

Abstract
Orally bioavailable SERDs may offer greater systemic drug exposure, improved clinical efficacy, and more durable treatment outcome for patients with ER-positive endocrine-resistant breast cancer. We report the design and synthesis of a boronic acid modified fulvestrant (5, ZB716), which binds to ERα competitively (IC50 = 4.1 nM) and effectively downregulates ERα in both tamoxifen-sensitive and tamoxifen-resistant breast cancer cells. Furthermore, It has superior oral bioavailability (AUC = 2547.1 ng·h/mL) in mice, indicating its promising clinical utility as an oral SERD.
Graphical abstract
INTRODUCTION
Fulvestrant, the only FDA approved selective estrogen receptor downregulator (SERD), is indicated for estrogen receptor positive (ER+), metastatic breast cancer in postmenopausal women following progression on prior endocrine therapy.1–3 Because of its poor oral bioavailability, fulvestrant is administered by intramuscular injection (im) at a monthly dose of 250 mg, which takes over 3 months to reach steady-state serum concentration and has negatively impacted its widespread clinical applications.4 Moreover, even at a later approved dose of 500 mg, the peak blood concentration of fulvestrant remains below a modest 25 ng/mL in the FINDER1 and FINDER2 clinical trials,3,5 suggesting that its optimal efficacy in patients may not have been achieved.2 In addition, in the 500 mg regimen, the steady-state fulvestrant concentration in patient blood still takes one month to establish. However, the promising clinical utility of fulvestrant and the increasing understanding of its mechanism of action have motivated the development of orally bioavailable SERDs.
Attempts to make orally bioavailable steroidal SERDs have been reported where fulvestrant-like compounds were designed and synthesized with modifications made primarily to the long alkyl chain to increase polarity and solubility.6–8 However, such designs failed to address the main problem that is responsible for the poor bioavailability of fulvestrant. It has been well-documented that fulvestrant undergoes rapid and extensive O-glucuronidation9,10 and O-sulfation11,12 to form polar phase II metabolites that are inactive and water-soluble. Four UDP-glucuronosyltransferase 1A (UGT1A) enzymes have been identified that glucuronidate fulvestrant at position 3, accounting for over 90% of metabolic inactivation and clearance of fulvestrant via glucuronidation.9 Likewise, sulfotransferases SULT1A and SULT1C were found to catalyze sulfate conjugation with fulvestrant at C-3 position, also leading to the rapid excretion of fulvestrant as sulfate conjugate from systemic circulation.11 Meanwhile, nonsteroidal SERDs with oral bioavailability have also been reported, as evidenced by two recent candidates (GDC-810, (E)-3-(4-((E)-2-(2-chloro-4-fluorophenyl)-1-(1H-indazol-5-yl)but-1-en-1-yl)phenyl)acrylic acid) and AZD9496, (E)-3-(3,5-difluoro-4-((1R,3R)-2-(2-fluo-ro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic acid) being evaluated in phase I clinical trials.13,14 These molecules share a common cinnamic acid moiety with efficacy and safety profiles eagerly awaited from the course of phase I and II clinical trials.15,16
Indeed, phase II metabolic inactivation and clearance are common biotransformation pathways that often render drugs like fulvestrant inaccessible to target tissues.12,17–19 On the basis of this understanding and previous studies in our laboratory,20–22 we envisioned a solution in which the 3-OH group of fulvestrant is replaced by a boronic acid group (Figure 1). We have observed that the boronic acid derivatives of antiestrogenic compounds such as 4-hydroxytamoxifen and endoxifen significantly reduced first pass metabolism of hydroxylated drug molecules, leading to their enhanced bioavailability in the systemic circulation.21,22 Therefore, we anticipate that this modification can minimize glucuronidation and sulfation while retaining sufficient binding affinity of the steroidal moiety of fulvestrant to confer its SERD properties. We report the synthesis and comprehensive evaluation of fulvestrant-3 boronic acid (5, ZB716), a novel, orally bioavailable steroidal SERD. We show that 5 binds to ER with high affinity and exerts its antiestrogenic effect on ER-expressing breast cancer cells. We also demonstrate that in both tamoxifen-sensitive and tamoxifen-resistant breast cancer cells, 5 potently inhibits cell proliferation and effectively degrades the hormone receptor protein in a dose-dependent manner. Moreover, 5 is shown to have far superior oral bioavailability in mice when compared to fulvestrant by subcutaneous injection (sc), suggesting that it can serve as a promising drug for breast cancer patients with progressing disease after prior endocrine therapy.
Figure 1.
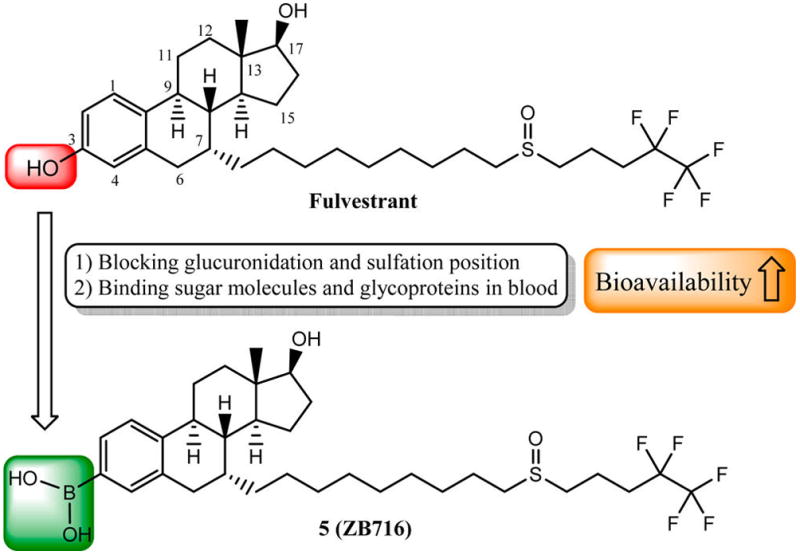
Design strategy of fulvestrant-3 boronic acid (5).
RESULTS
Chemistry
Starting from 17-acetyl S-deoxo fulvestrant (1), the target compound 5 was prepared through a four-step synthetic route (Scheme 1). Briefly, esterification of 1 with triflic anhydride provided its triflate (2), which was reacted with bis(pinacolato)diboron in the presence of palladium(II) acetate and tricyclohexylphosphine to produce 3-pinacolyl boronate ester (3). After removal of 17-acetyl group under basic condition, deacetyl boronate ester (4) was oxidized with mCPBA to give the final product 5 as colorless crystals.
Scheme 1. Synthetic Route for Fulvestrant-3 Boronic Acida.

a(a) Triflic anhydride, pyridine in DCM, −10 °C; (b) bis(pinacolato)-diboron, Pd(OAc)2, and tricyclohexylphosphine in CH3CN, 80 °C; (c) KOH in CH3OH and THF; (d) meta-chloroperoxybenzoic acid in DCM, 0 °C.
Compound 5 is a Potent Antiestrogen that Blocks ER Signaling in ER Positive Breast Cancer Cells
To determine if 5 acts as an antiestrogen, we used the T47D-kb-Luc stably transfected human breast cancer cell reporter gene assay23 where the antiestrogenic potency of 5 was determined by its ability to inhibit the activity of estradiol (E2). Data were normalized relative to the activity of E2 control. The T47D-kb-Luc cells are stably transfected with an artificial gene from the firefly that is only induced if estrogens bind and activate the ER to induce the gene product (luciferase). As shown in Figure 2, the inhibitory effect of 5 on E2-induced luciferase demonstrates a dose-dependent manner, completely blocking the estrogen response element (ERE) activity at 1 nM. Moreover, 5 was found comparable to fulvestrant in its potency as an antagonist of ER.
Figure 2.
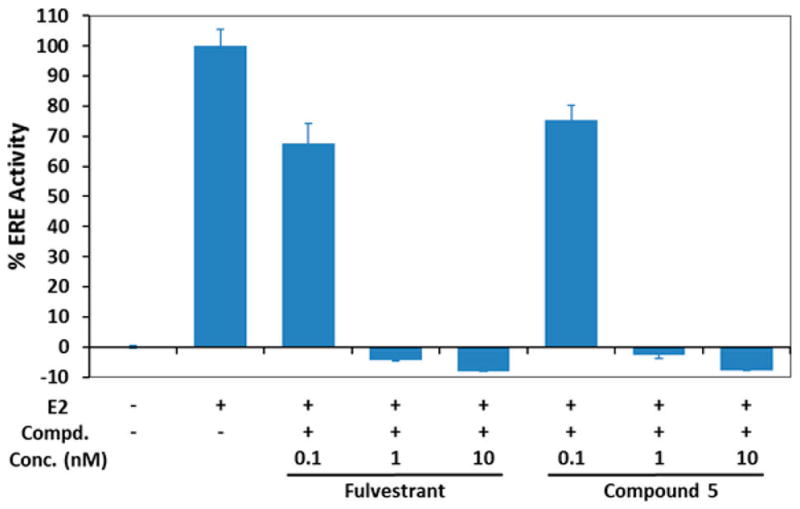
Antiestrogenic effects of 5 on T47D-kb-Luc cells in the presence of 0.01 nM of estradiol (E2). The concentrations of tested samples are 0.1, 1, and 10 nM, respectively. Data is represented as the mean ± SD (%) of three independent experiments.
To further elucidate the effects of 5 on estrogen signaling, we performed a real-time PCR study on five ER regulated genes, namely EGR3, PDZK1, PGR, TFF1, and WISP1. These genes are well-known to be responsive to the ER.24–31 As shown in Figure 3, the marked upregulation of these genes upon E2 treatment and the near-complete suppression of their expression upon treatment of 5 demonstrate its potent antagonistic activity, which is very similar to fulvestrant in comparison.
Figure 3.
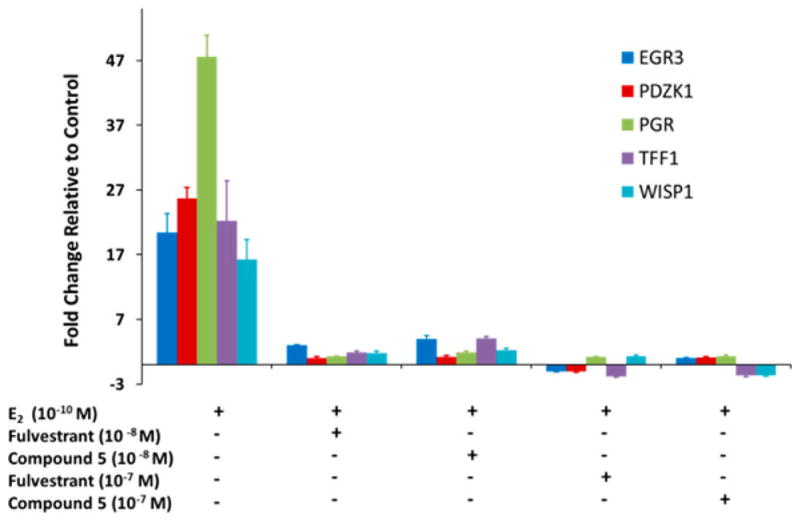
Effect of 5 on estrogen regulated genes, EGR3, PDZK1, PGR, TFF1, and WISP1.
Compound 5 is Effective in Blocking Tamoxifen-Resistant Breast Cancer Cell Growth
To test the potency of 5 against hormone-resistant breast cancer cells, we determined its IC50 values in MCF-7, T47D, and their tamoxifen-resistant variants, MCF-7/TamR and T47D/PKCα. MCF-7/TamR, has been maintained in our lab by prolonged treatment of MCF-7 with 4-hydroxytamoxifen (4-OHT),32 and T47D/PKCα cells were characterized previously.33,34 Cells were treated with vehicle or six different concentrations, ranging from 10−10 to 10−5 M of 4-OHT, fulvestrant, or 5 for 5 days before counting of survived cells. Results in Table 1 demonstrate that 5 shows the nM level potency as fulvestrant or 4-OHT against the proliferation of MCF-7 and T47D cells (dose-dependent cell inhibition curves can be found in Supporting Information). Like fulvestrant, 5 is highly active against the tamoxifen-resistant variants MCF-7/TamR and T47D/PKCα, in contrast to 4-OHT, which is much less effective in the resistant variants.
Table 1.
Effects of 5 on Proliferation of Breast Cancer Cells
| cell line | IC50 (μM) | ||
|---|---|---|---|
|
| |||
| 4-OHT | fulvestrant | 5 | |
| MCF-7 | 0.0033 | 0.0015 | 0.0032 |
| T47D | 0.024 | 0.0012 | 0.0061 |
| MCF-7/TamR | 22 | 0.044 | 0.069 |
| T47D/PKCα | 0.54 | 0.042 | 0.037 |
Compound 5 Binds to ERα with High Affinity (IC50 = 4.1 nM)
To determine the binding affinity of 5 to estrogen receptor alpha (ERα), a competitive displacement binding assay was performed in which 5 competes with a fluomone ligand and the percent displacement was quantitatively correlated to the fluorescence intensity from the displaced tracer. Figure 4 shows the competitive binding curves of 5, fulvestrant, and 17β-estradiol, with IC50 values measured at 4.1 nM for 5 and 3.0 nM for fulvestrant. As measured by the ability to displace endogenous ER ligand, 5 exhibits a near equal binding affinity toward ER as compared to fulvestrant.
Figure 4.
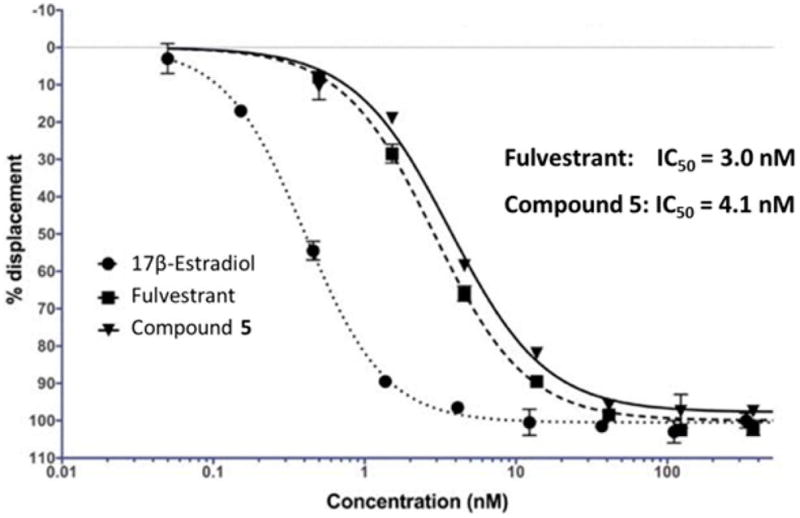
Competitive binding curves of estradiol, fulvestrant, and 5 to ERα.
Compound 5 Downregulates ERα with an IC50 of 12.7 nM
We next determined its ability to degrade ERα as compared to fulvestrant. T47D and its tamoxifen-resistant variant T47D/PKCα cells were treated with increasing concentrations of 5 or fulvestrant. In the dose range of 0.03–1μM both 5 and fulvestrant downregulated ER at greater than 50% level in the two cell lines tested (Figure 5). To measure the dose required to downregulate 50% of ER expression level, the treatment dose range was then lowered to 1–27 nM. In T47D cells, 5 downregulated ERα expression at an IC50 of 7.8 nM vs fulvestrant at 9.3 nM. In the tamoxifen-resistant T47D/PKCα cells, IC50 of 5 was 12.7 vs 8.5 nM of fulvestrant. These results confirm that 5 is as potent as fulvestrant in its action as a SERD (Supporting Information, page S8).
Figure 5.

Western blots showing ERα protein expression dramatically downregulated by fulvestrant (A) and 5 (B) in a dose-dependent manner.
Compound 5 is Orally Bioavailable in Mice
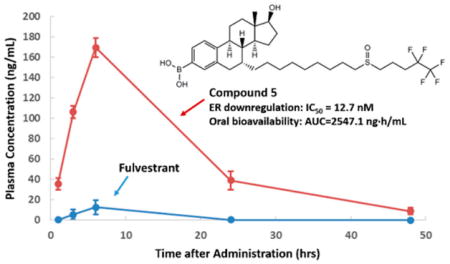
To verify if oral administration of 5 can achieve a therapeutically effective drug concentration that is systemically available, we conducted pharmacokinetic studies of 5 in mice. After a single oral dose of 8.3 mg/kg, blood samples were collected from mice and resulting plasma were analyzed for concentration of 5 at 1, 3, 6, 24, and 48 h after drug administration. Shown in Figure 6 are plasma total drug concentrations achieved after oral administration. 5 afforded over 160 ng/mL peak concentration, a level far exceeding that achieved by fulvestrant when given by sc injection to mice, providing definitive evidence that oral bioavailability of 5 is superior.
Figure 6.
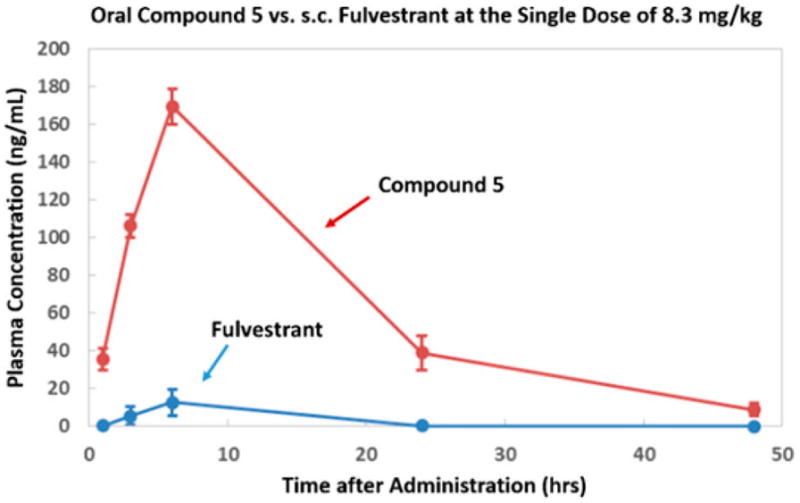
Pharmacokinetics of 5 in mice.
It is important to note that, upon the basis of our previous studies with the boronic derivatives of 4-hydroxytamoxifen and endoxifen,21,22 we initially anticipated a significant amount of 5 to be converted to fulvestrant both in vitro and in vivo. However, PK studies revealed that 5 remained as the predominant form in mice plasma, with fulvestrant accounting for only about 10% of the total active ingredients (Table 2). Its metabolic stability was later confirmed in vitro when the compound was dissolved first in ethanol then diluted in culture media for an extended period of time. These observations suggest that the bulk of the SERD activities of 5 is primarily attributable to itself, not its metabolite, fulvestrant.
Table 2.
Pharmacokinetic Parameters of Orally Administered 5 in Mice
| PK parameters | 5 | fulvestrant |
|---|---|---|
| t1/2 (h) | 23.5 | 14.0 |
| Cmax (ng/mL) | 169.8 | 15.2 |
| AUC (ng·h/mL) | 2547.1 | 158.4 |
DISCUSSION AND CONCLUSIONS
Compound 5 has been tested in key in vitro assays to confirm that it has all the major characteristic activities as a SERD. The ERE luciferase assay using a stably transfected ER+ breast cancer cell line, T47D-kb-Luc, demonstrates that 5 acts as a potent antiestrogen. In the presence of estradiol at 10−11 M, 5 inhibited the estrogen mediated gene expression activities, as measured by the relative luciferase unit, in a dose-dependent manner. This represents the first critical test that determined whether 5 can counteract estrogen in blocking ER regulations in breast cancer cells. Notably, the ability to do so by 5 closely matches that of fulvestrant (Figure 2), confirming that the modified compound retains the antiestrogenic activity of the steroidal SERD.
Considering that patients who experience disease progression upon tamoxifen or aromatase inhibitor (AI) therapy can still benefit from SERD therapy, it is important to ascertain whether 5 is efficacious in endocrine-resistant breast cancer cells. To this end, we prepared two tamoxifen-resistant variants of ER+ breast cancer cell lines. MCF-7/TamR was derived from the wide-type MCF-7 cells after long-term exposure to 4-hydroxytamoxifen over 12 months.24 The PKCα overexpressing cell line was derived from T47D and had been demonstrated to be resistant to antiestrogens.25,26 The fact that 5 strongly inhibited endocrine-resistant variants of breast cancer cell lines clearly demonstrates the lack of cross-resistance to this SERD.
By design, 5 was intended to possess comparable, if slightly reduced binding affinity to the estrogen receptor. In the ERE luciferase assays, analysis of ER regulated gene expression at the mRNA level and the antiproliferative assays in tamoxifen-resistant breast cancer cells, there were preliminary signs of 5 showing modestly attenuated potency, as reflected in its IC50 values as compared to fulvestrant. Thus, a reliable determination of the competitive binding activity of 5 is critical to confirming its mode of action. Indeed, it has largely retained the high ERα binding affinity with an IC50 value of 4.1 nM, indicating that replacing the 3-hydroxy group with the boronic acid, the main site of glucuronidation and sulfate conjugation, did not significantly diminish the binding capacity.
The hallmark of a SERD is its ability to downregulate the estrogen receptor through binding to the ER and marking it for ubiquitination and proteasomal degradation. This necessary property of SERDs, however, may not coincide with their proven antiestrogenic activities. The next assay on ER downregulation by Western blot confirms that 5 is not only effective in degrading the estrogen receptor in tamoxifen-sensitive breast cancer cells (T47D) but also in tamoxifen-resistant ones (T47D/PKCα) in a dose-dependent manner, with potencies comparable to fulvestrant.
The ultimate measure of the potential clinical utility of 5 as an oral SERD rests on its oral bioavailability. A pharmacokinetic study in mice provided critical data demonstrating a high plasma concentration of 5 after a single oral dose. Compared to fulvestrant, the drug exposure of 5 as measured by total plasma level is over 10 times higher than fulvestrant administered by subcutaneous injection, suggesting that 5 may offer high oral bioavailability in the clinical setting. This dramatic enhancement in oral bioavailability is clearly furnished by the boronic acid moiety that substituted the 3-hydroxyl group of fulvestrant. We have shown in previous studies21,22 that replacement of a phenolic hydroxyl group with a boronic acid or its precursor pinacolate significantly increased oral bioavailability by avoiding first pass metabolism. While the exact mechanism by which boronic acid prevents rapid clearance is not completely understood, previous studies suggest that boronic acids have a strong tendency to form reversible complexes with 1,2- and 1,3-diol groups common in sugar molecules and glycoproteins.35,36 Such reversible, covalent complexes between boronic acid and a diol group not only facilitate the enrichment in plasma due to the abundance of molecules containing the diol groups but also make the boronic acid moiety inaccessible to glucuronidation, thereby markedly reducing first pass metabolic clearance. Indeed, this unique property of boronic acid has recently been exploited for an enhanced delivery of insulin for treatment of diabetes.37
Taken together, our drug design strategy and the results from specific biological and pharmacokinetic studies allow us to conclude that the structural modification on C-3 position of fulvestrant by introducing a boronic acid moiety, designed to block first pass metabolism, has largely retained the pharmacological properties of fulvestrant, as it is shown to behave as a potent antiestrogen that inhibits ER-dependent breast cancer cell growth through tight binding to ER. Moreover, 5 also targets tamoxifen-resistant breast cancer cells by a combination action of distinct ER binding and ER downregulation as a SERD. More importantly, with all the distinct features of a SERD, 5 has proven to afford high oral bioavailability as an effective steroidal oral SERD for further evaluation in the clinic.
EXPERIMENTAL SECTION
Chemicals and Reagents
All reagents, solvents, and analytical standards were purchased from Sigma-Aldrich (St. Louis, MO), Fisher Scientific (Fairfield, NJ), AK Scientific (Union City, CA), and CombiPhos Catalysts (Princeton, NJ) and were used as received. 1H NMR spectra were recorded on a Bruker Fourier 300 MHz FT-NMR spectrometer. 13C NMR spectra were obtained on an Agilent 400-MR NMR spectrometer, and the data was processed using MestReNova NMR software (School of Chemistry, University of Bristol, Bristol, UK). Chemical shifts are reported as parts per million (ppm) relative to TMS. Specific rotation was determined with an AUTOPOL III polarimeter (Rudolph Research Analytical, NY). HRMS spectra data were collected on a Thermo LTQ Orbitrap-XL mass spectrometer in positive ion mode. The tested compound was confirmed to be >95% pure by HPLC.
(7R, 8R, 9S, 13S, 14S, 17S )-13-Methyl-7-(9-((4,4,5,5,5-pentafluoropentyl)thio)nonyl)-3-(((trifluoromethyl)sulfonyl)oxy)-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]-phenanthren-17-yl Acetate (2)
Starting from 1, the intermediate 2 was prepared with triflic anhydride and pyridine in DCM at −10 °C, which has been reported recently.38
(7R, 8R, 9S, 13S, 14S, 17S)-13-Methyl-7-(9-((4,4,5,5,5-pentafluoropentyl)thio)nonyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxa-borolan-2-yl)-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta-[a]phenanthren-17-yl acetate (3)
This procedure has also been reported in our previous work.38
(7R, 8R, 9S,13S, 14S, 17S)-13-Methyl-7-(9-((4,4,5,5,5-pentafluoropentyl)thio)nonyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxa-borolan-2-yl)-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta-[a]phenanthren-17-ol (4)
To a solution of 0.60 g (0.81 mmol) of boronate ester (3) in 4 mL of CH3OH/THF (1:1, v/v), 0.15 g (2.68 mmol) of KOH in CH3OH (2 mL) was added slowly at 0 °C. The resultant mixture was stirred at room temperature for 4 h. After neutralization with acetic acid to pH 7.0, the solvent was removed under vacuum. The residue was purified by a flash column chromatography (pet. ether/ethyl acetate) to give 0.44 g of 4 (yield, 78%) as colorless crystals. 1H NMR (DMSO-d6, 300 MHz): 7.39 (d, J = 7.8 Hz, 1H), 7.35 (s, 1H), 7.28 (d, J = 7.8 Hz, 1H), 4.49 (br, 1H), 3.54 (t, J = 8.4 Hz, 1H), 2.85–2.70 (m, 2H), 2.59–2.26 (m, 4H), 2.29 (m, 4H), 1.95–1.05 (m, 38H), 0.84 (m, 2H), 0.66 (s, 3H). 13C NMR (DMSO-d6, 100 MHz): 143.4, 136.7, 134.9, 132.0, 125.8, 83.8, 80.5, 46.5, 43.3, 41.8, 39.0, 37.2, 34.3, 33.1, 31.2, 30.4, 30.3, 29.7, 29.5, 29.4, 29.3, 28.9, 28.8 (t, J = 21 Hz), 28.5, 27.9, 27.2, 25.5, 25.1, 25.0, 22.7, 20.7, 11.7. ESI/MS (m/z): 701.4 (M + H)+.
((7R,8R,9S,13S,14S,17S)-17-Hydroxy-13-methyl-7-(9-((4,4,5,5,5-pentafluoropentyl)sulfinyl)nonyl)-7,8,9,11,12,13,14,15,16,17-deca-hydro-6H-cyclopenta[a]phenanthren-3-yl)boronic Acid (5)
To a solution of 0.40 g (0.57 mmol) of 4 in 10 mL of DCM (10 mL), 0.13 g (0.75 mmol) of mCPBA was added at 0 °C. The resultant mixture was stirred at 0 °C until the starting material completely disappeared on TLC. The reaction solution was diluted with 50 mL of DCM, washed with saturated Na2CO3, and dried with MgSO4. After filtration and concentration under vacuum, the residue was purified by a flash column chromatography (DCM/CH3OH) to give final product 5 (yield, 70%) as colorless crystals. It decomposed at higher than 230 °C. [α]D20 = 73 (c = 0.43, CH3OH). 1H NMR (DMSO-d6, 300 MHz):7.87 (s, 2H), 7.50 (d, J = 7.8 Hz, 1H), 7.45 (s, 1H), 7.23 (d, J = 7.8 Hz, 1H), 4.50 (br, 1H), 3.55 (t, J = 8.4, 1H), 2.89–2.63 (m, 6H), 2.45–2.24 (m, 4H), 1.95–1.10 (m, 26H), 0.87 (m, 2H), 0.67 (s, 3H). 13C NMR (DMSO-d6, 100 MHz): 141.7, 136.4, 134.1, 131.8, 125.2, 80.5, 51.4, 49.7, 46.5, 43.4, 41.9, 39.0, 37.3, 34.5, 33.2, 30.3, 29.8, 29.4, 29.2, 29.1, 29.0 (t, J = 21 Hz), 28.5, 28.0, 27.2, 25.5, 22.7, 22.5, 14.5, 11.7. HRMS (ESI+): calcd for C32H49BF5O4S (M + H)+ 635.3365, found 635.3364.
Cell Culture
The MCF-7 and T47D cell lines were originally purchased from ATCC (Manassas, VA) and were routinely maintained in phenol red-free DMEM medium supplemented with 5% FBS, 4 mM glutamine, 1 mM sodium pyruvate, 100 IU/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin. Cultures were maintained in 5% carbon dioxide at a temperature of 37 °C. For growth assays in the presence of E2 and either fulvestrant, 4-OHT, or 5, MCF-7, MCF-7/TamR, T47D, and T47D/PKCα cells were plated in six-well plates at a density of 50000/well in 5% FBS DMEM medium. The cells were then treated with fulvestrant, 4-OHT, or 5 separately at six different doses ranging from 10−10 to 10−5 M for 5 days, while equal volumes of DMSO were used as vehicle controls. Viable cell numbers were counted with a Z series Coulter Counter instrument (Beckman-Coulter) following manufacturer’s instructions. The ratio of drug treated viable cell numbers to vehicle treated viable cell numbers was defined as survival ratio where the control has the survival ratio of 100%. IC50 values were obtained from dose–response curves for all treatments.
Luciferase Assay
T47d-kb-Luc stably transfected human breast cancer cell reporter gene assay was used to determine the antiestrogenic activities of 5. The T47d-kb-Luc cells are stably transfected with an artificial gene from the firefly that is only induced in the cells if estrogens bind and activate the ER to induce the gene product (Luciferase) that is then measured with a quantitative enzyme assay that produces light. Antagonist activities were measured by the samples ability to inhibit the activity of estradiol, the natural estrogen. Data were then normalized relative to the activity of the estradiol control, and determinations were performed for three concentrations of the samples in quadruplicate in at least three separate experiments.
ER Binding Assay
Estrogen receptor binding determinations are performed using the LanthaScreen TR-FRET assay from Life Technologies. This method is a version of the Life Technologies Polar Screen ER FP method that uses recombinant ER and competition with a fluormone ligand. Binding assays will include 6–10 concentrations of test compounds that span the IC50 and are done in quadruplicate.
Western Blot of ER Downregulation
T47D and T47D/PKCα were plated at a density of 200000 cells/60 mm dish. Media containing the same drug concentrations as the growth curve assay were added on the day following plating (day 0) and allowed to incubate for 5 days for Western blot. Media with the tested compound was changed every other day. Cells were lysed, snapped frozen in liquid nitrogen, and stored at −80 °C until assay for ERα. Media were removed and dishes were washed with 1× DPBS. Lysates were made by adding 150 μL of complete lysis solution and scraping cells into a 1.5 mL micro-centrifuge tube. Lysates were placed on a rotisserie at 4 °C for 30 min and then spun at 4 °C at 12000 rcf for 10 min. Supernatants were assayed for protein content, snap-frozen, and stored at −80 °C if not run immediately. Then 50 μg of protein was subjected to Western blot protocol. Membranes were blocked and then incubated with 1:200 dilution of ERα antibody at 4 °C overnight followed by 1:10000 dilution of secondary antibody for 1 h at room temperature. They were then imaged on a LICOR infrared scanner.
Pharmacokinetic Study (Sampling and Analysis)
Female C57BL/6 mice were used for the pharmacokinetic study on 5. Mice (n = 5) were given oral gavage containing PBS and ethanol-dissolved 5 or fulvestrant by sc injection at a single dose of 8.3 mg/kg. After drug administration, blood samples were collected from the orbital sinus of the mice at various time points with each group of mice subjected to only one sampling. Murine blood was collected with a capillary into 1.5 mL microcentrifuge tubes containing 0.1 mL of 10% EDTA anticoagulant. Plasma was then separated from cell pellets by centrifugation in a refrigerated centrifuge at 4 °C and transferred to a separate tube. Plasma samples were frozen at −80 °C until analysis.
HPLC-MS/MS Analysis of Plasma Samples
Plasma samples were extracted with chloroform/methanol (2:1) using traditional Folch method for lipid extraction. Methanol (1 mL) and chloroform (2 mL) were added to each plasma sample followed by addition of 5 ng trans-Tamoxifen-13C2, 5N to each sample as the internal standard. The mixtures were stored at −20 °C overnight. Next the samples were sonicated for 5 min and centrifuged with a Thermo Scientific Heraeus Megafuge 16 centrifuge. The top layer was transferred to another test tube. The bottom layer was washed with 1 mL of chloroform/methanol (2:1) and centrifuged, and the solvent was transferred and combined with previous washings. HPLC grade water (0.8 mL) was added to the extracts. After vortexing, the mixture was centrifuged. The bottom layer was dried out with nitrogen and resuspended in 100 μL of HPLC grade acetonitrile. An aliquot of 10 μL sample was injected onto a Hypersil Gold column (50 mm × 2.1 mm; particle size 1.9 μm, Thermo Scientific) on a Dionex Ultimate 3000 UPLC system equipped with a TSQ Vantage triple quadrupole mass spectrometer for analysis. A binary mobile phase (A, water with 0.05% formic acid; B, acetonitrile with 0.05% formic acid) was used to achieve the gradient of initial 30% B for 1 min and then to 80% B at 8 min, to 100% B at 9 min, and returned to 30% B for 4 min. The flow rate was controlled at 0.6 mL/min. The settings of HESI source were as follows: spray voltage (3200 V), vaporizer temperature (365 °C), sheath gas pressure (45 psi), auxiliary gas pressure (10 psi), capillary temperature (330 °C). Nitrogen was used as the sheath gas and axillary gas. Argon was used as the collision gas.
RT-PCR Analysis
MCF-7 cells were seeded onto 6-well plates in DMEM supplemented with 5% FBS. On the following day, media were replaced with phenol red-free DMEM supplemented with 5% CS-FBS for 48 h. Cells were then treated with vehicle, E2, 5 + E2, or fulvestrant + E2 for 48 h. Total RNA was extracted from the cells and 4 μg of RNA was reverse-transcribed into cDNA in the presence of gene-specific oligonucleotide primers, and quantitative RT-PCR was performed using the manufacturer’s protocols for the Human Breast Cancer and Estrogen Receptor Signaling RT2 Profiler PCR Array (Qiagen). Relative gene expressions were calculated by using the 2−ΔΔCt method, in which Ct indicates the fractional cycle number where the fluorescent signal reaches detection threshold. The normalized ΔCt value of each sample, calculated using a total of five endogenous control genes (18S rRNA, HPRT1, RPL13A, GAPDH, and ACTB), was used to determine fold change values for genes in treated relative to control samples.
Ethical Considerations and Statistical Analysis
All animal experiments were approved by Xavier’s Institutional Animal Care and Use Committee. The facilities and laboratory animals program of Xavier University are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. Statistical analyses were performed using Microsoft excel software. Pharmacokinetic data analyses were performed using the PK software.39
Supplementary Material
Acknowledgments
This study was supported by an NIH grant 2G12MD007595 from NIMHD, and by Louisiana Cancer Research Consortium.
ABBREVIATIONS USED
- ER+
estrogen receptor positive
- E2
estradiol
- ERE
estrogen response element
- SERD
selective estrogen receptor down-regulator
- DCM
dichloromethane
- mCPBA
meta-chloroper-oxybenzoic acid
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b00753.
1H, 13C NMR spectra and high resolution mass spectrum of 5 (PDF)
Molecular formula strings (CSV)
References
- 1.Vergote I, Robertson JF. Fulvestrant is an effective and well-tolerated endocrine therapy for postmenopausal women with advanced breast cancer: results from clinical trials. Br J Cancer. 2004;90(Suppl 1):S11–S14. doi: 10.1038/sj.bjc.6601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robertson JF, Lindemann J, Garnett S, Anderson E, Nicholson RI, Kuter I, Gee JM. A good drug made better: the fulvestrant dose-response story. Clin Breast Cancer. 2014;14:381–389. doi: 10.1016/j.clbc.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Pritchard KI, Rolski J, Papai Z, Mauriac L, Cardoso F, Chang J, Panasci L, Ianuli C, Kahan Z, Fukase K, Lindemann JP, Macpherson MP, Neven P. Results of a phase II study comparing three dosing regimens of fulvestrant in postmenopausal women with advanced breast cancer (FINDER2) Breast Cancer Res Treat. 2010;123:453–461. doi: 10.1007/s10549-010-1022-9. [DOI] [PubMed] [Google Scholar]
- 4.Robertson JF. Fulvestrant (Faslodex) – how to make a good drug better. Oncologist. 2007;12:774–784. doi: 10.1634/theoncologist.12-7-774. [DOI] [PubMed] [Google Scholar]
- 5.Ohno S, Rai Y, Iwata H, Yamamoto N, Yoshida M, Iwase H, Masuda N, Nakamura S, Taniguchi H, Kamigaki S, Noguchi S. Three dose regimens of fulvestrant in postmenopausal Japanese women with advanced breast cancer: results from a double-blind, phase II comparative study (FINDER1) Ann Oncol. 2010;21:2342–2347. doi: 10.1093/annonc/mdq249. [DOI] [PubMed] [Google Scholar]
- 6.Yoneya T, Tsunenari T, Taniguchi K, Kanbe Y, Morikawa K, Yamada-Okabe H, Lee YH, Lee MH, Kwon LS. Effects of CH4893237: a new orally active estrogen receptor downregulator, on breast cancer xenograft models with low serum estrogen levels. Oncol Rep. 2009;21:747–755. [PubMed] [Google Scholar]
- 7.Kanbe Y, Kim MH, Nishimoto M, Ohtake Y, Yoneya T, Ohizumi I, Tsunenari T, Taniguchi K, Kaiho S, Nabuchi Y, Araya H, Kawata S, Morikawa K, Jo JC, Kwon HA, Lim HS, Kim HY. Newly discovered orally active pure antiestrogens. Bioorg Med Chem Lett. 2006;16:4959–4964. doi: 10.1016/j.bmcl.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 8.Jiang Q, Zheng S, Wang G. Development of new estrogen receptor-targeting therapeutic agents for tamoxifen-resistant breast cancer. Future Med Chem. 2013;5:1023–1035. doi: 10.4155/fmc.13.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoneya T, Taniguchi K, Tsunenari T, Saito H, Kanbe Y, Morikawa K, Yamada-Okabe H. Identification of a novel, orally bioavailable estrogen receptor downregulator. Anti-Cancer Drugs. 2005;16:751–756. doi: 10.1097/01.cad.0000171515.27439.de. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann J, Bohlmann R, Heinrich N, Hofmeister H, Kroll J, Künzer H, Lichtner RB, Nishino Y, Parczyk K, Sauer G, Gieschen H, Ulbrich HF, Schneider MR. Characterization of new estrogen receptor destabilizing compounds: effects on estrogen-sensitive and tamoxifen-resistant breast cancer. J Natl Cancer Inst. 2004;96:210–218. doi: 10.1093/jnci/djh022. [DOI] [PubMed] [Google Scholar]
- 11.Chouinard S, Tessier M, Vernouillet G, Gauthier S, Labrie F, Barbier O, Belanger A. Inactivation of the pure antiestrogen fulvestrant and other synthetic estrogen molecules by UDP-glucuronosyltransferase 1A enzymes expressed in breast tissue. Mol Pharmacol. 2006;69:908–920. doi: 10.1124/mol.105.015891. [DOI] [PubMed] [Google Scholar]
- 12.Wu B, Kulkarni K, Basu S, Zhang S, Hu M. First-pass metabolism via UDP-glucuronosyltransferase: a barrier to oral bioavailability of phenolics. L Pharm Sci. 2011;100:3655–3681. doi: 10.1002/jps.22568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ClinicalTrialsgov. U.S. National Institutes of Health; Bethesda, MD: Apr 28, 2016. A Study of ARN-810 (GDC-0810) in postmenopausal women with locally advanced or metastatic estrogen receptor positive breast cancer. NCT01823835. http:/clinicaltrials.gov. [Google Scholar]
- 14.ClinicalTrialsgov. U.S. National Institutes of Health; Bethesda, MD: Jul 12, 2016. AZD9496 first time in patients ascending dose study. NCT02248090. http:/clinicaltrials.gov. [Google Scholar]
- 15.Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, Douglas K, Sensintaffar J, Lu N, Lee KJ, Aparicio A, Kaufman J, Qian J, Shao G, Prudente R, Moon MJ, Joseph JD, Darimont B, Brigham D, Grillot K, Heyman R, Rix PJ, Hager JH, Smith ND. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem. 2015;58:4888–4904. doi: 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]
- 16.De Savi C, Bradbury RH, Rabow AA, Norman RA, de Almeida C, Andrews DM, Ballard P, Buttar D, Callis RJ, Currie GS, Curwen JO, Davies CD, Donald CS, Feron LJ, Gingell H, Glossop SC, Hayter BR, Hussain S, Karoutchi G, Lamont SG, MacFaul P, Moss TA, Pearson SE, Tonge M, Walker GE, Weir HM, Wilson Z. Optimization of a novel binding motif to (E)-3-(3,5-difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetra hydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)-acrylic Acid (AZD9496), a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. L Med Chem. 2015;58:8128–8140. doi: 10.1021/acs.jmedchem.5b00984. [DOI] [PubMed] [Google Scholar]
- 17.Edavana VK, Penney RB, Yao-Borengasser A, Williams S, Rogers L, Dhakal IB, Kadlubar S. Fulvestrant up regulates UGT1A4 and MRPs through ERalpha and c-Myb pathways: a possible primary drug disposition mechanism. SpringerPlus. 2013;2:620. doi: 10.1186/2193-1801-2-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hui Y, Luo L, Zhang L, Kurogi K, Zhou C, Sakakibara Y, Suiko M, Liu MC. Sulfation of afimoxifene, endoxifen, raloxifene, and fulvestrant by the human cytosolic sulfotransferases (SULTs): A systematic analysis. L Pharmacol Sci. 2015;128:144–149. doi: 10.1016/j.jphs.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Edavana VK, Yu X, Dhakal IB, Williams S, Ning B, Cook IT, Caldwell D, Falany CN, Kadlubar S. Sulfation of fulvestrant by human liver cytosols and recombinant SULT1A1 and SULT1E1. Pharmacogenomics Pers Med. 2011;4:137–145. doi: 10.2147/PGPM.S25418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Q, Zhong Q, Zhang Q, Zheng S, Wang G. Boron-based 4-hydroxytamoxifen bioisosteres for treatment of de novo tamoxifen resistant breast cancer. ACS Med Chem Lett. 2012;3:392–396. doi: 10.1021/ml3000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, Zhong Q, Zhang Q, Zheng S, Miele L, Wang G. Boronic prodrug of endoxifen as an effective hormone therapy for breast cancer. Breast Cancer Res Treat. 2015;152:283–291. doi: 10.1007/s10549-015-3461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong Q, Zhang C, Zhang Q, Miele L, Zheng S, Wang G. Boronic prodrug of 4-hydroxytamoxifen is more efficacious than tamoxifen with enhanced bioavailability independent of CYP2D6 status. BMC Cancer. 2015;15:625. doi: 10.1186/s12885-015-1621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson VS, Bobseine K, Gray LE., Jr Development and characterization of a cell line that stably expresses an estrogen-responsive luciferase reporter for the detection of estrogen receptor agonist and antagonist. Toxicol Sci. 2004;81:69–77. doi: 10.1093/toxsci/kfh180. [DOI] [PubMed] [Google Scholar]
- 24.Inoue A, Omoto Y, Yamaguchi Y, Kiyama R, Hayashi SI. Transcription factor EGR3 is involved in the estrogen-signaling pathway in breast cancer cells. J Mol Endocrinol. 2004;32:649–661. doi: 10.1677/jme.0.0320649. [DOI] [PubMed] [Google Scholar]
- 25.Kim H, Abd Elmageed ZY, Ju J, Naura AS, Abdel-Mageed AB, Varughese S, Paul D, Alahari S, Catling A, Kim JG, Boulares AH. PDZK1 is a novel factor in breast cancer that is indirectly regulated by estrogen through IGF-1R and promotes estrogen-mediated growth. Mol Med. 2013;19:253–262. doi: 10.2119/molmed.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bundred N. Preclinical and clinical experience with fulvestrant (Faslodex) in postmenopausal women with hormone receptor-positive advanced breast cancer. Cancer Invest. 2005;23:173–181. doi: 10.1081/cnv-50480. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi S, Yamaguchi Y. Estrogen signaling pathway and hormonal therapy. Breast Cancer. 2008;15:256–261. doi: 10.1007/s12282-008-0070-z. [DOI] [PubMed] [Google Scholar]
- 28.Nunez AM, Jakowlev S, Briand JP, Gaire M, Krust A, Rio MC, Chambon P. Characterization of the estrogen-induced pS2 protein secreted by the human breast cancer cell line MCF-7. Endocrinology. 1987;121:1759–1765. doi: 10.1210/endo-121-5-1759. [DOI] [PubMed] [Google Scholar]
- 29.Xie D, Nakachi K, Wang H, Elashoff R, Koeffler HP. Elevated levels of connective tissue growth factor, WISP-1, and CYR61 in primary breast cancers associated with more advanced features. Cancer Res. 2001;61:8917–8923. [PubMed] [Google Scholar]
- 30.Thomas C, Gustafsson JÅ. Progesterone receptor-estrogen receptor crosstalk: a novel insight. Trends Endocrinol Metab. 2015;26:453–454. doi: 10.1016/j.tem.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Inadera H. Estrogen-induced genes, WISP-2 and pS2, respond divergently to protein kinase pathway. Biochem Biophys Res Commun. 2003;309:272–278. doi: 10.1016/j.bbrc.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 32.Zhou C, Zhong Q, Rhodes LV, Townley I, Bratton MR, Zhang Q, Martin EC, Elliott S, Collins-Burow BM, Burow ME, Wang G. Proteomic analysis of acquired tamoxifen resistance in MCF-7 cells reveals expression signatures associated with enhanced migration. Breast Cancer Res. 2012;14:R45. doi: 10.1186/bcr3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yun J, Pannuti A, Espinoza I, Zhu H, Hicks C, Zhu X, Caskey M, Rizzo P, D’Souza G, Backus K, Denning MF, Coon J, Sun M, Bresnick EH, Osipo C, Wu J, Strack PR, Tonetti DA, Miele L. Crosstalk between PKCalpha and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis. 2013;2:e60. doi: 10.1038/oncsis.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tonetti DA, Chisamore MJ, Grdina W, Schurz H, Jordan VC. Stable transfection of protein kinase C alpha cDNA in hormone-dependent breast cancer cell lines. Br J Cancer. 2000;83:782–791. doi: 10.1054/bjoc.2000.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis GA, Palte MJ, Raines RT. Boronate-mediated biologic delivery. J Am Chem Soc. 2012;134:3631–3634. doi: 10.1021/ja210719s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang W, Gao X, Wang B. Boronic acid compounds as potential pharmaceutical agents. Med Res Rev. 2003;23:346–368. doi: 10.1002/med.10043. [DOI] [PubMed] [Google Scholar]
- 37.Chou DH, Webber MJ, Tang BC, Lin AB, Thapa LS, Deng D, Truong JV, Cortinas AB, Langer R, Anderson DG. Glucose-responsive insulin activity by covalent modification with aliphatic phenylboronic acid conjugates. Proc Natl Acad Sci U S A. 2015;112:2401–2406. doi: 10.1073/pnas.1424684112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang G, Zhong Q, Zheng S. Boron-based prodrug strategy for increased bioavailability and lower-dosage requirements for drug molecules containing at least one phenol (or aromatic hydroxyl) group. 2016/004166. Patent WO. 2016 Jan 7;
- 39.Zhang Y, Huo M, Zhou J, Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99:306–314. doi: 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.