

Abstract
Early blight, caused by the fungus Alternaria solani, is a devastating foliar disease of tomatoes, causes massive yield loss each year worldwide. Molecular basis of the compatible host–pathogen interaction was elusive. We adopted next generation sequencing approach to decipher miRNAs and mRNAs that are differentially expressed during Alternaria-stress in tomato. Some of the interesting findings were also validated by alternative techniques. Our analysis revealed 181 known-miRNAs, belonging to 121 miRNA families, of which 67 miRNAs showed at least 2-fold change in expression level with the majority being downregulated. Concomitantly, 5,450 mRNAs were significantly regulated in the same diseased tissues. Differentially expressed genes were most significantly associated with response to stimulus process, photosynthesis, biosynthesis of secondary metabolites, plant–pathogen interaction and plant hormone signal transduction pathways. GO term enrichment-based categorization of gene-functions further supported this observation, as terms related to pathogen perception, disease signal transduction, cellular metabolic processes including oxidoreductase and kinase activity were over represented. In addition, we have discovered 102 miRNA–mRNA pairs which were regulated antagonistically, and careful study of the targeted mRNAs depicted that multiple transcription factors, nucleotide-binding site leucine-rich repeats, receptor-like proteins and enzymes related to cellular ROS management were profoundly affected. These studies have identified key regulators of Alternaria-stress response in tomato and the subset of genes that are likely to be post-transcriptionally silenced during the infection.
Keywords: tomato, Alternaria solani, miRNA, transcriptome, next generation sequencing
1. Introduction
Early blight (EB) is one of the most devastating diseases of tomato (Solanum lycopersicum) caused by Alternaria solani (Ellis & Martin) Sorauer, a necrotrophic fungus. The disease largely affects the older leaves, stem and fruits of tomato plants.1 Cultivars highly resistant to EB are not known.1 Progress in breeding for EB resistance has been, however, limited by the lack of effective resistance genes in cultivated tomato, and by polygenic inheritance of the resistance.2 Thus, there is an urgent need for identifying new sources of resistance or host factors that are altered during the pathogenesis process and could be modulated for coordinating the resistance response.
Plants try to defend themselves against phytopathogens by coordinating defence-oriented transcriptional reprogramming of the affected cells. Several signalling and transcription factors are the key elements in this process. Expressions of some of these genes are regulated by miRNAs.3 It is expected that during tomato–Alternaria interaction expression of a set of miRNAs and mRNAs will be altered, however, such studies are lacking. Moreover, limited information is available about molecular signalling involved in tomato–necrotrophic pathogen interaction.
Recently, the next generation sequencing (NGS) technology has been used to obtain comprehensive sequencing data for the detection and study of alteration of the expression levels of miRNAs or mRNAs in various plant species under different stress conditions including fungus–stress.4–9 The identified miRNAs primarily controlled the expression of genes encoding disease-resistance proteins, serine/threonine protein kinases and transcription factors. In a recent publication, microarray-based profiling of tomato mRNA during the early stage of A.solani infection has been reported,10 however, a detailed analysis is not available.
For the proper understanding of the regulatory action of miRNA, an integrated analysis of miRNA and mRNA transcriptome is a requisite. Using plant as the model system, only three such studies with other pathogen or treatment have been reported; these include rice–rice stripe virus (RSV) interaction, rice and Sedum alfredii-cadmium treatment.11–13 Interestingly, rice–RSV study revealed expression of 22 up-regulated miRNAs targeted 24 repressed mRNAs which are linked to disease resistance pathways.
The best way of bestowing disease resistance to a plant is to potentiate its natural resistance mechanism. As mentioned earlier, transcriptional reprogramming and regulation of a set of gene/regulator would determine a plant’s response towards the pathogen. Deliberate alteration of expression of these factors potentially should control a plant’s response towards a pathogen. To appreciate fully the factors needed to be altered, the regulators of host processes during the pathogenesis and disease manifestation have to be unravelled. In this regard, a comparative analysis of control vs infected susceptible-plant is advantageous over studying the tolerant plant because multiple modes of tolerance are feasible; however, it is more likely that susceptibility response would follow a unique path. We hypothesized that several miRNAs have the crucial regulatory role in the transcriptional reprogramming process during Alternaria infection, and set out to uncover by genome-wide analysis of miRNA–mRNA relationships specifically regulated during the stress in tomato. To the best of our knowledge, this is the first report of integrating miRNA and mRNA transcriptome analysis data to endorse miRNA-mediated regulation of gene expression during tomato–A.solani interaction. This combined analysis facilitated in gaining significant insights into the regulatory network of susceptible response against an agronomically important necrotrophic pathogen of tomato.
2. Materials and methods
2.1. Plant material and treatment condition
Tomato (S.lycopersicum L.) cultivar Pusa Ruby (Indian Agricultural Research Institute) was used in this study and is known to be susceptible to EB. Plants were grown in pots containing Soilrite (Keltech, India) at 25°C and under natural light with addition of Hoagland solution at regular interval. A.solani (obtained from IIVR, India) was cultured in potato dextrose agar plate at 25°C for 5 days in dark. Leaves of one and a half month old plants were deliberately inoculated with a drop (∼10 µl, 106 CFU/ml water) of mycelial and spore suspension of A. solani on the dorsal surface of leaf and were kept in humid chamber until symptoms appeared (∼3 days) or as mentioned in individual experiments. Control or mock inoculated plants received only a drop of water as inoculum and maintained under similar condition.
2.2. Biochemical assays
2.2.1. Callose detection
Plant leaflets cut from their petiolule were immersed in alcoholic lactophenol (1:1:1:1 ratio of phenol:glycerol:lactic acid:water and 2 volumes of absolute ethanol) and warmed gently to remove the chlorophyll. The dechlorophyllized leaves were rinsed with water and stained with 0.01% toluidine blue solution in 150-mM disodium phosphate buffer (pH 9.5) for 30 min in dark. Leaves were then stained with 0.01% aniline blue for another 30 min in dark, and samples were observed under fluorescent microscope (Carl Zeiss Axioskop 40 FL) with an excitation and emission wavelengths of 470 and 515 nm, respectively.
2.2.2. Nitric oxide detection
Leaf sections (∼0.5 cm2) encompassing the fungal inoculation site were used. The leaf cuttings were gently vacuum-infiltrated with 10 mM DAF-2DA dissolved in 10 mM Tris–HCl (pH 7.5) at room temperature for 1 h. Fluorescence of the tissues was monitored under a fluorescent microscope (excitation/emission are 495/515 nm).
2.2.3. Detection of ROS accumulation
Detection of intracellular ROS, particularly hydrogen peroxide, using 3,3′-diaminobenzidine (DAB) was performed. Leaflets cut at their petiolule were incubated in DAB staining solution (DAB at 1 mg/ml of 10 mM Na2HPO4), in dark, overnight at 28°C with constant agitation. DAB staining solution was replaced with bleaching solution (ethanol:acetic acid:glycerol = 3:1:1) and incubated in boiling water for 10–15 min to get rid of chlorophyll, the process was repeated till all of the chlorophyll was bleached away, and then the leaflets were visualized under white light and photographed.
2.3. Scanning electron microscopy (SEM)
Infected and control tomato leaves were cut into ∼1-cm2 size pieces and fixed with 3% glutaraldehyde in 0.2-M sodium phosphate buffer (pH 7.4) overnight at 4°C. After rinsing the samples in same buffer, they were dehydrated in increasing concentration of ethanol (50, 70, 85 and 95%) for 30 min in each solution and were kept in absolute ethanol until attaching on SEM stubs. The prepared specimens were directly examined in a SEM (FEI Quanta 200 FEG MKII) at 5–10 kV.
2.4. RNA extraction and cDNA synthesis
Visible fungal mass and dead tissues from the Alternaria-infected leaves were gently removed and samples (∼300 mg) were collected only from the infected zone. Total RNA was prepared from both infected and mock-inoculated (control) leaves by TRIzol reagent (Invitrogen) following manufacturer’s protocol. RNA was resolved in a 0.8% denaturing formaldehyde gel to check its integrity. Only high-quality RNA was used for downstream processing. Typically, 3 µg of total RNA was reverse transcribed to cDNA using RevertAid Reverse Transcriptase (ThermoFischer scientific) and random hexamer primer at 42°C for 60 min. cDNA was stored at −20°C if not immediately used.
2.5. Real time PCR analysis
Only 2.5% of the cDNA reaction mix was used as template in each 20 μl reaction containing SYBR green PCR mix (Thermo Scientific) and gene specific primers (0.2 µM each). PCR and analysis were performed using Applied Biosystems 7500 FAST machine. For each sample of cDNA three replicates were analysed. Data were normalized based on the expression level of EF1α transcripts. Amplification of specific products were confirmed by obtaining a single peak in melt curve analysis, first derivative of fluorescence (dF/dT) vs temperature plot and by agarose gel electrophoresis (1.5% agarose-TAE gel) of PCR end-products. Relative expression levels of transcripts were determined using the 2−ΔΔcT method. All experiments were repeated with three independent RNA preparations.
2.6. Small RNA library preparation and sequencing
Total RNA for NGS was isolated following the method as mentioned above. Integrity of RNA (RIN >7) was further assessed using Bioanalyzer 2100 (Agilent Technologies) on an RNA 6000 Nanochip. These RNAs were also subjected to RT-PCR based analysis for detection of fungal RNA contamination using A. solani-specific primers for AltA1, GAPDH genes and ITS sequence (Supplementary Table S10). Small RNA libraries for sequencing were constructed using 1 µg of total RNA, according to the Illumina TruSeq Small RNA library preparation protocol (Illumina, USA). In brief, after adapter ligation at both ends the ligated products were reverse transcribed with Superscript II Reverse transcriptase (RT) by priming with adapter-specific RT-primers, PCR amplified and 140–190-bp size fragments were gel purified. The purified high-quality cDNA library was sequenced (Genotypic Pvt. Ltd., India) on an Illumina Genome AnalyserIIx (GAIIx).
2.7. Small RNA analysis
High-quality reads (Q-phred ≥30) obtained from four different samples (Controls 1 and 2, Infected 1 and 2, GEO accession: GSE75922) were subjected to adapter removal and size fractionation to collect only 18–25 nt reads. Probable fungal miRNA contaminants were discarded. Remaining reads were Blast searched against annotated plant miRNAs gathered from miRBase (release 21), Tomato Functional Genomics Database (TFGD) and Tomato genomics resources database (TGRD) to obtain the known miRNAs. Alignments with no gap and ≤2 mismatches between the query sequences and known miRNAs (subject) were only considered. The identified miRNAs were grouped into families based on sequence similarity. The reads which did not match to any known miRNAs were further processed to discover novel miRNAs using Mireap v0.2 software. Deseq was used to obtain the differentially expressed miRNAs. Outline of the analysis pipeline and a detailed protocol are presented in Supplementary Figs S1 and S2 and Supplementary Document S1.
2.8. Preparation of DNA oligonucleotide probes
About 10 pmol of DNA oligo was used for end labelling with 15 U of T4 PNK (NEB) and 40 µCi γ-32P-ATP (BRIT, India) in a total volume of 15 µl according to enzyme manufactures (NEB) protocol. The labelled probes were then purified using illustraTM MicroSpin G-25 column (GE Health care), and specific activity was determined after scintillation counting of the purified probe. Probes with specific activity of about 20 × 107 cpm/µg oligo was routinely obtained and used in subsequent applications.
2.9. Northern blot hybridization analysis of miRNAs
Total RNA, 40 μg per sample was resolved in a 7-M urea/10% polyacrylamide/TBE gel at 80 V and transferred to nylon membrane HybondTM–XL (GE healthcare) using Semi-Dry Transfer Cell (TE 77 PWR, GE Healthcare Amersham™) at 150 V for 1 h. RNA was fixed using UV cross linker (UVC 500 UV Crosslinkers, GE Healthcare Amersham™) at 15,000 J/cm2 for 2 min. The membrane was incubated for 30 min in pre-hybridization buffer (50% Deionized formamide, 5× SSPE buffer, 5× Denhardt’s solution, 0.5% SDS, supplemented with 0.02 mg/ml denatured Salmon sperm DNA) at 42°C, and then hybridization with γ-32P labelled DNA oligo (∼20 × 106 cpm) in the same prehybridization buffer was carried out in a heat seal bag by incubating overnight at 42°C with occasional shaking. Membrane was washed using a standard protocol, exposed to phosphor screen and image was captured at 100 µm pixel in Typhoon Trio+ (GE healthcare).
2.10. Detection of miRNA by polyadenylation-mediated PCR
Small RNA population from total RNA was enriched using PureLink® miRNA Isolation Kit (Invitrogen) according to manufacturer’s protocol. Quality was examined by estimating the integrity of tRNA band loaded in denaturing 7% urea PAGE gel. 500 ng of small RNA was polyadenylated, and reverse transcribed to cDNA according to the protocol of NCode™ VILO™ miRNA cDNA Synthesis Kit (Invitrogen). Desired miRNA was amplified from the cDNA pool by using mature miRNA-specific forward primer and universal qPCR reverse primer provided in the kit. Relative intensity of the U6 snRNA-specific product band or tRNA band in the same amount of small RNA samples that were resolved in a urea-PAGE gel was monitored as normalizing factor.
2.11. mRNA library construction, Illumina sequencing and transcriptome data analysis
Total RNA from Control 1 and Infected 1 samples were also used for mRNA library preparation and sequencing (Illumina Genome AnalyserIIx) according to the Illumina TruSeq RNA library protocol (Supplementary Document 1). The paired end raw reads were quality checked using SeqQC (a proprietary tool developed in Genotypic Technology Pvt. Ltd, India) and processed by Genotypic in-house script for adapters and low-quality bases trimming towards 3′-end. Sequence files are deposited in GEO database under the accession number GSE75923. Tophat-2.0.72 and Cufflinks-2.0.14 tools were used for assembling transcripts and estimating their abundances. Data were normalized as fragments mapped per kilobase of exon per million reads mapped. Cuffdiff (included in the cufflinks package) was used to find significant changes in transcript expression. Outline of the analysis pipeline is presented in Supplementary Fig. S3. Transcripts having log2 Fold Change (log2FC) at least ±1 and P ≤ 0.05 were used for further analysis.
2.12. Gene Ontology (GO) and the KEGG pathway annotation of Alternaria responsive transcripts
The differentially expressed mRNAs were subjected to GO terms and pathway enrichment analyses. The GO terms were extracted for the regulated genes from Sol Genomics Network (SGN) ITAG2.4.go.csv. GO term enrichment was done using Cytoscape plugin: BiNGO of Cytoscape v3.2.1 software with Benjamini and Hochberg false discovery rate (FDR) setting of ≤0.05. BlastKOALA was used for K number assignment to the differentially expressed genes (DEGs) using SSEARCH computation against a non-redundant set of KEGG genes, so that the gene could be put under a pathway. Pathway enrichment was done in a similar manner as for GO using Cytoscape v3.2.1 using the same parameters.
2.13. Prediction of miRNA targets
Two different softwares were used to predict the putative targets of differentially expressed known and novel miRNA candidates. The putative target sites of miRNA candidates were identified by aligning the miRNA sequences with tomato unigene library, using the plant small RNA Target Analysis Server, psRNATarget, with default parameters except a more relaxed cut-off threshold (5.0) of maximum expectation for higher prediction coverage. In addition to it, we used target prediction function of TFGD with default parameters to predict the putative targets. Expression levels of targets were taken from list of differentially expressed mRNAs identified in our mRNA transcriptome data analysis. GO terms were assigned to these targets using the similar method that has been utilized for assigning terms to DEGs as described in previous section.
2.14. Target cleavage validation by using modified 5′RLM RACE
Total RNA from tomato leaves were isolated, an RNA adapter was directly ligated to 3 μg of RNA using T4 RNA ligase (NEB). RNA was reverse transcribed using reverse transcriptase (Fermentas) with random hexamers. To amplify RACE products, first PCR was done with 1% of the RT reaction. Initial PCR reactions were done with an adaptor primer and complementary gene specific primers (Supplementary Table S10). PCR cycles were 95°C for 30 s, 58°C for 30 s and 72°C for 45 s for 35 cycles. The nested PCRs were performed on 2.5 μl of initial reactions with the nested adaptor primer and complementary gene-specific internal primers. The resulting PCR products were gel purified, cloned in pGEM-T Easy vector (Promega) and sequenced.
2.15. Statistics
Each RNA sample for NGS was obtained from multiple leaves collected from different plants. Except radioactive-based assays, in all other experiments at least three different plants of same age group were used. Three different RNA preparations were used for real-time PCR analysis; reactions were also done in triplicates. Q-PCR results are expressed as means ± standard error (SE). Two-tail Student’s t-test was used and P < 0.05 was considered significant for RT-PCR-based differential gene expression analysis.
3. Results
3.1. Plant infection and disease characterization
Leaves of tomato plants were inoculated with A.solani using droplet inoculation method, and chlorosis around the inoculums appeared within 24 h post-inoculation (hpi now onwards) followed by the development of typical blight symptoms at 72 hpi (Fig. 1A). Leaves were succumbed to the disease within 120 hpi. To confirm the disease establishment and to decide on the time of sampling for RNA preparation we did light microscopic, SEM and multiple biochemical assays, followed by analysis of the expression level of stress-related marker genes. Microscopic studies have confirmed fungal invasion through stomatal openings (Fig. 1B). Assays for detection of callose, nitric oxide (NO) and reactive oxygen species (ROS) accretion indicated that all three showed maximum accretion from 48 to 96 hpi with a peak at 72 hpi (Fig. 1C–E). Further analysis of expression level of pathogenesis-related PR1b and Chitinase genes (Fig. 1F) also showed significant up-regulation occurred during this time. Positive results in biochemical assays for callose deposition, ROS and NO generation together with up-regulated expression of PR1b and chitinase confirmed disease establishment and significant progression within 72 hpi. Hence, RNA was isolated from tomato leaves infected for ∼72 h for further analyses.
Figure 1.

Symptoms in deliberately infected tomato leaves, and characterization of the disease progression. (A) Manifestation of different stages of disease development during Alternaria infection. (B) Trypan blue staining displaying hyphal invasion (red arrow) through stomata (yellow arrow) into the leaf (left panel), right panel shows SEM of infected leaf surface confirming Alternaria hyphal (red arrow) invasion through stomatal opening (yellow arrow). (C) Detection of callose deposition in infected region. Control (mock inoculated, left panel) and infected (right panel) leaf fluorescence images after staining for the detection of callose deposition, showing accumulation of callose indicated by green fluorescence (black arrow) around hyphal mat, as a response to early plant defence. (D) Visualization of nitric oxide (yellow arrow) production at infection site (white arrow) by diaminofluorescein diacetate (DAF-2DA) staining. (E) Assays for detection of time-dependent accumulation of ROS at infection site. Brown coloration around the infection site resulted from DAB staining confirming ROS generation. Production of maximum ROS at 72 hpi is apparent. (F) Quantitative real-time PCR data showing expression level of selected marker genes for biotic stress. Both PR1 and chitinase transcript levels were upregulated compared with the control. Expression level of EF1α was used for normalization. *Significantly changed.
3.2. Differential expression of diverse sRNA population is associated with Alternaria-infection in tomato
Only high-quality RNAs devoid of detectable fungal RNA contamination were used for library construction. Four small RNA libraries were constructed and sequenced using NGS system to investigate the expression of small RNAs in tomato upon Alternaria stress. A total of 18.5, 11, 13.4 and 9.8 million reads ranging in size from 16 to 36 nt were retrieved from Control 1, Control 2, Infected 1 and Infected 2 libraries, respectively. After removing low-quality reads and adapter trimming, the remaining reads were size filtered from 18 to 25 nt to enrich the sample with reads corresponding to the size of typical miRNAs. These filtered sequences represented 1,009,952 (Control 1), 1,163,133 (Control 2), 636,784 (Infected 1) and 218,018 (Infected 2) unique sequences (Supplementary Table S1).
The small RNA length distribution among the four libraries showed maximum reads in the size range of 21–24 nt (65.86 and 75.17% for total and unique reads, respectively, in the average of four samples) which corresponds to typical dicer-derived products (Supplementary Fig. S4). In total reads 24 nt sRNAs dominated the sRNA transcriptome in a control plant which is consistent with the typical size distribution of sRNAs found in other plants.4,5 However, in infected plants, total reads of 21 nt sRNAs increased (average 16.4 vs. 23.5% in control vs. infected), and the 24-nt population decreased markedly (average 24.8 vs. 14.5% in control vs. infected) (Supplementary Fig. S4A). Among the unique reads, the majority of sRNAs were 24 nt in length (39.08%, average of four samples) followed by 23 nt, however, 21 and 22-nt long reads were comparatively equally distributed (Supplementary Fig. S4B). Distribution of 24 vs. 21 nt unique reads in control vs. infected plants also followed the similar pattern as in total reads. In addition, the 21 and 22 nt class sRNAs showed the highest redundancy, whereas the 24-nt class showed the lowest redundancy (Supplementary Fig. S4A and B).
3.3. Both conserved and species-specific microRNAs are Alternaria-stress responsive in tomato
3.3.1. Profiling of conserved and species-specific miRNAs
The cleaned and size fractionated reads acquired above were used in BlastN search against a collection of all the annotated plant miRNAs, obtained from multiple databases comprising miRBase (Release 21), TFGD and TGRD. On the basis of sequence similarity (minimum alignment length 18, maximum two mismatches and no gap), our analysis revealed 181 known miRNAs, which belonged to 121 miRNA families. Most of the identified miRNAs belonged to Solanaceae-specific families (75/121 families). Others (24/121 families) were conserved miRNAs for a plant, while few were reported from a specific plant species only (Supplementary Fig. S5).
We analysed the identified families and found that they included a varying number of miRNA members. All miRNA families with at least five raw read counts for a member in at least one sample have been listed in Supplementary Table S2. Among the conserved miRNAs detected, the miR156 family was the largest, having seven members that were distinguished by the specific differences in their nucleotide sequences, followed by other abundant miRNAs such as miR166, miR171and miR482 families comprising of six members each. MiR167 included five members and miR319, miR396, miR9471 and miR393 contained four members each. Of the remaining 112 miRNA families, 2 families included 3 members, 19 families contained 2 members each and rest 91 miRNA families were each represented by a single member only (Supplementary Table S2).
The frequencies of miRNA reads in a family varied from 1 (miR5658, miR4414, miR7717, etc.) to 1,01,449 (miR166), indicating that miRNA families are expressed in a widely variable frequency within the tissue. Of all the identified miRNA families, 10 families were represented by more than 1,000 normalized reads in the tomato dataset (Supplementary Table S2). In addition to miR166, miR159 (50,644 reads) and miR398 (13,132 reads) were among the major abundant families in the library. On average in a control tomato plant about 6.62% of total miRNAs are represented by highly abundant family members. On the contrary, less than 10 reads were detected for species specific and many solanaceae-specific miRNAs such as miR9472, miR8032, miR5083 and miR1873, etc. (Supplementary Table S2). Analysis of the sequenced data also showed that the relative abundance of members of the miRNA families varied greatly (Supplementary Fig. S6, Supplementary Tables S3 and S4), suggesting differential expression from different miRNA genes of the same family in tomato contributed to the final number of a mature miRNA produced in the tissue.
3.3.2. Differential expression analysis of conserved and species-specific miRNAs
miRNAs with at least five raw read counts from two equivalent libraries were selected for further analysis to determine differentially expressed miRNAs between Alternaria-treated and mock-treated samples using DEseq. Out of 181 identified miRNAs, 100 were differentially expressed. Next, to identify miRNAs that were notably changed in expression level after infection, we applied another selection criterion of log2FC to be at least ±1. This stringent selection yielded 67 differentially expressed miRNAs, and that the majority of them (44 out of 67) were down-regulated (Fig. 2A, Supplementary Table S5). Among the differentially regulated miRNAs, sly-miR9524, sly-miR397 and sly-miR9513 were highly down-regulated with a log2FC values of −4.99 (P = 0.04), −4.39 (P = 0.01) and −4.08 (P = 0.02), respectively, and sly-miR482a, sly-miR8175 and sly-miR6300 were highly up-regulated with log2FC values of 3.13 (P = 0.04), 3.28 (P =0.05) and 2.65 (P= 0.01), respectively (Fig. 2B).
Figure 2.

Differentially expressed miRNAs between Alternaria infected and mock-treated tomato plants. (A) Scatter plot of miRNA expression pattern. Log2 value of read count of mock treated and Alternaria-infected samples are plotted. Green dots (grey dots in print) represent down-regulated, red circles (grey circles in print) indicate upregulated and black dots represent unchanged miRNAs. (B) Bar plot showing expression pattern of highly regulated miRNAs having log2 fold change at least ±2. Majority of the miRNAs were found to be down-regulated.
3.4. Several novel and previously uncharacterized miRNAs are also differentially expressed during Alternaria-infection
3.4.1. Profiling of novel miRNAs
We next inquired whether this cultivar of tomato also expressed any novel or previously uncharacterized miRNAs. Approximately 2,582,051 unannotated sequences (accumulated from four samples), not matching to known miRNAs or any other ncRNA such as tRNA, rRNA, snRNA, snoRNA and repeats, were aligned to the tomato genome (Sol Genomics Network). The reads aligned to tomato genome were used as input in the miRNA prediction software Mireap to obtain the putative novel miRNAs with their predicted hairpin precursors. To further increase the confidence of claiming the identified sequences as novel miRNAs, we applied the three Ambros criteria with slight modification. In addition, a sequence with minimum five read counts in at least one sample was taken for novel miRNA analysis. We, thus, recognized 60 potential novel miRNA candidates from all 4 samples (Supplementary Table S6). To further confirm precursor stem loop structure of these predicted novel miRNAs, the putative precursor sequence was subjected to folding analysis using RNAfold software. All these novel miRNA candidates’ precursor sequences were folded into a hairpin-like structure apparently similar to other known miRNAs (Fig. 3A and Supplementary Fig. S7). Moreover, the miRNA* of eight miRNAs were readily detected in our dataset. The precursor sequence of these new miRNA candidates varied from 63 to 101 nt in length and the MFE of precursor hairpins ranged from −25 to −50 kcal/mol. Of the 60 new miRNAs 52 are situated in the intergenic region, and 8 are located in the intronic regions of genes (Supplementary Table S6). The mature novel miRNAs could be found in both 5′ and 3′ arms of the predicted precursors and their read number varied from 1 to 20,631. However, some of these candidates could not be detected in all samples.
Figure 3.

Predicted precursor structure and validation of expression of novel miRNAs. (A) Predicted secondary structures of the precursor of selected novel miRNAs as determined using RNAfold. Mature miRNA and miRNA star sequences—both detected by sequencing—are denoted in red (dark grey in print) and green (light grey in print), respectively. (B) Image showing expression pattern of three novel miRNAs as detected by northern blot hybridization analysis. (C) Poly A tailing mediated amplification and detection of selected novel miRNAs. (D) Semi-quantitative RT-PCR analysis for validation of expression of precursor sequence of two of the novel miRNAs. RT-dependent PCR samples are loaded in +RT lanes and −RT lanes contained samples of PCR performed with mock cDNA.
3.4.2. Differential expression analysis of novel miRNAs
Differential expression analysis using DESeq was performed to determine the differential expression level of predicted novel miRNAs. Among the differentially expressed novel miRNAs, 52 miRNAs had log2FC at least ±1. Among these, 39 and 13 were down and up-regulated, respectively, and only 7 novel miRNAs were found to be significantly (P ≤ 0.05) differentially expressed (Supplementary Table S7). Both known and novel miRNAs expression data showed a majority of the differentially expressed miRNAs to be down-regulated as a response to Alternaria-stress.
3.5. Differential expressions of both known and novel miRNAs could be validated using alternative techniques
3.5.1. Validation of expression of known miRNAs
To validate the expression pattern of selected miRNAs, having at least 10 reads in any sample, we performed Poly A tailing mediated miRNA-amplification and northern blot analysis (Fig. 4). The highly up-regulated miRNAs, sly-miR482b, sly-miR168, sly-miR6300, sly-miR156f, sly-miR403-3p and sly-miR9519, and highly down-regulated miRNAs, sly-miR408b-3p, sly-miR398a-3p, sly-miR397 and sly-miR9513 were selected for testing along with sly-miR156a/b, sly-miR159, sly-miR4376, sly-miR166 and sly-miR6024 which were not significantly differentially expressed.
Figure 4.
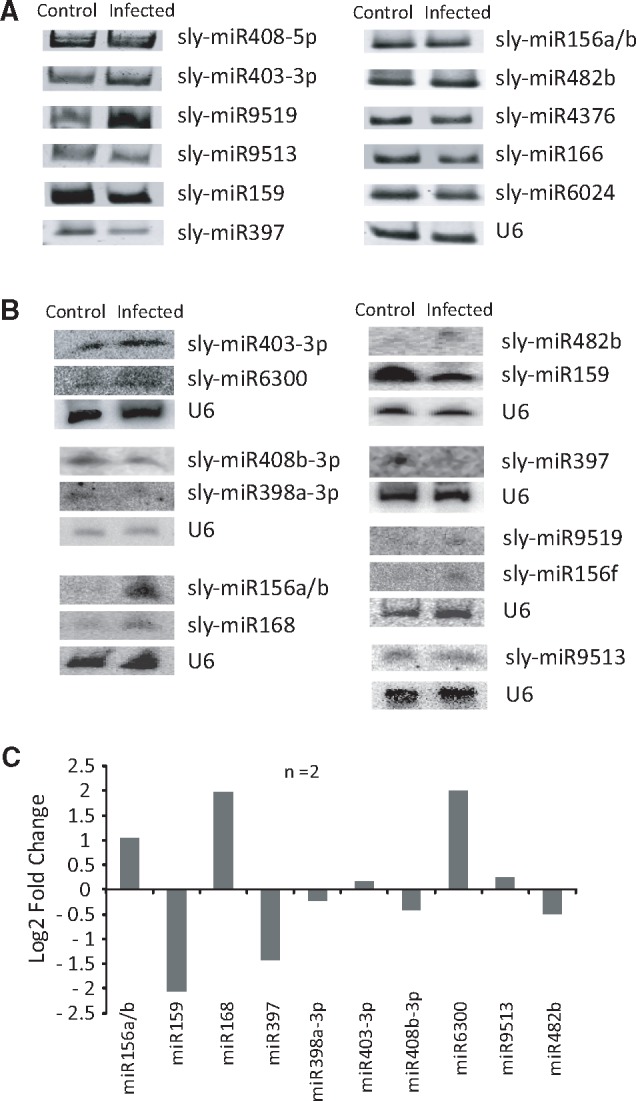
Validation of expression level of miRNAs using alternative approaches. (A) PAGE photograph of poly A tailing mediated RT-PCR analysis of selected known miRNAs. U6 snRNA expression was monitored as normalization factor. (B) Images of northern blot hybridization analysis of selected known miRNAs. (C) Quantification of band intensity obtained (using ImageQuant ID GEL quantification software) in northern blot analysis (n = 2). Data presented as log2 fold change (normalized intensity of infected/control).
PolyA tailing semiquantitative PCR (n = 3) analysis showed that out of 11 miRNAs tested 8 were differentially expressed (Fig. 4A). Northern blot analyses (n = 2) (Fig. 4B) indicated that expression of most of the miRNAs tested (10/12 miRNAs) matched with the trend of regulation observed in NGS analysis, however, the extent of regulation differed (Fig. 4C, Supplementary Fig. S8). Noticeable difference in expressions of sly-miR156a/b, sly-miR397, sly-miR482b, sly-miR159, sly-miR168 and sly-miR156f were also observed. Two conserved miRNAs, miR159 and miR156a/b, which did not fulfil log2FC criteria (at least ±1) in NGS analysis showed considerable alteration in expression in Northern blot analysis.
Next, we investigated whether the expression level of certain miRNAs was also dependent on the infection progression. Samples from different time points post infection (8–72 hpi) were collected for Northern blot analysis. Visible difference in expression of miRNAs tested was apparent only at 72 hpi (Supplementary Fig. S9). Expressions of miR168 and miR403 were upregulated, whereas the expression of miR397 was downregulated and the changes observed at 72 hpi are similar to NGS as well as to poly-A tailing mediated RT-PCR validation analyses. Thus the expression of these miRNAs was regulated only during the development of blight symptom.
3.5.2. Validation of expression of novel miRNAs
We selected four novel miRNAs (Fig. 3A) for validation of their expression. These miRNAs had at least 10 raw read counts, and miRNA* sequence of three of these miRNAs (sly-miRPR11, sly-miRPR26 and sly-miRPR29) could be detected in the NGS data. Using Northern blot analysis (n = 2), sly-miRPR1, sly-miRPR26 and sly-miRPR29 could be detected in both the samples (Fig. 3B). The novel miRNAs, sly-miRPR11, sly-miRPR26 and sly-miRPR29 with detectable star sequences, were also subjected to PolyA tailing mediated RT-PCR analysis for further validation. All three were readily detected in both the samples and sly-miRPR29 exhibited significant change in expression level during the infection (Fig. 3C) which is similar to the pattern observed in NGS analysis. Also, expression of probable precursor sequences of two of these novel miRNAs (sly-miRPR11 and sly-miRPR29) was confirmed by RT-PCR analysis performed with RNAs from both control and infected plants (Fig. 3D).
3.6. Genes belonging to stress-related pathways are targeted by differentially expressed miRNAs
miRNAs mediate their function in the cell by triggering cleavage or translational inhibition of specific target mRNAs, with which it has sequence complementarity. We searched two online databases psRNATarget and TFGD, to find predicted targets of 67 differentially expressed known miRNAs. The databases predicted 7,025 putative targets cumulatively (6,576 miRNA–mRNA pairs in psRNATarget and 1,055 pairs in TFGD, 606 common between the two). The discrepancy in the number of targets predicted by two softwares was due to the difference in the scoring matrix of the two databases. To better understand the group of genes targeted by the differentially expressed miRNAs during Alternaria stress, GO-based functional annotation and enrichment analysis of the predicted targets were performed utilizing Hypergeometric test applying Benjamini & Hochberg FDR correction value ≤0.05, with all predicted targets. Under the molecular function category of the GO classification, genes showing kinase activity (GO:0016301), receptor activity (GO:0004872), transferase activity (GO:0016740), transporter activity (GO:0005215), ATPase activity (GO:0016887), catalytic activity (GO:0003824), oxidoreductase activity (GO:0016724), signal transducer activity (GO:0004871) were highly enriched as targets of the differentially expressed miRNAs. Under the biological process category genes involved in cell death (GO:0008219), RNA metabolic process (GO:0016070), tRNA processing (GO:0008033) were targeted by miRNAs and only thylakoid membrane related genes were found to be enriched for the cellular component category as targets (Supplementary Fig. S10). In further analysis of the 606 target mRNAs simultaneously predicted by both software, no specific pathway was enriched, however, ‘biosynthesis of secondary metabolites’, ‘biosynthesis of amino acids’ and ‘carbon metabolism’ pathways are highlighted, as more genes of these pathways seem to be targeted. It is worth mentioning that some amino acids are precursors of phytohormones and it has been noted earlier that hormone biosynthesis/signalling pathways were affected during the infection.
3.7. mRNA expression profiles in tomato in response to Alternaria stress
In order to detect the alteration in expression of genes during Alternaria-stress RNA-seq analysis was performed for two tomato cDNA libraries, Control 1 and Infected 1, constructed with the same RNA preparation that has been used for small RNA transcriptomics. Sequencing generated 75.92 and 78.34 million reads, and after removing the low-quality reads (<20 Phred score) and trimming off the adapter sequences, 70.27 and 72.56 million clean reads remained for Control 1 and Infected 1 samples, respectively. Mean read length obtained was 99 bp (maximum and minimum were 100 and 50 bp). The sequenced reads were aligned to the tomato reference genome (Sol Genomics Network), which showed 81.23 and 80.85% matched sequences for Control 1 and Infected 1 libraries, respectively. Next, these sequences were assigned to transcripts and a total of 23,011 and 22,997 assembled transcripts (identified by their respective gene ID) from Control 1 and Infected 1 samples, respectively, were obtained (Supplementary Table S8). Finally, differential gene expression analysis revealed 5,450 genes showed at least 2-fold change in expression level (P ≤ 0.05). This analysis also revealed that relatively more genes were up-regulated (52.53%) compared with 47.46% genes which were down-regulated (Supplementary Fig. S11, Supplementary Table S9). These DEGs were subjected to further analysis to identify the pathways they were representing.
3.8. Validation of mRNA transcriptome analysis data
To validate the NGS expression data, we randomly selected 18 differentially expressed mRNAs (log2FC at least ±1, P ≤ 0.05) and validated their relative expression levels with qRT-PCR, using gene-specific primers (Supplementary Table S10). Data presented in Fig. 5A show that 17 mRNAs were differentially expressed and the expression pattern matched with NGS data analysis, however, the extent of differential expression differed.
Figure 5.

Experimental validation of expression level of selected genes. (A) Quantitative RT-PCR analysis data of randomly selected Alternaria-responsive 18 mRNAs. (B) Pearson correlation scatter plot of comparisons of differential expression level as measured by NGS analysis (log2FC) and experimentally validated data (log2FC) for mRNAs and miRNAs. ‘r’ indicates the Pearson correlation coefficient.
3.9. Correlation of NGS analysis and validation data
The intensity of bands in miRNA Northern blots was determined using ImageQuant ID GEL quantification software, from which normalized fold change in expression of miRNAs was derived. Fold change values of mRNA expression from quantitative RT-PCR analysis was also collected. Correlation between the relative expression level detected by these validated data and NGS sequencing data were then calculated. We obtained a highly significant Pearson correlation coefficient of r = 0.84 (Fig. 5B), which strongly supported that our NGS transcriptome data were valid.
3.10. Functional and pathway annotation of Alternaria–stress responsive transcripts
To better understand the function of the DEGs (P ≤ 0.05), GO terms and pathway enrichment analyses (KEGG analysis) were performed. We found that 3,154 genes were involved with 17 major molecular functions, 62 genes were part of 3 different cellular components and 57 genes participated in 1 biological process (Supplementary Fig. S12A). The GO terms predominantly enriched midst the regulated genes were related to molecular function including ‘catalytic activity’ (GO:0003824), ‘transferase activity’ (GO:0016740) ‘kinase activity’ (GO:0016301), ‘oxidoreductase activity’ (GO:0016491) represented by 1382, 539, 265 and 272 DEGs. Among the ‘kinase activity’ group majority belonged to ‘protein kinase activity’ (GO:0004672, 211 genes), and ‘transferase activity’ group had majority genes belonged to ‘transferring phosphorous-containing groups’ (GO:0016772, 283 genes). Other GO terms that were enriched including ‘transmembrane receptor protein kinase activity’ (GO:0019199), ‘transmembrane receptor activity’ (GO:0004888), ‘receptor activity’ (GO:0004872), ‘chitinase activity’ (GO:0004568), ‘lipoxygenase activity’ (GO:0016165), ‘oxygen binding’ (GO:0019825), ‘antioxidant activity’ (GO:0016209) are related to pathways involved in a stress response. Genes associated with ‘extracellular region’ (GO:0005576), ‘photosystem’ (GO:0009521) and ‘thylakoid’ (GO:0009579) were most significant (represented by 30, 18 and 9 genes, respectively) among the cellular component GO terms. In the GO biological process class, there was a higher number of DEGs annotated to ‘response to stimulus’ (GO:0050896, 63 genes), more specifically ‘response to biotic stimulus’ (GO:0009607, 14 genes).
To elucidate the exact biological processes the DEGs may participate in during Alternaria stress, we analysed the allotment of a DEG to a particular KEGG pathway. We were able to allot 5,080 genes in 334 different pathways. Next, we performed enrichment analysis, which revealed 24 important pathways were significantly (P ≤ 0.05) enriched in response to Alternaria stress (Supplementary Fig. S12B). It was noticeable that genes associated with photosynthesis particularly photosynthesis–antenna proteins (PATH:ko00196) were significantly enriched among the DEGs, suggesting plant photosynthesis is affected during infection owing to the chlorosis and blight progression during the disease. In addition, genes involved in the biosynthesis of secondary metabolites (PATH: ko01110) and phenylpropanoids (PATH:ko00940), unsaturated fatty acids, zeatin, flavonoid, cutin suberin and wax, stilbenoid, diarylheptanoid and gingerol and steroids were enriched. Moreover, genes for metabolism of phenylalanine, porphyrin and chlorophyll, nitrogen, linoleic and alpha–linoleic acid and starch and sucrose were enriched. Also, DEGs involved in valine, leucine and isoleucine degradation, or apoptosis were enriched. And lastly, plant–pathogen interaction (PATH: ko04626) and plant hormone signal transduction (PATH: ko04075) pathways were enriched, signifying, substantial regulation of genes contributing in the modulation of plant hormone signalling and plant defence occurred during the infection.
3.11. Differential expression of pathogenesis/disease development-related genes occur during Alternaria-stress
A close inspection of genes belonging to a particular pathway that were differentially regulated during the infection revealed that genes, (i) involved in plant hormone biosynthesis/signal transduction for auxin, ethylene (ET), salicylic acid (SA), jasmonic acid (JA) and abscisic acid (ABA) were regulated, (ii) for ROS or NO generation/scavenging were significantly regulated and (iii) which may have direct role in stress response such as RLKs, NB–LRRs and WRKY transcription factors were also significantly regulated (Supplementary Table S9 and Supplementary Fig. S13).
Usually, necrotrophic pathogens are benefitted from SA-mediated hypersensitive response due to added cell death, and in a resistance response, SA pathway is down regulated. Isochorismate synthase is a major gene in SA biosynthetic process, which is usually upregulated during a resistance response to a necrotrophic pathogen infection and systemic acquired resistance, however, it was downregulated (log2FC = −2.5, although not P significant) in our compatible interaction study. Concomitantly, several Phenylalanine ammonia–lyase (PAL) genes, of the SA–biosynthesis secondary pathway, were significantly upregulated together with the expression of pathogenesis-related PR-1, and PR-5 (log2FC= +6.19 and log2FC = +5.49, respectively) genes. These data support the view that SA-mediated defence pathway was suppressed in this compatible interaction. Although a role of PAL genes in this process is unclear, probably SA-mediated cell death would still occur as these genes’ expression was increased, which might benefit the fungus during this susceptible interaction.14
During necrotrophic pathogen infection, the plant’s defence response consists of synergistic activation of ET and JA pathways.15 In line with this, the majority of the genes associated with these pathways were noticeably upregulated. Out of 106 genes associated with the ET pathway that were recognized, 76 were upregulated including 34 genes responsible for ET biosynthesis (1-aminocyclopropane-1-carboxylate synthase (ACS), 1-aminocyclopropane-1-carboxylate oxidase (ACO) and 43 genes (out of 62) responsible for ET signal transduction (ethylene-responsive transcription factors, ERFs). Moreover, 16 genes (among 23 genes detected) involved in jasmonic acid biosynthesis (Allene oxide cyclase and lipoxygenase) and signal transduction (Jasmonate ZIM domain, JAZ) were also found to be upregulated. Suggesting both ET and JA pathways were also activated as the host was continuously trying to resist the pathogen (Supplementary Fig. S13).
The role of Auxin in a stress response has recently been appreciated, and the hormone’s role in Alternaria–stress response is found to be complicated. Auxin conjugating genes such as GH3.8 were both up and down regulated. On the other hand, a majority of the repressors of auxin signalling pathway including Aux/IAA1, 2, 5, 7, 9, 10 and 11 were significantly downregulated, indicating the onset of Auxin signalling also occurred during the infection. We have also noticed that one of the marker genes for auxin signalling pathway, proteinase inhibitor II (Pin2), was markedly upregulated (log2FC = +10.03), supporting the view of activation of auxin signalling pathway during Alternaria-infection in tomato. However, 75% of the auxin effector genes SAUR were down-regulated. Therefore, a detailed analysis is required to comprehend the exact role of auxin in this process.
In plant defence response, ROS and NO are thought to regulate programmed cell death through the establishment of the hypersensitive reaction.16 Genes related to ROS generation/scavenging systems were highly upregulated including respiratory burst oxidase (RBOH), peroxidise (POD), glutathione peroxidase (GPX), catalase (CAT), nitric oxide reductase, glutathione s transferase, ascorbate peroxidase. In addition, two classes of detoxifying genes UDP-glycosyltransferase (UDPG) (36/66 genes) and ABC transporters (17/25 genes) were upregulated many folds, probably to detoxify toxins generated by A.solani. Regarding ABA signalling, Out of seven ABA receptors showing differential expression, six depicted highly elevated expression, suggesting, ABA-signalling was also activated.
The sensors of Ca2+ flux in plants in response to stress, including 16 calmodulin-like protein (CAM), calcium-dependent protein kinase (CDPKs), 2 CBL-interacting protein kinase (CIPK) were upregulated many folds, however, membrane proteins like sodium/calcium exchanger (NCX), Calcium/proton exchanger are down-regulated, implying calcium accumulation is favoured within the cell which may serve as a second messenger in host to activate downstream gene regulation.
There were 183 DEGs that were different kinds of transcription factors (TFs), such as the WRKY family (i.e. WRKY16, 23, 29, 30, 37, 27, 73), ERF family (i.e. ERF2, 1b,1a, 2a,7,10), NAC, basic leucine zipper (bZIP) family, GRAS family, basic helix loop helix (bHLH) family and heat stress transcription factor (HSF), and master regulator myeloblastosis protein (MYB) family. Among these DEGs almost all the WRKY transcription factors were found to be upregulated (35/36 genes), ERF (43/63), NAC (25/38), MYB (36/54) and HSF (5/6) followed the same trend. However, the majority of the genes belonging to the bHLH group of transcription factor were found to be down-regulated (23/31).
Plant transmembrane receptor-like kinases (RLKs), receptor-like proteins (RLPs) and intracellular nucleotide-binding site leucine-rich repeat (NB-LRR) genes are surveyors of pathogen-associated molecular patterns and effector molecules for initiating stress signalling. Very interestingly, 219/326 significantly differentially regulated such genes were upregulated, indicating active signalling was prevalent inside the infected tissues. It will be intriguing to decipher the role of highly differentially regulated disease resistance related NB-LRR genes in the EB disease biology.
3.12. Combined expression analysis of miRNAs and cognate target mRNAs confirms pathogenesis/disease development-related genes are majorly targeted during Alternaria–tomato compatible interaction
We integrated our miRNA and mRNA sequencing data to get relative expression level of miRNA–mRNA pairs. To impose more stringency in the analysis we have considered only 606 common targets, among these 450 could be detected in our transcriptome analysis. We have further filtered these possible targets based on predetermined cut-off value of log2FC at least ±1 (P ≤ 0.05) in the transcriptome dataset and obtained 202 target genes. 102 (50.4%) among these showed significant opposite expression level compared with the expression of cognate miRNA (Supplementary Table S11). The oppositely regulated targets included (i) signalling/sensor molecules such as calmodulin binding protein (Solyc02g079040) and 14 NB-LRR disease resistance genes, 13 of them were cc–nbs–lrr type, (ii) transcription factors including MYB, Squamosa promoter binding like (SBP), AP2-ethylene-responsive and GRAS family, (iii) Nucleoredoxin 2 (Solyc05g005460), a thioredoxin-disulfide reductase and a Glutaredoxin family protein (Solyc08g082590) which might have role in oxidation-reduction process, (iv) multiple RLKs and RLPs and (v) genes related to ubiquitination process such as U-box (Solyc05g008230) and F-box proteins (Solyc01g106510) (Supplementary Table S11). However, it must be noted here that we are considering only genes which are likely to be cleaved by miRNAs, apart from this, translational inhibition of gene expression by miRNAs is also important, which could not be accounted in the present analysis.
Further, filtering of the targets using more stringent target prediction criteria, such that the target and the miRNA have almost perfect complementarity throughout its length, we obtained a list of 15 stringent miRNA–mRNA pairs showing opposite expression pattern during Alternaria–stress (Fig. 6A). Tomato degradome analysis data (Supplementary Table S11) confirmed nine of these interactions, and our 5′-RLM RACE analysis also confirmed some of these interactions during stress (Fig. 6B, Supplementary Fig. S14). The majority of these targets has roles in stress-related gene or pathway regulation, suggesting miRNA-mediated regulation of transcriptome is an essential process in Alternaria–stress response in tomato.
Figure 6.

Scheme showing some of the key Alternaria-responsive miRNAs and their targets in tomato. (A) Regulated miRNAs with their corresponding targets which are inversely correlated in our NGS analysis. The arrows indicate positive regulation and the hammer head represents negative regulation. (B) miRNA cleavage sites in some mRNAs as determined by modified 5′-RLM RACE. Arrows indicate the position of the cleavage.
4. Discussion
4.1. miRNAs in Alternaria-disease biology
Recent publications have demonstrated that miRNAs are involved in plant defence. The advent of NGS technique has expanded the list of miRNAs discovered in plant and their differential expression in different plants exposed to various biotic stresses. These studies also include analysis of tomato miRNA regulation during biotrophic pathogen interaction, such as tomato–cucumber mosaic virus and tomato-ToLCNDV17; and necrotrophic interactions, such as tomato-Botrytis cineria4 and tomato-Fusarium.18 Essential role of tomato miRNAs targeting NB-domain protein in conferring resistance to Fusarium fungal attack18 has been established. However, which miRNAs and genes are precisely regulated during Alternaria-stress in tomato was not known. Hence in this study, we adopted high throughput sequencing approaches to performing a combined expression profiling analysis of miRNAs and mRNAs in the same sample to provide an enhanced understanding of potential miRNA regulatory network in tomato subjected to Alternaria-stress. In fact, an extensive search of databases only yielded one reference related to whole transcriptome analysis during plant–pathogen interaction, describing small RNA and mRNA transcriptome during rice–RSV interaction.11 Thus, comprehensive study of the miRNA and mRNA expression profiles and correlating miRNA–mRNA expression in tomato inoculated with and without A.solani is well timed.
We initiated this study with the identification of the optimum stage of disease development which was selected for isolating total RNA. We considered following factors, (i) the disease should be progressed sufficiently, (ii) the tissue should not be completely blighted as these tissues will contain more dead cells and degraded RNA and (iii) the plant should show prominent biochemical or molecular response towards the disease, as it will indicate activation of signalling components. Our observations prompted us to use leaves of stage 3 (∼72 hpi) of the disease (Fig. 1A). The tissues indeed showed heightened expression of marker genes, PR1b and Chitinase, for biotic stress response (Fig. 1F), confirming appropriate diseased samples were used for comparison purpose. Concurrently, we had ensured that (i) high-quality of RNA were used, (ii) no fungal RNA contaminated our samples and (iii) only high-quality sequence reads were analysed with sufficient stringency wherever required.
The recent publications on tomato miRNA transcriptome analysis also described a number of conserved and novel miRNAs, however, it is difficult to combine all these information to make a comprehensive database for comparison purpose. In the miRBase, only a few miRNAs are listed as originated from tomato. Thus to eliminate the probability of excluding any significant miRNA from our analysis, we have gathered information about all plant-specific miRNAs available in the miRBase, and tomato miRNAs in TFGD and TGRD databases for making a comprehensive list of miRNAs. This list was used for the identification of miRNAs in our dataset. A perfect match was named according to the name of the matching miRNA found in any of these databases; however, a miRBase match received the priority during naming. An unmatched but otherwise fulfilling pre-set criteria for identification of miRNAs was called as a novel miRNA, and its corresponding sRNA number was also looked for in the TFGD database. This arrangement has avoided unnecessary new naming of a miRNA with known sequence information and eased the tracking of already available information regarding the miRNAs identified here.
Overall analysis of small RNA data revealed two interesting facts, (i) during the infection the total reads number of 21 nt sRNAs increased and 24-nt population decreased markedly (Supplementary Fig. S4) and (ii) tomato genome encodes more types of Solanaceae/species-specific miRNAs compared with conserved miRNAs (Supplementary Table S2 and Supplementary Fig. S5). The heterochromatic siRNAs are 24-nt long, having a possible role in DNA methylation whereas miRNAs or secondary siRNAs are of 21 nt.19 Thus it will be interesting to investigate if the infection has caused reduced methylation at some specific loci leading to the expression of A.solani responsive genes.
We have meticulously analysed the expression of each member of a miRNA family. Members to a family were assigned based on the sequence similarity among miRNAs and assumed minor mismatches might have aroused due to the expression from different precursor sequences, and different products that might have generated due to the irregular processing of the same precursor are placed in a same family-member group. We believe this is the best method for identifying a family and its members. However, we have also noticed that the numbers of designated family members differed from the numbers reported in previous studies,20,21 although some of the abundant expresser families (miR166, miR168, miR156, miR 6027) were also identified in these reports.
The expression level of different members of the same miRNA family showed noticeable variation among them (Supplementary Fig. S6 and Supplementary Table S4). Interestingly, upon infection the expression pattern of isoforms also altered within a family (Supplementary Fig. S6 and Supplementary Table S4). Visible changes were noticed for miR167, miR171, miR393, miR319, miR397 and miR166 among the conserved miRNA families. In line with this observation, members of the miR166 family were found to have different expression pattern in wheat in response to powdery mildew infection and heat stress.5
Differential miRNA expression analysis indicated that both conserved and Solanaceae-specific miRNAs were differentially regulated during Alternaria stress, which is analogous to observations with other plant–pathogen interaction studies.4,18,22,23 Our stringent analysis demonstrated that majority of miRNAs were down-regulated upon Alternaria-stress. Similar, down-regulation of the majority of miRNAs were observed in tomato–Phytophthora infestans,24 brinjal–Verticillium dahliae6 interactions. However, during Botrytis cinerea, CMV25 and Fusarium oxysporum18 stresses tomato plant responded by up-regulating majority of miRNAs. Hence, it seems the pattern of regulating the expression of miRNAs during stress is dependent on a specific plant and pathogen interaction, and thus it is difficult to generalize a rule of regulation of miRNAs during stress.
Some recent studies conducted using different fungus and plants showed the importance of specific miRNAs in making a plant susceptible or resistant against a particular pathogen.4,18 miR166, miR169 and miR397 were reported to be down-regulated in susceptible soybean variety upon Phakopspora pachyrhizi infection.26 Our data showed that during tomato–Alternaria interaction miR169 and miR397 were down-regulated many fold but expression of miR166 was not regulated significantly. A close inspection of the miRNAs regulated as an effect of necrotrophic fungi Botrytis cinerea4 and A.solani (this study) in tomato we found that 13 miRNAs were commonly regulated by both the stresses in tomato. Eight miRNAs (sly-miR156d-3p, sly-miR6300, sly-miR156f, sly-miR482a, sly-miR482b, sly-miR160a and sly-miR168a/b were up-regulated and sly-miR396a-3p was down-regulated) showed similar trend of regulation among the two stresses, however, five miRNAs (sly-miR171e, sly-miR169b/d/e, sly-miR172a/b, sly-miR390a-5p and sly-miR399a) showed opposite regulation. A similar comparison of regulated miRNAs between biotrophic pathogen CMV-stress25 and Alternaria-stress in tomato showed 11 miRNAs to be differentially regulated in common. Among them, seven miRNAs showed the similar trend in regulation (sly-miR167c, sly-miR6024b, sly-miR168a/b were up-regulated, and sly-miR166k, sly-miR397, sly-miR408b-3p and miR395a/b were down-regulated in both the cases). However, remaining four miRNAs (sly-miR393a-3p, sly-miR390a-3p, sly-miR398a-3p and miR408b-5p) showed opposite regulation. Thus, it seems there are certain common response genes that are probably regulated by miRNAs during a biotic stress; however, the specific response is governed by regulation of a specific set of miRNAs, reinstating our previously stated notion.
In addition to known miRNAs, 60 novel putative miRNAs were also discovered (Supplementary Table S6). The novel miRNAs are in general low expressing as compared with known miRNAs. Although we were able to validate the expression of four arbitrarily chosen putative novel miRNAs using alternative techniques, further experimentations are essential to declare them as real miRNAs. However, the precursors of at least two of these novel miRNAs (sly-miRPR11 and sly-miRPR29) were detectable and showed variable expression in infected tissues, thus it is safe to conclude that these two miRNAs are biologically active.
4.2. Transcriptomics of EB diseased leaves
Profiling of differentially expressed transcripts during Alternaria-stress, and subsequent functional and pathway enrichment analyses (Supplementary Fig. S12A and B) provided a comprehensive view of response regulators during the compatible interaction. We have also delved into more profiling details to find out (i) which GO terms are represented by up and downregulated genes, and (ii) the expression status of genes of an enriched pathway. These analyses provided an enhanced perspective to the overall expression data.
Our data suggest that enriched GO-terms ‘chlorophyll binding’ and ‘tetrapyrrole binding’ were represented by down-regulated genes in the molecular function category. Under the cellular component category, the majority of the terms enriched were against down-regulated genes, which also included terms related to photosynthesis, more specifically photosystem reaction centre I and thylakoid. Moreover, pathway annotation also showed mostly down-regulated genes enriched the terms related to photosynthesis process, especially photosynthesis–antenna proteins, porphyrin and chlorophyll metabolism, starch and sucrose metabolism and carbon fixation (Supplementary Fig. S12B), further supporting photosynthesis was severely affected during Alternaria stress, which could be explained by chlorosis and blight symptom owing to the disease. Other plants are also known to be affected by down-regulation of photosynthesis during a pathogen attack,7,27 however, this could help the plant to cope the stress.
A biotic stress is perceived by the plant at the membrane, followed by initiation of a signalling cascade in which RLK, RLP and NB-LRR proteins play crucial roles, and subsequently, a set of stress-management-related genes are expressed. Our analysis suggested that multiple such proteins were differentially regulated. In line with this, the major categories that are GO-enriched and represented by mostly up-regulated genes belonged to ‘transferase activity’, ‘kinase activity’ and ‘catalytic activity’; and especially ‘phosphotransferase activity’ and ‘protein kinase activity’. This indicates, events like phosphorylation and de-phosphorylation increased in the cell during the stress, leading to the activation of various signalling intermediates thus altering signalling pathways. KO terms for plant pathogen interaction pathway have also been found to be enriched and represented by upregulated genes. These results prove that an active signalling cascade was primed during the stress. Accordingly, under biological process category terms related to response to stimuli and specifically biotic stimuli were enriched.
The diseased plant responds by activation of genes that help in scavenging the toxins produced inside the tissues or by producing secondary metabolites that might check the pathogen growth to cope the stress. An important category which was enriched among the up-regulated genes was ‘glutathione transferase (GST) activity’. GSTs are known for their ability to detoxify toxins, protect a plant from oxidative stress and regulating cell proliferation and death by interfering with MAPK pathway.28Alternaria-infection is associated with oxidative burst (Fig. 1), which could be induced by the necrotrophic pathogen to kill the cells, and cellular scavenging enzymes such as GSTs thus are activated to mitigate the stress. Upregulated genes were also involved in secondary metabolite biosynthesis pathways including phenylalanine and phenylpropanoid biosynthesis. Phenylpropanoid metabolism pathway generates diverse kind of secondary metabolites which can protect plants against pathogens. Thus it can be inferred, an increase in the expression of genes responsible for phenylalanine metabolism is providing enough precursor (phenylalanine) for phenylpropanoid metabolism, which in turn forms basic units of many secondary metabolite biosynthesis.29 In summary, genes involved in three crucial pathways such as activation of, stress-signal transduction, cellular detoxification machinery and metabolic pathways including biosynthesis of secondary metabolites were altered upon Alternaria infection in tomato.
4.3. Integrating miRNA–mRNA transcriptomics
Within a cell, if the level of a miRNA increases, decreased level of the cognate target mRNA indicates increased cleavage activity of the miRNA, on the other hand, a decrease in the level of miRNA results in increased level of target mRNAs. Hence, a transcriptome-wide combined analysis of miRNA and mRNA expression level was carried out to identify the miRNA–mRNA pairs which were regulated during tomato–A.solani compatible interaction. This has enabled us to understand which pathways and biological processes of the cell were most likely regulated by miRNAs during the stress.
We used considerably differentially expressed miRNAs to identify putative targets using two separate target prediction databases. A comparison between the GO-based functional enrichment of these predicted targets with the differentially expressed transcripts (Supplementary Figs. S10 and S12A) revealed the biological activities that were likely regulated by miRNAs during the stress. For example in both the analyses, (i) majority of the enriched genes were categorized under ‘molecular function’; (ii) activities like ‘catalytic’ and ‘transferase’ were most enriched, and (iii) ‘kinase activity’, ‘molecular transducer’, ‘signal transducer’ and ‘receptor activities’ were also substantially reflected. Such observations indicate that genes representing the above-mentioned ontologies are likely targeted by miRNAs. However, genes showing ‘oxidoreductase’, ‘UDP-glycosyltransferase’ and ‘oxygen binding’ activity were enriched only in regulated transcripts, indicating although genes involved in above processes were differentially expressed during Alternaria stress but this regulation was not due to the miRNA-mediated cleavage of transcripts. Similarly, ‘response to stimulus’ especially ‘response to biotic stimulus’ was enriched in the ‘biological process’ category only amid the regulated transcripts. On the contrary, ‘cell death’ and ‘RNA and ncRNA metabolism’ and ‘processing’ terms were enriched under the same category for the predicted targets only. These observations suggesting only a few specific biological processes were regulated by miRNAs during the stress.
Under the ‘cellular component’, genes associated to thylakoid were common between the two analyses, but no genes related to photosynthesis was enriched among the predicted targets although photosynthetic pathway was found to be affected as a whole during Alternaria stress, implying photosynthetic genes are majorly regulated transcriptionally and may not be significantly affected by miRNA-mediated silencing pathway.
During the infection ROS is overproduced (Fig. 1), thus we set out to detect whether genes related to ROS generation/scavenging pathway were indeed targets of miRNAs. Our analysis indicated that genes involved in oxidation-reduction processes and detoxification during oxidative stress were targeted by miRNAs and their expression was inversely correlated to that of targeting miRNAs, these include stu-miR398a-3p-Nucleoredoxin 2 (Solyc05g005460), sly-miR9518-glutaredoxin family protein (Solyc08g082590), sly-miR408b-3p-blue copper protein–plastocyanin (Solyc01g104400), sly-miR9474-5p-nudix hydrolase 2 (Solyc05g016690) and sly-miR169b-UDP-glucosyltransferase (Solyc06g062290).
We were able to validate some of the predicted targets by analysing recently published tomato degradome NGS data21,30 and 5′-RACE analysis (Fig. 6b). Among 606 common predicted targets, we found 44 mRNAs to be cleaved at the exact position as predicted in our analysis. Next, a closer inspection of transcriptomics data showed that 38 miRNAs formed 102 miRNA–mRNA interaction pairs and showed opposite regulation (Supplementary Table S11). It was noticed that majority of the 102 oppositely expressed targets with respect to their cognate miRNA, could be clustered into specific groups including transcription factors, NB–LRRs and ubiquitination related proteins. NB–LRR–miRNA interactions have been well documented18,31 and F-box proteins are known targets of miR393 and miR394.32 As late infection inflicts massive cell death, it is imminent that cellular general metabolic processes will be largely affected. We have shown earlier that several metabolism-related GO-terms were enriched, and miRNA–mRNA paired expression analysis revealed some genes involved in the basic metabolism of cell like Phosphofructokinase family protein, Citrate synthase, Fructose-bisphosphate aldolase, etc. were also targeted and expression was negatively affected (Supplementary Table S11).
Further stringent analysis by imposing more selective criteria has led to the identification of specific genes that are most likely targeted by miRNAs (Fig. 6). Many of these interactions are also validated in independent studies18,21,30,33,34 (Supplementary Table S11, Fig. 6b). Hence, in this study key genes and miRNA regulators responsible for mediating Alternaria–stress response in tomato has been identified, and we have confirmed Alternaria-stress-related regulation of miRNA expression as one of the important regulatory events that occur to reprogramme gene expression cascade to cope the stress.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Supplementary Material
Acknowledgements
This study was supported by a DBT grant to P.K. Authors acknowledge Bose Institute CIF for technical helps and are thankful to Dr. Sujoy Saha, IIVR, India, and Dr. Subrata Dutta, BCKV, India for assistance with pathogen studies. Sudhriti Ghosh Dostidar has helped in some data analysis.
Accession numbers
Conflict of interest
None declared.
Funding
This study was supported by a grant, BT/PR12942/AGR/36/652/2009, from Department of Biotechnology, Government of India to PK.
References
- 1. Peralta I.E., Knapp S., Spooner D.M.. 2005, New species of wild tomatoes (Solanum Section Lycopersicon: Solanaceae) from Northern Peru, Syst. Bot., 30, 424–34. [Google Scholar]
- 2. Chaerani R., Groenwold R., Stam P., Voorrips E.R.. 2007, Assessment of early blight (Alternaria solani) resistance in tomato using a droplet inoculation method. J. Gen. Plant Pathol., 73, 96–103. [Google Scholar]
- 3. Mallory A.C., Bartel D.P., Bartel B.. 2005, MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant cell, 17, 1360–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin W., Wu F.. 2015, Characterization of miRNAs associated with Botrytis cinerea infection of tomato leaves. BMC Plant Biol., 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xin M., Wang Y., Yao Y., et al. 2010, Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol., 10, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang L., Jue D., Li W., Zhang R., Chen M., Yang Q.. 2013, Identification of MiRNA from eggplant (Solanum melongena L.) by small RNA deep sequencing and their response to Verticillium dahliae infection. PLoS One, 8, e72840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Windram O., Madhou P., McHattie S., et al. 2012, Arabidopsis defense against Botrytis cinerea: chronology and regulation deciphered by high-resolution temporal transcriptomic analysis. Plant Cell, 24, 3530–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan G., Liu K., Kang J., et al. 2015, Transcriptome analysis of the compatible interaction of tomato with Verticillium dahliae using RNA-sequencing. Front. Plant Sci., 6, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manzo D., Ferriello F., Puopolo G., et al. 2016, Fusarium oxysporum f.sp. radicis-lycopersici induces distinct transcriptome reprogramming in resistant and susceptible isogenic tomato lines. BMC Plant Biol., 16, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Upadhyay P., Rai A., Kumar R., Singh M., Sinha B.. 2015, Microarray analyses during early stage of the tomato/Alternaria solani interaction. Genom. Data, 6, 170–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang J., Zhang F., Li J., Chen J.P., Zhang H.M.. 2016, Integrative analysis of the microRNAome and transcriptome illuminates the response of susceptible rice plants to rice stripe virus. PLoS One, 11, e0146946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han X., Yin H., Song X., et al. 2016, Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol. J., 14, 1470–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tang M., Mao D., Xu L., Li D., Song S., Chen C.. 2014, Integrated analysis of miRNA and mRNA expression profiles in response to Cd exposure in rice seedlings. BMC Genom., 15, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dempsey D.A., Vlot A.C., Wildermuth M.C., Klessig D.F.. 2011, Salicylic acid biosynthesis and metabolism. The Arabidopsis Book/American Society of Plant Biologists, 9, e0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu Z., An F., Feng Y., et al. 2011, Derepression of ethylene-stabilized transcription factors (EIN3/EIL1) mediates jasmonate and ethylene signaling synergy in Arabidopsis. Proc. Natl Acad. Sci. U. S. A., 108, 12539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y., Loake G.J., Chu C.. 2013, Cross-talk of nitric oxide and reactive oxygen species in plant programed cell death. Front. Plant Sci., 4, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pradhan B., Naqvi A.R., Saraf S., Mukherjee S.K., Dey N.. 2015, Prediction and characterization of Tomato leaf curl New Delhi virus (ToLCNDV) responsive novel microRNAs in Solanum lycopersicum. Virus Res., 195, 183–95. [DOI] [PubMed] [Google Scholar]
- 18. Ouyang S., Park G., Atamian H.S., et al. 2014, MicroRNAs suppress NB domain genes in tomato that confer resistance to Fusarium oxysporum. PLoS Pathogens, 10, e1004464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Axtell M.J. 2013, Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol., 64, 137–59. [DOI] [PubMed] [Google Scholar]
- 20. Cao X., Wu Z., Jiang F., Zhou R., Yang Z.. 2014, Identification of chilling stress-responsive tomato microRNAs and their target genes by high-throughput sequencing and degradome analysis. BMC Genomics, 15, 1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Candar-Cakir B., Arican E., Zhang B.. 2016, Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J., 14, 1727–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang W., Luan Y.. 2015, The advance of tomato disease-related microRNAs. Plant Cell Rep., 34, 1089–97. [DOI] [PubMed] [Google Scholar]
- 23. Chen J., Feng J., Liao Q., et al. 2012, Analysis of tomato microRNAs expression profile induced by Cucumovirus and Tobamovirus infections. J. Nanosci. Nanotechnol., 12, 143–50. [DOI] [PubMed] [Google Scholar]
- 24. Luan Y., Cui J., Zhai J., Li J., Han L., Meng J.. 2015, High-throughput sequencing reveals differential expression of miRNAs in tomato inoculated with Phytophthora infestans. Planta, 241, 1405–16. [DOI] [PubMed] [Google Scholar]
- 25. Feng J., Liu S., Wang M., Lang Q., Jin C.. 2014, Identification of microRNAs and their targets in tomato infected with Cucumber mosaic virus based on deep sequencing. Planta, 240, 1335–52. [DOI] [PubMed] [Google Scholar]
- 26. Kulcheski F.R., de Oliveira L.F., Molina L.G., et al. 2011, Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genomics, 12, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bilgin D.D., Zavala J.A., Zhu J., Clough S.J., Ort D.R., DeLucia E.H.. 2010, Biotic stress globally downregulates photosynthesis genes. Plant Cell Environ., 33, 1597–613. [DOI] [PubMed] [Google Scholar]
- 28. Laborde E. 2010, Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Different., 17, 1373–80. [DOI] [PubMed] [Google Scholar]
- 29. Vogt T. 2010, Phenylpropanoid biosynthesis. Mol. Plant, 3, 2–20. [DOI] [PubMed] [Google Scholar]
- 30. Karlova R., van Haarst J.C., Maliepaard C., et al. 2013, Identification of microRNA targets in tomato fruit development using high-throughput sequencing and degradome analysis. J. Exp. Bot., 64, 1863–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shivaprasad P.V., Chen H.M., Patel K., Bond D.M., Santos B.A., Baulcombe D.C.. 2012, A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell, 24, 859–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones-Rhoades M.W., Bartel D.P., Bartel B.. 2006, MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol., 57, 19–53. [DOI] [PubMed] [Google Scholar]
- 33. Jeong D.H., Park S., Zhai J., et al. 2011, Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell, 23, 4185–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baldrich P., Campo S., Wu M.T., Liu T.T., Hsing Y.I., San Segundo B.. 2015, MicroRNA-mediated regulation of gene expression in the response of rice plants to fungal elicitors. RNA Biol., 12, 847–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.