed in RIPA and equal protein concentration of lysate was in incubated with 100 μM of HDAC substrate (Active Motif), followed by developer solution containing 2 μM of trichostatin A (TSA) to stop the reaction. Fluorescence was measured using a fluorescent plate reader (Biotek Synergy 2; excitation:340nm, emission:460nm) and plotted relative to naïve samples.
Flow cytometry
Large intestine was shaken in 5 mM EDTA/1 mM DTT for 10 min and then transferred to fresh 5 mM EDTA/1 mM DTT to incubate for 10 more minutes to obtain IEL fractions. IEL pellets were mixed in 30% isotonic Percoll and layered onto 70% isotonic Percoll. Cells were collected from Percoll gradients following centrifugation at 1800 rpm for 20 min and washed in RPMI-1640. Cells were stained with a combination of the following fluorescence-conjugated mAbs: PE–CF594 anti-CD4 (BD), PE-Cy7 anti-TCRβ (eBioscience), PE anti-CD326 (EpCAM) (Miltenyi Biotec), eFluor450 anti-CD8β (eBioscience), eFluor450 anti-TCRγδ (eBioscience), APC anti-IFNγ (eBioscience), FITC anti-CD45 (eBioscience), PerCP-Cy5.5 anti-CD8α (eBioscience), BV650 anti-IL-17 (BD), APC anti-IL-22 (BioLegend). Dead cells were excluded with Fixable Aqua Dead Cell Stain kit (Invitrogen). Samples were acquired on LSR Fortessa II and analyzed with FlowJo software (v10.0; TreeStar).
Histology, immunohistochemistry, immunofluoresence
Sections were fixed in 4% paraformaldehyde or methanol carnoy (60%methanol, 30% chloroform, 10% acetic acid), paraffin embedded, sectioned, and stained with hematoxylin and eosin or periodic acid-schiff/Alcian blue. For immunofluorescence, tissue was stained with anti-GFP (ThermoFisher), anti-IL-18 (Rockland), or HCS CellMask™ Red Stain (ThermoFisher) and DAPI. For histological scoring, H&E-stained colonic tissue sections were blindly graded by a board-certified pathologist on a scale of 1–5 for each of the following C. rodentium-associated histologic parameters: (a) mural edema (1–5), (b) epithelial lesions (crypt hyperplasia, elongation, erosion) (1–5), and (c) leukocyte infiltration (1–5) (Giacomin et al., 2015; Jain et al., 2015; Liu et al., 2012; Osborne et al., 2013). Each parameter was scored at 12 days post infection on a scale ranging from 1–5; allowing for maximum total histologic score of 15. 1 corresponded to no inflammation and 2, 3, 4, and 5 corresponded to minimal, mild, moderate, and severe, respectively.
Statistics
Results are shown as mean ± s.e.m. Mice of the indicated genotypes were assigned at random to groups. Mouse studies were not performed in a blinded fashion. All inclusion/exclusion criteria were pre-established. Statistical significance was determined with the t-test. All data meet the assumptions of the statistical tests used. Results were considered significant at *p ≤ 0.05; **p ≤ 0.01. Statistical analyses were performed using Prism version 7.0 (GraphPad).
Supplementary Material
Acknowledgments
We thank members of the Alenghat, Way, Haslam, Qualls, and Deshmukh labs for scientific input, reagents, and critical reading of the manuscript. We also thank K. Steinbrecher and S. Divanovic for technical assistance and D. Artis for useful discussions. We thank CCHMC Veterinary Services, Pathology Research Core, Research Flow Cytometry Core, and Confocal Imaging Core. This research is supported by the NIH (DK093784), a Crohn’s and Colitis Foundation of America/Janssen/AGA award to T.A., and by operating grants to B.A.V. from the Canadian Institutes of Health Research (CIHR) (MOP-126051) and from NSERC. T.A. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trust. B.A.V. is the Children with Intestinal and Liver Disorders (CH.I.L.D.) Foundation Research Chair in Pediatric Gastroenterology. S.S.W. is supported by the HHMI Faculty Scholars program, and an Investigator in the Pathogenesis Award from the Burroughs Wellcome Fund. This project is supported in part by PHS grant P30 DK078392 and the CCHMC Trustee Award and Procter Scholar’s Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
T.A. and N.N. designed the studies. N.N., J.W., S.W., V.W., and J.M. carried out experiments. M.B.J., B.A.V., and S.S.W. provided bacterial and mouse strains, advice or technical expertise and T.A. and N.N. analyzed the data and wrote the manuscript.
References
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37:158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CG, Mullican SE, Choi I, Grunberg S, Sinha R, Wynosky-Dolfi M, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature. 2013;504:153–157. doi: 10.1038/nature12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson A, Pifer R, Behrendt CL, Hooper LV, Yarovinsky F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe. 2009;6:187–196. doi: 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010;6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RE. Epidemiology of diarrhoeal disease: implications for control by vaccines. Vaccine. 1993;11:100–106. doi: 10.1016/0264-410x(93)90002-f. [DOI] [PubMed] [Google Scholar]
- Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JM, Bhinder G, Sham HP, Ryz N, Huang T, Bergstrom KS, Vallance BA. CD4+ T cells drive goblet cell depletion during Citrobacter rodentium infection. Infect Immun. 2013;81:4649–4658. doi: 10.1128/IAI.00655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111:2247–2252. doi: 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S, Natoli G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012;109:E2865–2874. doi: 10.1073/pnas.1121131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol. 2011;11:445–456. doi: 10.1038/nri3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciorba MA, Bettonville EE, McDonald KG, Metz R, Prendergast GC, Newberry RD, Stenson WF. Induction of IDO-1 by immunostimulatory DNA limits severity of experimental colitis. J Immunol. 2010;184:3907–3916. doi: 10.4049/jimmunol.0900291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P, Sansonetti PJ. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 2004;304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- DuPont HL. Clinical practice. Bacterial diarrhea. N Engl J Med. 2009;361:1560–1569. doi: 10.1056/NEJMcp0904162. [DOI] [PubMed] [Google Scholar]
- DuPont HL. Acute infectious diarrhea in immunocompetent adults. N Engl J Med. 2014;370:1532–1540. doi: 10.1056/NEJMra1301069. [DOI] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman BE, Hammarlund E, Raue HP, Slifka MK. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc Natl Acad Sci U S A. 2012;109:9971–9976. doi: 10.1073/pnas.1203543109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12-and IL-15-responsive IFN-gamma-producing cells. Immunity. 2013;38:769–781. doi: 10.1016/j.immuni.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. 2012;12:503–516. doi: 10.1038/nri3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity. 2012;37:171–186. doi: 10.1016/j.immuni.2012.05.020. [DOI] [PubMed] [Google Scholar]
- Giacomin PR, Moy RH, Noti M, Osborne LC, Siracusa MC, Alenghat T, Liu B, McCorkell KA, Troy AE, Rak GD, et al. Epithelial-intrinsic IKKalpha expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J Exp Med. 2015;212:1513–1528. doi: 10.1084/jem.20141831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Srinivasan N, Pott J, Schiering C, Krausgruber T, Ilott NE, Maloy KJ. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol. 2015;8:1226–1236. doi: 10.1038/mi.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Jain U, Cao Q, Thomas NA, Woodruff TM, Schwaeble WJ, Stover CM, Stadnyk AW. Properdin provides protection from Citrobacter rodentium-induced intestinal inflammation in a C5a/IL-6-dependent manner. J Immunol. 2015;194:3414–3421. doi: 10.4049/jimmunol.1401814. [DOI] [PubMed] [Google Scholar]
- Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Nunez G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koroleva EP, Halperin S, Gubernatorova EO, Macho-Fernandez E, Spencer CM, Tumanov AV. Citrobacter rodentium-induced colitis: A robust model to study mucosal immune responses in the gut. J Immunol Methods. 2015;421:61–72. doi: 10.1016/j.jim.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell. 2015;163:1428–1443. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zaki MH, Vogel P, Gurung P, Finlay BB, Deng W, Lamkanfi M, Kanneganti TD. Role of inflammasomes in host defense against Citrobacter rodentium infection. J Biol Chem. 2012;287:16955–16964. doi: 10.1074/jbc.M112.358705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, Feng D, Steger DJ, Schug J, Artis D, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011;25:2480–2488. doi: 10.1101/gad.175950.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. 2005;7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, Low JS, Harman CC, Graham M, Elinav E, et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell. 2015;163:1444–1456. doi: 10.1016/j.cell.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LC, Joyce KL, Alenghat T, Sonnenberg GF, Giacomin PR, Du Y, Bergstrom KS, Vallance BA, Nair MG. Resistin-like molecule alpha promotes pathogenic Th17 cell responses and bacterial-induced intestinal inflammation. J Immunol. 2013;190:2292–2300. doi: 10.4049/jimmunol.1200706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF, Jr, Foley E, Moskaluk CA, Bickston SJ, Cominelli F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol. 1999;162:6829–6835. [PubMed] [Google Scholar]
- Ramanan D, Cadwell K. Intrinsic Defense Mechanisms of the Intestinal Epithelium. Cell Host Microbe. 2016;19:434–441. doi: 10.1016/j.chom.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid-Yu SA, Small CL, Coombes BK. CD3(−)NK1.1(+) cells aid in the early induction of a Th1 response to an attaching and effacing enteric pathogen. Eur J Immunol. 2013;43:2638–2649. doi: 10.1002/eji.201343435. [DOI] [PubMed] [Google Scholar]
- Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, Wu X, Thomas SR, Lee WL, Turkoz M, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14:937–948. doi: 10.1038/ni.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi H, Masuda A, Nishiumi S, Nishida M, Takagawa T, Shiomi Y, Kutsumi H, Blumberg RS, Azuma T, Yoshida M. Gamma interferon produced by antigen-specific CD4+ T cells regulates the mucosal immune responses to Citrobacter rodentium infection. Infect Immun. 2010;78:2653–2666. doi: 10.1128/IAI.01343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund B. Interleukin-18 in intestinal inflammation: friend and foe? Immunity. 2010;32:300–302. doi: 10.1016/j.immuni.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, Frankel G, Dougan G, MacDonald TT. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J Immunol. 2002;168:1804–1812. doi: 10.4049/jimmunol.168.4.1804. [DOI] [PubMed] [Google Scholar]
- Sivakumar PV, Westrich GM, Kanaly S, Garka K, Born TL, Derry JM, Viney JL. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut. 2002;50:812–820. doi: 10.1136/gut.50.6.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) Lymphoid Tissue-Inducer Cells Promote Innate Immunity in the Gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H, Kanai T, Okazawa A, Kishi Y, Sato T, Takaishi H, Inoue N, Ogata H, Iwao Y, Hoshino K, et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand J Gastroenterol. 2003;38:837–844. doi: 10.1080/00365520310004047. [DOI] [PubMed] [Google Scholar]
- Wei OL, Hilliard A, Kalman D, Sherman M. Mast cells limit systemic bacterial dissemination but not colitis in response to Citrobacter rodentium. Infection and immunity. 2005;73:1978–1985. doi: 10.1128/IAI.73.4.1978-1985.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045–1059. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.