

Abstract
Control over the integrative self‐sorting of metallo‐supramolecular assemblies opens up possibilities for introducing increased complexity and function into a single self‐assembled architecture. Herein, the relationship between the geometry of three ligand components and morphology of three self‐sorted heteroleptic [Pd2 L 2 L′2]4+ cages is examined. Pd‐mediated assembly of two bis‐monodentate pyridyl ligands with native bite angles of 75° and 120° affords a cis‐[Pd2 L 2 L′2]4+ cage while the same reaction with two ligands with bite angles of 75° and 60° gives an unprecedented, self‐penetrating structural motif; a trans‐[Pd2(anti‐L)2 L′2]4+ heteroleptic cage with a “doubly bridged figure eight” topology. Each heteroleptic assembly can be formed by cage‐to‐cage conversion of the homoleptic precursors and morphological control of [Pd2 L 2 L′2] cages is achieved by selective ligand displacement transformations in a system of three ligands and at least six possible cage products.
Keywords: cage compounds, morphological control, self-sorting, supramolecular chemistry, topology
Nature makes excellent use of integrative self‐sorting to regulate the structural morphology of multi‐component protein complexes through recognition pathways that ensure each subunit is combined with precise positional control. Inspired by these elegant processes and the potential to advance molecular complexity, chemists seek to control multi‐component self‐assembly in artificial systems. So far, most of the reported self‐assembled structures have rather symmetric structures composed of multiple copies of identical components. Recently, efforts have been directed toward increasing structural and functional complexity through the rational design of multi‐component systems.1
In metallo‐supramolecular chemistry, control over the integrative self‐sorting of discrete heteroleptic assemblies has been achieved by encoding specific information within their components.2 For example, steric3 and topological4 constraints have been used to control the assembly of multi‐component structures, such as tweezers,5 grids,6 prisms,7 nanotubes,8 and more.2 For nano‐sized coordination cages, however, morphological control of multi‐component structures is still a growing area. For example, Zheng, Stang, and others reported “charge separation” as an approach to achieve a range of 3D heteroleptic assemblies from “capped” PdII or PtII cations and a combination of carboxylate and pyridine donors.2d, 9 Mukherjee and co‐workers utilized “capped” PdII cations in combination with imidazole and pyridine donor ligands to access a range of heteroleptic cage structures with accessible cavities.2d, 10
An even higher level of self‐sorting complexity is encountered in systems composed of “naked” PdII or PtII cations and multiple bis‐monodentate ligands,11 due to an increased number of accessible binding sites and greater number of statistically possible products. In this respect, [Pd2 L 4] coordination cages are an archetypal platform to study self‐sorting phenomena because of their regular arrangement of banana‐shaped ligands around a central cavity.12 Through modification of the ligand backbone, a notable line‐up of functional host compounds has already been prepared based on homoleptic versions of the [Pd2 L 4] framework.13 Consequently, interest in controlling the morphology of heteroleptic [Pd2 L 2 L′2] cages stems from the prospect of further regulating the rational combination and relative position of chosen pairs of engrafted functionalities. Corresponding strategies to access mixed‐ligand [Pd2 L 2 L′2] assemblies are currently in early stages of development in a number of research groups.14, 15 We have recently shown that combining shape complementary ligands with isoquinoline/pyridine donors is a viable route to a heteroleptic [Pd2 L 2 L′2]4+ cage (1) under thermodynamic control.16
Herein, we report on the geometrically constrained self‐sorting and interconversion of [Pd2 L 2 L′2] heteroleptic cage structures (Figure 1). In doing so, we introduce two new well‐defined heteroleptic cages; a cis‐[Pd2 LC 2 LP 2]4+ (2) and a trans‐[Pd2(anti‐LA)2 LC 2]4+ (3) assembly (Figure 1 a). Structure 3 is an unprecedented structural motif in which one of the ligands is forced to adopt an unusual anti‐conformation in order to obey a [Pd2 L 4] stoichiometry. Self‐sorting studies reveal that each heteroleptic structure can be readily accessed through clean cage‐to‐cage transformations of their homoleptic cage precursors. Moreover, morphological control of [Pd2 L 2 L′2] cages is demonstrated through ligand substitution reactions where selective displacement leads to a rearrangement to the more geometrically favored structure.
Figure 1.
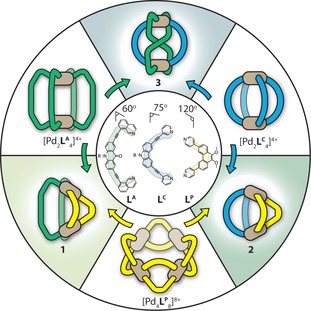
A representation of the homoleptic and heteroleptic structures; cis‐[Pd2 LA 2 LP 2]4+ (1), cis‐[Pd2 LC 2 LP 2]4+ (2) and trans‐[Pd2(anti‐LA)2 LC 2]4+ (3) are achieved by pairing geometrically distinct ligands LA, LC, and LP or by cage‐to‐cage integrative transformations.
The ligand components, based on either an acridone (LA), phenanthrene (LP), or carbazole backbone (LC) were synthesized as previously described.16, 17 Their respective Pd‐mediated homoleptic assemblies were prepared by heating a mixture of the ligand with 0.5 equivalents of [Pd(CH3CN)4](BF4)2 in CD3CN. 1H NMR spectroscopy and ESI‐MS analysis revealed cage products with the formula [Pd2 L 4]4+ for LA and LC,17 whilst for LP, a 1:2 mixture of products with the formula [Pd3 LP 6]6+ and [Pd4 LP 8]8+ was identified (see the Supporting Information).
So far, the assembly of template‐free [Pd2 L 2 L′2] cages has been achieved either by functionalization of the donor periphery15 or by a combination of different donor scaffolds.16 Therefore, we set out to examine whether a [Pd2 L 2 L′2]4+ assembly can be accessed solely from ligands bearing simple pyridine donors that are attached to different backbones with mutually compatible geometries. We chose ligands LC and LP as appropriate candidates, since preliminary modelling suggested that their respective backbone angles are well‐matched with a cis‐[Pd2 L 2 L′2]4+ architecture. Accordingly, we heated a 1:1:1 mixture of LC, LP, and [Pd(CH3CN)4](BF4)2 in CD3CN at 70 °C for 24 h, which resulted in the quantitative formation of a single product, 2, with a distinct 1H NMR spectrum relative to its ligand and homoleptic cage counterparts (Figure S10, S24 in the Supporting Information). The total amount of 12 aromatic signals indicated that each ligand maintains its twofold symmetry in the cage assembly.
Further evidence for a heteroleptic structure was provided by ESI‐MS, which revealed prominent signals at m/z 633.9 and 1039.3, consistent with the formula [Pd2 LC 2 LP 2+nBF4](4−n)+ (n=0, 1; Figure S12).
Compared to 1,16 the relational angle between the PdII planes in 2 is expected to be less acute, owing to the overall tilt created by the pyridine donors of LC in comparison with the 8‐isoquinoline donors of LA (Figure S38). Whilst the combination of donors LA and LP provided multiple NOE contacts to confidently assign a cis‐configuration of ligands for 1, the single NOE contact for 2 (Figure S14), observed between the pyridyl moieties of LC and LP, did not provide enough evidence for the expected cis‐arrangement. The final confirmation for the structure of 2 was afforded by X‐ray analysis of single crystals obtained by slow vapor diffusion of a 10:1 mixture of benzene/acetone into a CD3CN solution of 2. The heteroleptic structure crystallizes in the triclinic space group P with two, almost identical molecules of the cage, in the asymmetric unit (Figure S36). Indeed, each PdII center coordinates two LC and two LP ligands in a cis‐configuration, with LC being slightly bowed (Figure 3 a,b) to accommodate a Pd⋅⋅⋅Pd distance of 11.5 Å, which is considerably shorter than in [Pd2 LC 4]4+ (ca. 14.3 Å)17 but more in favor with the geometric preference of the free ligand (Figure S39). Despite the low crystallographic symmetry, the cis‐arrangement of ligands in a single cage molecule is consistent with a C 2h symmetry.
Figure 3.
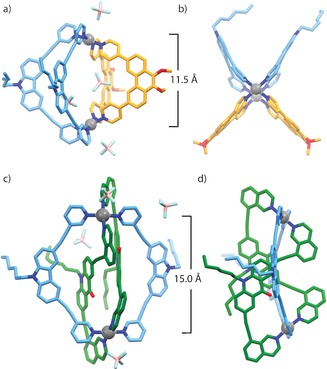
X‐ray crystal structures of cis‐[Pd2 LC 2 LP 2]4+ (2) and trans‐[Pd2(anti‐LA)2 LC 2]4+ (3): a) the structure of 2 showing the occupation of the cavity by two BF4 − counterions; b) top view of 2; c) perspective view of 3 showing the ligand‐occupied cavity; d) side view of 3 showing the trans/anti arrangement of LA. Pd⋅⋅⋅Pd distances are shown and hydrogen atoms are removed for clarity.25
Having realized the possibility of combining different ligand backbones in “bent” cis‐[Pd2 L 2 L′2] architectures with LP as the common component, we moved forth to examine the self‐sorting of LA with LC. Whilst ligand pairs LA/LP and LC/LP display shape complementary bite angles, LA and LC do not; both ligands deviate from a perfect rectangular concave shape by −30° and −15°, respectively (Figure 1). Based on other examples of self‐sorting where either the size or geometry of the ligands differ so significantly that no co‐assembly within the same structure could be observed,18 we expected that combining ligands LA and LC with PdII would result in a narcissistic self‐sorted mixture of their respective homoleptic cages.
Fascinatingly, the sample obtained from heating a 1:1:1 mixture of LA, LC, and [Pd(CH3CN)4](BF4)2 at 70 °C for 24 h gave a highly complex 1H NMR spectrum, consisting of 32 aromatic signals belonging to a new species, 3, as well as only minor proton signals originating from the homoleptic cages of LA and LC (Figure 2 a,b). Longer periods of heating (over 2 weeks) did not result in any notable changes in the 1H NMR spectrum, indicating that 3 is in equilibrium with its homoleptic cage counterparts in a ratio of 14:1.2:1 (3/[Pd2 LA 4]4+/[Pd2 LC 4]4+).
Figure 2.
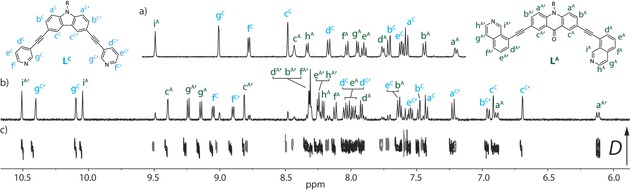
The aromatic region of the 1H NMR spectrum (500 MHz/CD3CN/25° C) of: a) a non‐equilibrated 1:1 mixture of [Pd2 LA 4]4+ and [Pd2 LC 4]4+; b) 3, obtained by heating a 1:1:1 mixture of LA, LC and PdII; c) the DOSY spectrum of (b) showing all of the assigned aromatic signals of 3 with the same diffusion coefficient (log D=−9.19, r=9.1 Å). Note: the DOSY peaks belonging to [Pd2 LA 4]4+ and [Pd2 LC 4]4+ are shown in gray.
DOSY analysis (Figure 2 c, Figure S22) confirmed that the 32 aromatic signals belong to a single species, indicating that a loss of symmetry has occurred and the proton signals are originating from a more topologically complex situation. Initially we postulated that such signal splitting was due to cage interpenetration,19 however, the hydrodynamic radius obtained from the DOSY spectrum (0.91 nm) suggested otherwise, being comparable to that of the homoleptic cages present in the mixture. In agreement with this, ESI‐MS (Figure S18) revealed signals originating from the major species at m/z 570.7, 789.9, and 1228.9, which were consistent with a heteroleptic monomeric cage of the formula: [Pd2 LA 2 LC 2+n BF4](4−n)+ (n=0–2).
This rather intriguing result led us to perform a full assignment of the 32 aromatic proton signals by COSY and NOESY NMR analysis (Figure S19–21). Examination of the 2D NMR spectra revealed that indeed, each ligand has lost the twofold symmetry in the assembly resulting in 18 and 14 proton signals for LA and LC, respectively. Furthermore, several curious NOESY contacts were identified between the two sets of proton signals originating from each ligand, as well as an intra‐ligand HiA′–HiC′ contact, suggesting an anti‐conformation for LA arising from a twist in one of the two isoquinoline moieties. Whilst 2D NMR spectroscopy offered useful hints regarding the structure of 3, X‐ray analysis provided the complete answer. Slow vapor diffusion of diisopropyl ether into a solution of the cage in CD3CN resulted in crystals suitable for single‐crystal X‐ray analysis. The structure of 3 is indeed topologically complex with respect to the normally encountered [Pd2 L 4] assemblies, resembling a “doubly bridged figure‐eight” structure (Figure 1, Figure 3 c,d). As suggested by the NOESY experiment, one isoquinoline moiety of each LA ligand is twisted, affording a very unique trans‐[Pd2(anti‐LA)2 LC 2]4+ topology. In contrast with the structure of 2, the alkyne bonds of LC in 3 adopt a considerably arched conformation to accommodate a much larger Pd⋅⋅⋅Pd distance of 15.0 Å. Furthermore, the potential cavity created by the [Pd2 LC 2] moiety is occupied by the backbones of two LA molecules which mutually participate in offset π stacking, thus forcing the BF4 − counterions to sit at the periphery of the structure. Interestingly, the figure‐eight motif of LA introduces structural chirality20 (Figure S34), which we note is a unique feature of a figure‐eight topology.21 Closer inspection of the structure revealed an S 2 symmetry, which is consistent with the signal splitting observed in the 1H NMR spectrum. Indeed, the doubly bridged figure‐eight structure of 3 is an unprecedented topology not only for [M2 L 4] cages, but metallo‐supramolecular assemblies in general. To our knowledge, X‐ray characterization of [Pd2 L 2 L′2]4+ species has so far been elusive and this example also demonstrates the conformational changes ligands can undergo in order to adapt to the strict square‐planar geometry of PdII in a cage assembly.
Transformations and rearrangements in systems involving discrete metallo‐supramolecular architectures have recently received much interest.22 Given that integrative transformations for coordination cages remain scarce23 and that the homoleptic assemblies of LA, LP, and LC are stable in one another's absence, we were curious whether cages 2 and 3 can be accessed through cage‐to‐cage transformations. Indeed, heating a 2:1 mixture of [Pd2 LC 4]4+ and [Pd4 LP 8]8+ led to the clean formation of 2 within 24 h according to 1H NMR spectroscopy (Figure 1 and Figure S24). We propose that the conversion of the higher nuclearity cage species of LP ([Pd3 LP 6]6++[Pd4 LP 8]8+) to the lower nuclearity product 2 may be driven by an additional entropic factor. In a similar manner, heating a 1:1 mixture of [Pd2 LC 4]4+ and [Pd2 LA 4]4+ gave 3 in a similar distribution to that obtained from the mixture of LA, LC, and PdII (Figure S25). In this case, the absence of an entropic benefit may contribute to the incomplete integrative self‐sorting. In both cases, longer heating periods did not affect the 1H NMR spectra, indicating that 2 and 3 are the thermodynamic minima of their respective system.
We furthermore examined heteroleptic cage transformations by ligand substitution reactions on 2 and 3. Heating a suspension of LA and 2 resulted in quick dissolution of the ligand and after heating at 70 °C for 6 h, the signals in the 1H NMR spectrum corresponding to 2 were completely consumed to give 1 and two equivalents LC (Figure 4). In a similar experiment, heating a 2:1 mixture LP and 3 resulted in complete conversion into 1, suggesting this process proceeds through a complete structural re‐organization. Furthermore, 1 appeared to be stable in the presence of LC when the mixture was heated at 70 °C (Figure S27). The thermodynamic stability of 1–3 was also compared by deconstruction experiments with [D5]pyridine (Figure S31–33), which revealed complete decomposition for 2 and 3 and incomplete decomposition for 1 under the same conditions. Thus, the ligand induced heteroleptic‐to‐heteroleptic cage conversion can be explained by the mutually ideal bite angle of LA and LP, driving the system to the particularly stable compound 1 in the presence of free ligand.
Figure 4.
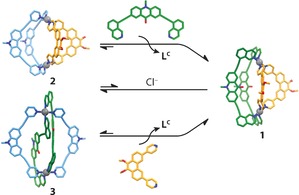
Ligand‐induced (top, bottom) and cage‐to‐cage (center) transformations of 2 and 3 into 1. In the cage‐to‐cage conversion, [Pd2 LC 4]4+ is formed as a by‐product.
Interestingly, however, heating a 1:1:1 mixture of the three homoleptic assemblies ([Pd2 LA 4]4+, [Pd4 LP 8]8+, and [Pd2 LC 4]4+) resulted in 3 as the major product.24 A similar result was obtained from the combination of the three ligands and PdII (Figure S26). To shed light on the observed self‐sorting, we investigated the reaction of a 1:1 mixture of 2 and 3. After heating the mixture for several hours, however, no notable ligand shuffling was observed. This kinetic problem could be somewhat modulated by the addition of 0.5 equivalents of Cl− ions as competing ligands,17 resulting again in conversion to 1, although only partially (Figure S29). The enthalpic benefit of the π‐stacked acridone cores in 3 as well as an already occupied cavity may contribute to a kinetically stabilized structure in this transformation.
In summary, morphological control of novel [Pd2 L 2 L′2] cage architectures has been achieved through the interplay of complementary ligand geometries. We demonstrated that both cis‐ and trans‐configured heteroleptic cages 2 and 3 can be accessed through clean cage‐to‐cage integrative transformations, and notably, the carbazole component can be selectively displaced in favor of a more geometrically suited ligand. Selectively displacing a particular ligand component in a mixture of three ligands and at least six possible cage products (Figure 1) demonstrates an unprecedented degree of structural control in the system. The chiral trans‐[Pd2(anti‐LA)2 LC 2]4+ cage represents a new topology for metallo‐supramolecular assemblies whilst also highlighting the conformational changes ligands can undergo in the presence of a geometrically demanding metal ion. Further studies are underway to utilize these novel architectures as platforms for positioning multiple functionalities at predetermined positions in the [Pd2 L 2 L′2] framework.
Dedicated to Professor Leonard Lindoy on the occasion of his 80th birthday
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Andreas Brockmeyer and Dr. Petra Janning (Max Planck Institute for Molecular Physiology, Dortmund) and Dr. Holm Frauendorf (Georg‐August University Göttingen) for mass spectra measurements. Dyanne Cruickshank from Rigaku Oxford diffraction is thanked for assistance in collecting X‐ray data of 3. W.M.B. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. We thank the Fonds der Chemischen Industrie and the European Research Council (ERC Consolidator grant 683083, RAMSES) for their support.
W. M. Bloch, J. J. Holstein, W. Hiller, G. H. Clever, Angew. Chem. Int. Ed. 2017, 56, 8285.
References
- 1.
- 1a. Hou X., Ke C., Stoddart J. F., Chem. Soc. Rev. 2016, 45, 3766–3780; [DOI] [PubMed] [Google Scholar]
- 1b. He Z., Jiang W., Schalley C. A., Chem. Soc. Rev. 2015, 44, 779–789; [DOI] [PubMed] [Google Scholar]
- 1c. Mattia E., Otto S., Nat. Nanotechnol. 2015, 10, 111–119. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. De S., Mahata K., Schmittel M., Chem. Soc. Rev. 2010, 39, 1555–1575; [DOI] [PubMed] [Google Scholar]
- 2b. Saha M. L., De S., Pramanik S., Schmittel M., Chem. Soc. Rev. 2013, 42, 6860–6909; [DOI] [PubMed] [Google Scholar]
- 2c. Li H., Yao Z.-J., Liu D., Jin G.-X., Coord. Chem. Rev. 2015, 293–294, 139–157; [Google Scholar]
- 2d. Mukherjee S., Mukherjee P. S., Chem. Commun. 2014, 50, 2239–2248; [DOI] [PubMed] [Google Scholar]
- 2e. Smulders M. M. J., Riddell I. A., Browne C., Nitschke J. R., Chem. Soc. Rev. 2013, 42, 1728–1754; [DOI] [PubMed] [Google Scholar]
- 2f. Chakrabarty R., Mukherjee P. S., Stang P. J., Chem. Rev. 2011, 111, 6810–6918; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2g. Cook T. R., Stang P. J., Chem. Rev. 2015, 115, 7001–7045; [DOI] [PubMed] [Google Scholar]
- 2h. Safont-Sempere M. M., Fernández G., Würthner F., Chem. Rev. 2011, 111, 5784–5814. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Schmittel M., Ganz A., Chem. Commun. 1997, 999–1000; [Google Scholar]
- 3b. Yoshizawa M., Nagao M., Kumazawa K., Fujita M., J. Organomet. Chem. 2005, 690, 5383–5388; [Google Scholar]
- 3c. Yamanaka M., Yamada Y., Sei Y., Yamaguchi K., Kobayashi K., J. Am. Chem. Soc. 2006, 128, 1531–1539. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Sauvage J. P., Weiss J., J. Am. Chem. Soc. 1985, 107, 6108–6110; [DOI] [PubMed] [Google Scholar]
- 4b. Solladié N., Chambron J.-C., Sauvage J.-P., J. Am. Chem. Soc. 1999, 121, 3684–3692; [Google Scholar]
- 4c. Sleiman H., Baxter P., Lehn J.-M., Rissanen K., J. Chem. Soc. Chem. Commun. 1995, 715–716; [Google Scholar]
- 4d. Baxter P. N. W., Lehn J.-M., Baum G., Fenske D., Chem. Eur. J. 1999, 5, 102–112; [Google Scholar]
- 4e. Lindoy L. F., Park K.-M., Lee S. S., Chem. Soc. Rev. 2013, 42, 1713–1727. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Kishore R. S. K., Paululat T., Schmittel M., Chem. Eur. J. 2006, 12, 8136–8149; [DOI] [PubMed] [Google Scholar]
- 5b. Samanta S. K., Schmittel M., Org. Biomol. Chem. 2013, 11, 3108–3115. [DOI] [PubMed] [Google Scholar]
- 6. Schmittel M., Kalsani V., Fenske D., Wiegrefe A., Chem. Commun. 2004, 490–491. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Yoshizawa M., Nakagawa J., Kumazawa K., Nagao M., Kawano M., Ozeki T., Fujita M., Angew. Chem. Int. Ed. 2005, 44, 1810–1813; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 1844–1847; [Google Scholar]
- 7b. Klosterman J. K., Yamauchi Y., Fujita M., Chem. Soc. Rev. 2009, 38, 1714–1725. [DOI] [PubMed] [Google Scholar]
- 8. Schmittel M., Kalsani V., Michel C., Mal P., Ammon H., Jäckel F., Rabe J. P., Chem. Eur. J. 2007, 13, 6223–6237. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Bar A. K., Raghothama S., Moon D., Mukherjee P. S., Chem. Eur. J. 2012, 18, 3199–3209; [DOI] [PubMed] [Google Scholar]
- 9b. Zheng Y.-R., Zhao Z., Wang M., Ghosh K., Pollock J. B., Cook T. R., Stang P. J., J. Am. Chem. Soc. 2010, 132, 16873–16882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Bar A. K., Mostafa G., Mukherjee P. S., Inorg. Chem. 2010, 49, 7647–7649; [DOI] [PubMed] [Google Scholar]
- 10b. Samanta D., Shanmugaraju S., Joshi S. A., Patil Y. P., Nethaji M., Mukherjee P. S., Chem. Commun. 2012, 48, 2298–2300. [DOI] [PubMed] [Google Scholar]
- 11.We use the term “bis-monodentate ligand” to describe a monodentate bridging ligand able to bind two metal ions. See for example: Q.-F. Sun, S. Sato, M. Fujita, Angew. Chem. Int. Ed 2014, 53, 13510–13513; Angew. Chem 2014, 126, 13728–13731.
- 12.
- 12a. Han M., Engelhard D. M., Clever G. H., Chem. Soc. Rev. 2014, 43, 1848–1860; [DOI] [PubMed] [Google Scholar]
- 12b. Frank M., Johnstone M. D., Clever G. H., Chem. Eur. J. 2016, 22, 14104–14125. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Yazaki K., Noda S., Tanaka Y., Sei Y., Akita M., Yoshizawa M., Angew. Chem. Int. Ed. 2016, 55, 15031–15034; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15255–15258; [Google Scholar]
- 13b. Yazaki K., Sei Y., Akita M., Yoshizawa M., Chem. Eur. J. 2016, 22, 17557–17561; [DOI] [PubMed] [Google Scholar]
- 13c. Krick M., Holstein J., Würtele C., Clever G. H., Chem. Commun. 2016, 52, 10411–10414; [DOI] [PubMed] [Google Scholar]
- 13d. Kishi N., Li Z., Yoza K., Akita M., Yoshizawa M., J. Am. Chem. Soc. 2011, 133, 11438–11441; [DOI] [PubMed] [Google Scholar]
- 13e. Löffler S., Lübben J., Wuttke A., Mata R. A., John M., Dittrich B., Clever G. H., Chem. Sci. 2016, 7, 4676–4684; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13f. Johnstone M. D., Schwarze E. K., Ahrens J., Schwarzer D., Holstein J. J., Dittrich B., Pfeffer F. M., Clever G. H., Chem. Eur. J. 2016, 22, 10791–10795; [DOI] [PubMed] [Google Scholar]
- 13g. Han M., Michel R., He B., Chen Y.-S., Stalke D., John M., Clever G. H., Angew. Chem. Int. Ed. 2013, 52, 1319–1323; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1358–1362; [Google Scholar]
- 13h. Lewis J. E. M., Gavey E. L., Cameron S. A., Crowley J. D., Chem. Sci. 2012, 3, 778–784. [Google Scholar]
- 14.
- 14a. Johnson A. M., Hooley R. J., Inorg. Chem. 2011, 50, 4671–4673; [DOI] [PubMed] [Google Scholar]
- 14b. Yamashina M., Yuki T., Sei Y., Akita M., Yoshizawa M., Chem. Eur. J. 2015, 21, 4200–4204. [DOI] [PubMed] [Google Scholar]
- 15. Preston D., Barnsley J. E., Gordon K. C., Crowley J. D., J. Am. Chem. Soc. 2016, 138, 10578–10585. [DOI] [PubMed] [Google Scholar]
- 16. Bloch W. M., Abe Y., Holstein J. J., Wandtke C. M., Dittrich B., Clever G. H., J. Am. Chem. Soc. 2016, 138, 13750–13755. [DOI] [PubMed] [Google Scholar]
- 17. Zhu R., Lübben J., Dittrich B., Clever G. H., Angew. Chem. Int. Ed. 2015, 54, 2796–2800; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2838–2842. [Google Scholar]
- 18.
- 18a. Frank M., Krause L., Herbst-Irmer R., Stalke D., Clever G. H., Dalton Trans. 2014, 43, 4587–4592; [DOI] [PubMed] [Google Scholar]
- 18b. Johnson A. M., Wiley C. A., Young M. C., Zhang X., Lyon Y., Julian R. R., Hooley R. J., Angew. Chem. Int. Ed. 2015, 54, 5641–5645; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5733–5737; [Google Scholar]
- 18c. Ayme J.-F., Beves J. E., Campbell C. J., Leigh D. A., Angew. Chem. Int. Ed. 2014, 53, 7823–7827; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7957–7961; [Google Scholar]
- 18d. Yokoyama H., Ueda Y., Fujita D., Sato S., Fujita M., Chem. Asian J. 2015, 10, 2292–2295; [DOI] [PubMed] [Google Scholar]
- 18e. Jiménez A., Bilbeisi R. A., Ronson T. K., Zarra S., Woodhead C., Nitschke J. R., Angew. Chem. Int. Ed. 2014, 53, 4556–4560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4644–4648. [Google Scholar]
- 19. Freye S., Hey J., Torras-Galán A., Stalke D., Herbst-Irmer R., John M., Clever G. H., Angew. Chem. Int. Ed. 2012, 51, 2191–2194; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2233–2237. [Google Scholar]
- 20.Whilst the X-ray anomalous scattering data revealed that the measured crystal of 3 is composed of only the M,M enantiomer, we assume to have a racemic mixture of cage enantiomers in solution, as no external chiral information was provided to favor the assembly of one enantiomeric cage over the other.
- 21.
- 21a. Werner A., Michels M., Zander L., Lex J., Vogel E., Angew. Chem. Int. Ed. 1999, 38, 3650–3653; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 3866–3870; [Google Scholar]
- 21b. Strotmeyer K. P., Fritsky I. O., Pritzkow H., Krämer R., Chem. Commun. 2004, 28–29; [DOI] [PubMed] [Google Scholar]
- 21c. Setsune J., Tsukajima A., Okazaki N., Lintuluoto J. M., Lintuluoto M., Angew. Chem. Int. Ed. 2009, 48, 771–775; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 785–789; [Google Scholar]
- 21d. Katoono R., Tanaka Y., Kusaka K., Fujiwara K., Suzuki T., J. Org. Chem. 2015, 80, 7613–7625. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Wang W., Wang Y.-X., Yang H.-B., Chem. Soc. Rev. 2016, 45, 2656–2693; [DOI] [PubMed] [Google Scholar]
- 22b. McConnell A. J., Wood C. S., Neelakandan P. P., Nitschke J. R., Chem. Rev. 2015, 115, 7729–7793; [DOI] [PubMed] [Google Scholar]
- 22c. Han M., Luo Y., Damaschke B., Gómez L., Ribas X., Jose A., Peretzki P., Seibt M., Clever G. H., Angew. Chem. Int. Ed. 2016, 55, 445–449; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 456–460. [Google Scholar]
- 23.
- 23a. Ronson T. K., Roberts D. A., Black S. P., Nitschke J. R., J. Am. Chem. Soc. 2015, 137, 14502–14512; [DOI] [PubMed] [Google Scholar]
- 23b. Li J.-R., Zhou H.-C., Nat. Chem. 2010, 2, 893–898; [DOI] [PubMed] [Google Scholar]
- 23c. Fujita M., Fujita N., Ogura K., Yamaguchi K., Nature 1999, 400, 52–55. [Google Scholar]
- 24.This can be explained by the faster structural rearrangement of [Pd2 LA 4]4+ and [Pd2 LP 4]4+ into 3 compared with [Pd2 LA 4]4+ and [Pd4 LP 8]8+ into 1 (Figure S30).
- 25. CCDC 1537049 and CCDC-1537050 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary