


Abstract
Methylome-wide association studies are typically performed using microarray technologies that only assay a very small fraction of the CG methylome and entirely miss two forms of methylation that are common in brain and likely of particular relevance for neuroscience and psychiatric disorders. The alternative is to use whole genome bisulfite (WGB) sequencing but this approach is not yet practically feasible with sample sizes required for adequate statistical power. We argue for revisiting methylation enrichment methods that, provided optimal protocols are used, enable comprehensive, adequately powered and cost-effective genome-wide investigations of the brain methylome. To support our claim we use data showing that enrichment methods approximate the sensitivity obtained with WGB methods and with slightly better specificity. However, this performance is achieved at <5% of the reagent costs. Furthermore, because many more samples can be sequenced simultaneously, projects can be completed about 15 times faster. Currently the only viable option available for comprehensive brain methylome studies, enrichment methods may be critical for moving the field forward.
INTRODUCTION
Cells in the human brain appear to survive and function for decades (1). Molecular changes to existing cells are therefore essential to effectively respond to altered neuronal inputs, interactions with support cells or environmental changes. DNA methylation, an archetypal cellular information storage mechanism, has been heavily implicated as a primary mechanism for stable activity-dependent transcriptional alterations within the central nervous system (2). Indeed, methylation appears critical for normal cell functioning and aberrant methylation has been associated with a wide variety of psychiatric disorders (3–5).
To distinguish between different forms, we follow the custom of studies of multiple methylation types to omit the middle ‘p’ (symbolizing the phosphodiester bond between ‘C’ and ‘G’) and write ‘mCG’ rather than ‘mCpG’. This simplifies notation as it avoids including the ‘p’ when referring to other methylation types. In the vast majority of somatic tissues, DNA methylation occurs almost exclusively at CpG dinucleotides (6–8). However, in mammalian brain two additional common forms of methylation exist. First, widespread methylome reconfiguration occurs during fetal and adolescent development, resulting in an increase of de novo cytosine methylation outside the CG context (mCH, where H = A, C or T) (9–11). It is likely that these alterations are, for instance, relevant to memory and learning (11–13). Second, hydroxymethylation (hmC) (14) involves enzymatic oxidation of the methyl group base by the ten–eleven translocases (TET1, TET2, TET3) and occurs predominantly in the CG context (15,16). Studies suggest that hmC broadly impacts brain function and neural plasticity (5,17,18). Examples include positive correlations between hmC levels and cerebellar development (5), the association of fear extinction (an important form of reversal learning) to a dramatic genome-wide redistribution of hmC within the infralimbic prefrontal cortex (19) and the memory deficits exhibited by TET1 knockout mice (20).
Because detailed biological knowledge linking specific methylation sites to phenotypes is lacking, methylome-wide association studies (MWAS) will be critical to identify such links (21–23). This implies screening all potential methylation sites in brain. Current microarray technologies do not begin to approach this scope of coverage. For example, commonly used arrays typically cover only a small proportion of the 28 million common CGs and almost completely miss mCH and hmC, forms of methylation that seem of particular relevance for neuroscience. The obvious risk of the current reliance on such low coverage screening methods is the potential to miss numerous signals of critical etiological importance.
Technologies using next-generation sequencing can offer a more comprehensive view of the brain methylome (21–24). The most exhaustive approach requires sequencing the entire genome twice (Figure 1A), beginning with whole genome bisulfite (WGB) sequencing. The sodium bisulfite treatment prior to sequencing converts non-methylated cytosines to uracil but leaves methylated cytosines intact (25), yielding methylation estimates as the percentage of unconverted cytosines. WGB sequencing cannot discriminate between mC and hmC. The genome therefore needs to be sequenced a second time, using a sample preparation such as TET-assisted bisulfite (TAB) sequencing (26) that leaves only hydroxymethylated sites unconverted. WGB/TAB also requires sequencing the entire genome at higher than usual coverage due to the bisulfite conversion (i.e. the lower diversity of the DNA samples after bisulfite conversion makes sequencing and alignment of reads more challenging). As a result, WGB is currently not economically feasible for the sample sizes required for MWAS (21).
Figure 1.

Overview of methods to assay the brain methylome. (A) The bisulfite methods each start with separate aliquots of genomic DNA. In the case of whole genome bisulfite (WGB) sequencing, sodium bisulfite treatment prior to sequencing converts non-methylated cytosines to uracil but leaves methylated cytosines intact. This provides an estimation of methylation based on the number of unconverted cytosines sequenced. TET-assisted bisulfite (TAB) sequencing sample preparation leaves only hydroxymethylated sites unconverted and provides a direct estimate of hydroxymethylation (hmC). WGB does not discriminate between mC and hmC. However, by subtracting the TAB hmC estimates from the WBS data we obtain estimates of mC. Further distinction between (h)mCG and (h)mCH is achieved by examining the sequence context of each site. (B) With our enrichment panel, genomic DNA is fragmented and fragments are then captured by proteins or other molecules with high affinity for methylated DNA. Next, the non-methylated genomic fraction is removed and the methylation-enriched fraction sequenced. Different sample preparations will enrich for different forms of methylation. Our panel assays mCG using a methyl-CG binding domain (MBD) protein. The MBD protein strictly captures fragments that have methylated CG sites. To assay mCH, we used methylated DNA immunoprecipitation (MeDIP) on DNA fragments that were not captured by the MBD protein. We labeled this approach MBD-DIP. While anti-mC antibodies bind both mCG and mCH, pre-depletion of mCG leads predominantly to capture mCH. For hmC we used a separate aliquot of the genomic DNA, and used selective chemical labeling and capture (hMe-Seal) approach.
Enrichment methods represent an ‘older’ group of methods used to assay the methylome. With this approach, genomic DNA is first fragmented and the methylated fragments are then bound to proteins (27,28) or other molecules with high affinity for methylated DNA. The non-methylated fraction is washed away and the methylation-enriched fraction sequenced (28–31). Thus, whereas WGB/TAB is inefficient because it covers the entire genome, of which the majority is not methylated (32), enrichment methods sequence only the methylated portions. Successful examples of MWAS performed with enrichment methods already exist (3). The goal of this paper to further optimize enrichment assays and study whether they allow comprehensive, adequately powered and cost-effective genome-wide investigations of all three common methylation forms in brain.
Optimized enrichment panel
Several studies have compared methylation enrichment methods with WGB sequencing (31,33,34). However, a wide variety of enrichment methods exist that vary considerably in their quality, where performance may depend critically on the use of optimized protocols (35). Furthermore, evaluations of enrichment methods have been limited to mCG and little is known about their potential to capture hmC and mCH. Finally, rather than providing methylation information about individual sites, enrichment methods assay the total amount of methylation for a locus that is about the size of the extracted fragments. It is critical that this property is taken into account when evaluating these methods. For this study we first selected the best assays to create a panel of enrichment methods to assay the three common forms of DNA methylation and implemented additional optimizations to the selected laboratory protocols. Next, we evaluated the performance of this panel through comparisons with WGB and TAB sequencing while taking the local property of enrichment methods into account.
Figure 1B provides an overview of the enrichment methods that we evaluate with WGB and TAB sequencing (Figure 1B). To assay mCG we use a methyl-CG binding domain (MBD) protein. Specially, we used the DNA-binding domain of human MBD2 protein that has been shown to generate the highest quality data compared to other MBD proteins (35). Fragments with few methylated sites are more difficult to capture (31). Therefore, we increased the sensitivity of MBD enrichment to DNA fragments harboring very few mCG sites by using a low salt elution buffer.
The MBD2 protein strictly binds methylated cytosines in the CG context. To assay mCH, we used methylated DNA immunoprecipitation (MeDIP) on the DNA fragments that were not captured by MBD enrichment. We labeled this approach MBD-DIP. While anti-mC antibodies are able to bind both mCG and mCH, the pre-depletion of mCG now predominantly leads to capture of mCH. In contrast to the standard (single-step) MeDIP, this two-step approach allows us to assay mCH status of fragments not containing mCG. Furthermore, in this article we show that the sensitivity to detect both mCG and mCH is improved substantially using MBD-DIP rather than standard MeDIP alone.
In the third component of our panel, we enriched the hmC fraction via hMe-Seal (36), using a second aliquot of genomic DNA. The hMe-Seal method installs modified glucose moieties at hmC residues, which in turn are biotinylated to allow for affinity purification. This selective labeling and chemical capture approach compares favorably to hmC enrichment methods using proteins or antibodies (37). We substituted the enzyme in the manufacturer's kit and augmented incubation times to improve labeling performance.
Two additional optimization steps were used for the enrichment assays during library construction. First, we used the shortest possible DNA fragments compatible with the sequencing platform (∼140 bp) to further increase sensitivity (i.e. small, light fragments are easier to bind) and resolution. Second, because precision in bisulfite sequencing depends on the number of sequenced bases covering each putative methylation site, long paired-end reads are optimal for this assay. In contrast, the number of sequenced fragments determines the precision of enrichment methods. For this approach single-end and short reads are better because these libraries are both less expensive and faster to sequence thereby maximizing the number of sequenced fragments for a given budget.
MATERIALS AND METHODS
Human sample
Postmortem brain tissue (Brodmann area 10) was obtained from the Douglas Bell-Canada Brain Bank (DBCBB) (http://www.douglasbrainbank.ca). The tissue originated from a 49-year old Caucasian female. It was dissected by neuropathology technicians, snap-frozen and then stored at −80°C. The DBCBB obtained informed consent from next of kin.
DNA isolation
A total of 100 mg of human brain tissue was homogenized in Buffer RTL (Qiagen, # 79216) with a 1 ml syringe fitted with a 20 G needle. Genomic DNA was extracted from the homogenate using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen, # 80224). Genomic DNA from sorted nuclei was extracted using the Gentra Puregene Tissue Kit (Qiagen, # 158667). Purity and concentration of extracted DNA was assessed by NanoDrop.
Nuclear isolation and sorting
Using protocols described elsewhere (38,39) we isolated the nuclei of neurons from other cells (mainly glial) present in brain tissue. Post-mortem tissue (250 mg) was placed in 5 ml of lysis buffer (0.32 M sucrose, 5 mM CaCl2, 3 mM magnesium acetate, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 10 mM Tris–HCl, 1 mM dithiothreitol (DTT), 0.1% v/v Triton X-100) and thawed and dounce homogenized for 1 min on ice. Homogenate was then placed on top of a 9 ml sucrose cushion (1.8 M sucrose, 3 mM magnesium acetate, 1 mM DTT, 10 mM Tris–HCl) in ultracentrifuge tubes and centrifuged at 121 200 × g for 3 h at 4°C. The supernatant and layer of cellular debris was removed by pipette and the nuclear pellet dissociated by addition of 500 μl of 1× phosphate buffered saline (PBS) + 1% v/v bovine serum albumin (BSA) with a 20 min incubation on ice without agitation. Following dissociation, the nuclear pellet was fully re-suspended by gentle pipetting. A total of 25 μl of unlabeled nuclear suspension was retained for use as unstained control. A total of 25 μl of nuclear suspension was incubated with mouse IgGk-PE antibody (EMD Millipore, #FCMAB230P, 1:120) as negative control. The remaining nuclear suspension was incubated with Milli-Mark Anti-NeuN-PE antibody (EMD Millipore, # FCMAB317PE, 1:100). All samples were diluted with 1× PBS + 1% v/v BSA and incubated for 1 h at 4°C with end-over-end rotation. After incubation, nuclei were concentrated by centrifugation at 400 × g at 4°C for 4 min, supernatant discarded and re-suspended in 500 μl 1× PBS + 1% v/v BSA. Stained nuclei were immediately transported for sorting on ice and protected from light. Fluorescence-activated cell sorting (FACS) was performed at the VCU Massey Cancer Center Flow Cytometry Shared Resource. Following FACS, PE+ and PE− samples were re-analyzed by FACS to assess purity.
Enrichment panel
DNA fragmentation
Genomic DNA was sonicated to 100–150 bp using a Covaris S2 ultra-sonicator (10 cycles, 1 cycle per burst, 60 s, 10% duty cycle, intensity 5.0). Multiple sonications were performed for each sample (2 μg DNA maximum per 100 μl fragmentation). The fragmented aliquots from each sample were pooled together prior to any downstream assays.
MBD
We used the MethylMiner™ Kit (Invitrogen) to enrich for mCG via affinity purification with a MBD2 protein. For each sample, 15 μl of stock Dynabeads® M-280 Streptavidin (10 μl of beads per 1 μg of DNA input) was coupled to 5.25 μg of MBD-Biotin Protein according to the vendor supplied protocol. Prepared MBD-beads were added to each input sample of 1.5 μg of fragmented DNA and adjusted to a final volume of 200 μl in 1× bind/wash buffer. The MBD-beads were incubated with DNA on an orbital shaker at 650 rpm for 1 h at room temperature in a 96-well 1.2 ml square well plate. Following incubation the plate was placed on a magnetic rack and the supernatant containing non-captured, mCG-depleted DNA was retained for MBD-DIP. Beads were then washed three times by addition of 200 μl of 1× bind/wash buffer followed by 3 min incubations at room temperature on an orbital shaker at 650 rpm. Methylated DNA was eluted from the beads with three treatments with 200 μl low salt elution buffer (12.5% high salt elution buffer + 87.5% low salt elution buffer [v/v]; 500 mM NaCl final), following the same incubation paradigm as the wash steps. The combined 600 μl mCG-enriched eluate was purified by ethanol precipitation.
After optimization, we have empirically determined that a low salt elution improves the specificity of the assay for loci with moderate numbers of CpG sites, giving better methylome-wide representation (40). Additionally, shearing genomic DNA to as small fragments as the downstream sequencing protocol allows (100–150 bp) further increases both the resolution and sensitivity of the MBD enrichment assay (i.e. small, sparsely methylated fragments require less energy to be captured than larger fragments).
MBD-DIP
The mCG-depleted DNA fraction was retained after MBD enrichment and purified by ethanol precipitation. MeDIP was used to enrich for mCH from the mCG-depleted fraction of each sample. We used the MagMeDIP kit (Diagenode, # C02010021) with 1.0 μg of mCG-depleted DNA as input. Briefly, input DNA was heat denatured and incubated with anti-mC antibody and magnetic beads overnight at 4°C with end-over-end rotation. After incubation, beads were washed and mCH-enriched DNA liberated with proteinase K treatment. Reagent volumes and concentrations were prepared according to the vendor supplied protocol. The crude mCH enriched fractions were cleaned by column purification (ChIP DNA Clean & Concentrator, Zymo, # D5201) prior to library generation.
hMe-Seal
Selective chemical labeling and enrichment of hmC (hMe-Seal) (36) was performed using components of the Hydroxymethyl Collector™ kit (Active Motif). We substituted the enzyme in the kit with T4 β-glucosyltransferase from New England Biosciences (# M0357) to improve labeling performance. In our experience, using high-quality and high-activity enzyme is critical for successful hmC enrichment. A total of 1.5 μg of 150 bp fragmented gDNA was used as input for each assay. Briefly, T4 β-glucosyltransferase (10 U per sample, 16 h incubation at 30°C) was used to selectively label hmC residues with 150 μM final UDP-azide-glucose. Each azo-glucosylated hmC was next biotinylated via dibenzocyclooctyl click chemistry by addition of the provided Biotin Conjugate Solution (likely DBCO-S-S-PEG3-Biotin at 150 μM final) with a 2 h incubation at 37°C. After biotinylation, the labeled DNA was column purified, enriched with paramagnetic streptavidin beads, washed and eluted following the vendor supplied protocol, with the substitution of end-over-end rotation with agitation on an orbital shaker at 700 rpm in a 96-well 1.2 ml square well plate. In an independent study (37) this chemical capture-based approach compared favorably to hmC enrichment methods using proteins or antibodies.
MeDIP
MeDIP was performed with 1.0 μg fragmented gDNA using the MagMeDIP Kit (Diagenode, # C02010021). Capture protocol, purification, library preparation and sequencing were carried out identically to that of the MBD-DIP samples.
Sequencing
The MBD and hMe-Seal enriched fractions are used to create indexed sequencing libraries using the TruSeq Nano DNA HT Library Prep Kit (Illumina). For MBD-DIP and MeDIP enriched fractions, the Accel-NGS Methyl-Seq DNA Library Kit (Swift Biosciences) was used to generate indexed libraries directly from single-stranded DNA. Libraries were size-selected using SPRI beads to obtain a mean insert size of 150 bp. Libraries were pooled and 75-cycle single-end sequenced on an Illumina NextSeq 500 using v2 chemistry.
Data processing
Reads were aligned (build hg19/GRCh37) with Bowtie2 (41) using a seed-and-extend approach combined with local alignment while allowing for gaps. Specifically, we used a 20 bp seed with zero mismatches. Rather than considering the entire read, local alignment was used to improve sensitivity by finding the maximum similarity score between the reference sequence and a substring of the extension that may be ‘trimmed’ at both ends. Gaps were allowed to account for small indels.
We quality controlled (QC) multi- and duplicate-reads. Reads often map to multiple genomic locations but in most cases a single alignment can be selected because it is clearly better than the others. In the case of multi-reads, multiple alignments are about equally good. When Bowtie2 encounters a set of equally good alignments, it uses a pseudo-random number to select one primary alignment. Duplicate-reads are reads that start at the same nucleotide positions. When sequencing a whole genome, duplicate-reads often arise from template preparation or amplification artifacts. In our context of sequencing an enriched genomic fraction, duplicate-reads are increasingly likely to occur because reads align to a much smaller fraction of the genome. In instances where >3 (duplicate) reads start at the same position, we reset the read count to one implicitly assuming that these reads all tagged a single clonal fragment.
A natural way to quantify enrichment is to count the number of fragments covering a cytosine. In contrast to bisulfite sequencing where precision depends on the number of sequenced bases covering each putative methylation site, the number of sequenced fragments determines the precision of enrichment methods. For this reason, single-end libraries are in principle more cost-effective then paired end libraries. However, with single-end libraries the fragment sizes are not observed. Counting the number of reads covering the putative methylation site instead, seriously underestimates coverage as the fragment that generated the read is usually longer than the read. To remedy this, we estimated the fragment size distributions from the empirical sequencing data using isolated cytosines (42). To evaluate this approach, we showed that methylation estimates obtained from paired end data (where the fragment size distribution is observed) are almost identical to those obtained from single end data with our estimator (correlation was 0.999). The estimated fragment size distributions is used to calculate the probability that a sequenced fragment will cover the cytosine under consideration. Coverage for each cytosine was then calculated by taking the sum of probabilities for all fragments aligning within proximity of the cytosine. For example, this probability is 1.0 for fragments with reads starting within one read-length of the cytosine, but is ≤1.0 for fragments with reads starting more than one read-length away.
Coverage is also affected by the total number of used reads per sample that is a function of sequencer loading and output, rather than differences in methylation. Therefore, coverage estimates were standardized using the total number of reads that remained after quality control.
Bisulfite sequencing
DNA fragmentation
Genomic DNA was sonicated to 550 bp using a Covaris S2 ultra-sonicator (one cycle, two cycles per burst, 45 s, 10% duty cycle, intensity 2.0). Multiple sonications were performed for each sample (2 μg DNA maximum per 100 μl fragmentation). The fragmented aliquots from each sample were pooled together to meet the input needs of both WGB and TAB, prior to any downstream assays.
Bisulfite and TET1 treatment
For WGB sequencing the fragmented DNA was bisulfite treated using reduced conversion times with the EZ DNA Methylation-Lightning™ Kit (Zymo Research) to convert non-methylated cytosines to uracil. For TAB (43) sequencing we used the WiseGene kit to treat the fragmented DNA with T4 β-glucosyltransferase (16 h at 30°C) to protect hmCs from oxidation by installation of a glucose moiety. Treatment with TET1 then oxidized only mCs to 5-carboxylcytosine. Finally, the TET1-oxidized DNA is bisulfite treated using the EZ DNA Methylation-Lightning™ Kit (Zymo Research) to convert non-hydroxymethylated cytosines to uracil. To assess conversion and protection rates, fragmented genomic DNA was spiked with commercial methylated and hydroxymethylated non-mammalian control DNAs (WiseGene). The mC control consisted of SssI treated lambda DNA, which should theoretically contain 100% methylated CpGs and non-methylated non-CpG cytosines. In reality, we observed a 96.3% methylation level for CpGs in the mC control and likely the result of incomplete methylation during the production of the control. The hmC control consisted of a 1.64-kb amplicon from pUC19, intended to contain 100% hydroxymethylcytosines at all locations. The observed hmC level for the hmC control was also reduced at 91.1% but is not unexpected considering past evidence of contamination of commercial 5-hmCTP with dCTP (44,45). Conversion and protection rates are presented in Supplementary Table S1.
Sequencing
We used the Accel-NGS™ Methyl-Seq DNA Library Kit (Swift Biosciences) to create indexed Illumina libraries directly from the bisulfite and TET1-oxidized/bisulfite converted DNA. Libraries were pooled with PhiX (15%) to compensate for lowered cytosine signal and 2 × 150 bp paired-end sequenced on an Illumina NextSeq 500 using v2 chemistry.
Data processing
BS-Seeker2 (46) was used to align data and call methylation levels. BS-Seeker2 converts both reads and reference to align reads (build hg19/GRCh37) in a three-letter base space using Bowtie2 (41). We used local (i.e. removal of terminal mismatches from the reads) and gapped alignment (e.g. allowing small indels) to increase mapping rates (46). The local alignment algorithm also automatically truncates adapter sequences. The maximum number of mismatches allowed per read was three. Only successfully mapped read-pairs were used to call methylation levels with correction for overlaps between read-pairs. To avoid unreliable estimates, for the bisulfite data we used methylation calls that were based on five or more sequencing reads.
Analyses
Calculation of methylation sum at fragment-sized loci
Enrichment methods assay loci that are about the size of the extracted fragments. We used our bisulfite data to characterize methylation patterns for these fragment-sized loci (Figure 2). For example, the total amount of methylation of a target site Y was calculated as a weighted sum of the methylation at all sites in the region surrounding Y, with weights for each site equal to the probability that fragments covering site X also cover that neighboring site. For example, whereas sites close to X will have a weight close to one because they will almost always be covered by the same fragments, the weights will be zero for sites located at a distance larger than the maximum fragment size. To calculate the weights we need the fragment size distribution, which we estimated empirically using a method outlined elsewhere (42).
Figure 2.

Methylation patterns at enrichment loci. Enrichment methods assay loci that are about the size of the extracted fragments. We used our bisulfite data to characterize the methylation patterns at these loci. For example, the total amount of methylation of a target site Y (or methylation sum) was calculated as a weighted sum of the methylation at all sites flanking Y. The weight for a neighboring site i was equal to the probability that a fragment containing site Y also covered site i. Thus, whereas sites close to Y will have a weight close to one because they will almost always be covered by the same fragments, this probability will be zero for sites located at a distance larger than the maximum fragment size. Calculation of the weights/probabilities requires the fragment size distribution, which was estimated using a method outlined elsewhere (42). (A) The total number of putative methylation sites was comparable for mCG and hmCG (median ∼5 sites per fragment). For mCH there were many more sites (∼74) per fragment as this includes all cytosines outside the CG context. (B) Mean methylation levels per fragment showed a bimodal pattern for mCG and hmCG, with a clear inflection point (vertical line) separating the two peaks. The second peak for mCG suggests that if methylation occurs, it tends to be substantial (∼0.7–0.9 range). In contrast, for hmCG the second peak suggests much more modest levels of methylation (∼0.2–0.5 range). For both mCG and hmCG, the first peak was located at zero suggesting that a proportion of fragments did not contain methylated sites. For mCH we observed very low levels of methylation. Inspection of region close to zero (insert) showed that mCH distribution was unimodal (i.e. no peak at zero). This implies that almost every locus contained some mCH methylation. (C) Variance was very low for all three forms of methylation, suggesting sites across the fragment-sized loci have similar methylation statuses. (D) The sum of methylation of all sites within the fragment-size loci was largest for mCG (median 2.4). For hmCG the methylation sum was much smaller (0.9). Although methylation levels outside the CG context were low, mCH methylation sum still reached substantial levels (1.9) due to the much larger number of possible methylation sites (Figure 3A).
Validation of enrichment methods using bisulfite data
To validate the enrichment methods we calculated the sensitivity (proportion of methylated loci correctly detected by enrichment methods), specificity (proportion of non-methylated loci correctly identify by enrichment methods) and the overall agreement (proportion of times the enrichment methods arrived at the same conclusion regarding the methylation status as the bisulfite data). For this purpose, the bisulfite data were first used to determine whether loci were methylated or non-methylated. For this purpose we plotted the mean methylation levels of all loci (Figure 2B). These methylation levels showed a bimodal pattern for mCG and hmCG. The first modus was located at zero suggesting that a proportion of fragments did not contain methylated sites. We estimated the inflection point (i.e. minimum value between the two modi) using the R package ‘inflection’. For mCH, distribution was unimodal (i.e. no peak at zero) as almost every locus contained some methylated sites. To determine mCH agreement we used an arbitrary threshold of one, meaning the locus had to have at least one fully methylated site to be considered methylated.
Sensitivity/specificity/agreement was calculated as the mean of the four possible duplicate combinations (e.g. enrichment assay 1 or 2 versus bisulfite assay 1 or 2). Importantly, bisulfite data is not perfectly reliable and will arrive at erroneous conclusions regarding the methylation status for a subset of the loci. Therefore, to obtain a benchmark of comparison for the enrichment results, we calculated the sensitivity/specificity/agreement for the bisulfite duplicates. For the bisulfite data we used methylation calls that were based on five or more sequencing reads. This yielded conservative estimates of the relative performance of the enrichment methods, as the bisulfite methods were not penalized for sites omitted for not passing this QC filter.
Methylation profiles across genomic features
We studied methylation across a variety of genomic features downloaded from the UCSC genome annotation database. Using the method described in the previous paragraph we first classified loci as methylated versus non-methylated. Next, we calculated odds ratios to study whether sites located in the studied feature were more likely to be methylated compared to sites not in this feature. In these analyzes, it is important to select the genomic region used for comparisons carefully. To illustrate the reasoning, assume testing whether CGs in exons are methylated using the ‘rest of the genome’ as the background. Assume that methylation is more likely to occur in repeats, which make up a large part of the genome. Under this assumption we may observe relatively lower methylation rates in exons. However, this finding will be driven by the fact that the repeat rich background against which exons are tested shows high methylation levels. Furthermore, all features tested against this background would be affected and suggest relatively lower methylation rates. Although due to the non-experimental nature we can never completely rule out such confounders, the choice of a proper background is important to reduce dependency between comparisons. We distinguish three scenarios: (i) features that do not overlap with other features, (ii) features that are nested in other features, (iii) features that partially overlap with other features.
Features that do not overlap with other features were tested against the rest of the genome from which all tested features were excluded to reduce dependence and select a ‘reference’ category. For a feature A that is nested in feature B, we used the part of feature B that was not overlapping with feature A as the background. For example, exons are nested in genes. To test for enrichment of methylated sites in exons, we tested all CG/C's in exons versus all CG/C's that were in genes minus the exon CG/C's. To determine which features were nested, we calculated Index(A⊆B) defined as (number overlapping bases features A and B)/(total number of bases feature A). If feature A is, for example, completely nested in feature B: Index(A⊆B) = 1. Thresholds used to determine nesting was Index(A⊆B) > 0.9. Finally, features A and B may only partially overlap (e.g. although conserved regions may overlap with genes, this is not always the case). To handle this scenario we first created three separate categories (i) regions where A and B overlap, (ii) regions covered by A but not overlapping with B and (iii) regions covered by B but not overlapping with A. Next, we choose one category as the background and the other two categories are tested against this reference category. To determine overlap we determined whether either 0.5 < Index(A⊆B) < 0.9 or 0.5 < Index(B⊆A) < 0.9. A matrix of values for Index(A⊆B) as well as the full list of tested features and their background is provided in Supplementary Table S5.
RESULTS
Descriptive statistics (Supplementary Tables S1 and 2) show that for bisulfite (WGB/TAB) sequencing we obtained a median of 774 million 150 bp reads (387 million read pairs) per sample, and for the enrichment methods 82.9 million 75 bp reads per sample. After bisulfite sequencing, 66.9% of the read pairs mapped successfully with a mappable mean read depth of 27.3×. The alignment rate for the enrichment methods was 99.0%. For the bisulfite methods, non-methylated cytosines showed a bisulfite conversion rate of 99.3% (WGB/TAB) and our normalized hmC protection rate was 91.4% (TAB). Both our bisulfite coverage and conversion rates were comparable to, or better than, those reported elsewhere for studies of post-mortem brain samples (9,47).
Our bisulfite data indicated substantial amounts of both mC and hmC in the CG context (Supplemental Figure S1). This underscored the need to subtract TAB from WGB estimates to determine correct mCG levels. Outside the CG context, only a very small proportion of the ∼1 billion cytosines were methylated. In agreement with previous findings (9), non-CG methylation was predominantly of the mCH rather than hmCH type. Therefore, in the remainder of this article we only consider mCH. Furthermore, to avoid reducing the reliability of the mCH estimates caused by subtracting the TAB hmCH estimates that were typically close to zero (i.e. often subtracting ‘noise’), we estimated mCH levels directly from the WGB data.
Enrichment methods extract fragments with one or more methylated sites. The number of sequenced fragments covering a putative methylation site is then used to estimate the amount of methylation. Thus, rather than providing methylation information about a single site, enrichment coverage is affected by the total amount of methylation of a locus that is about the size of the extracted fragments. We labeled this quantity the ‘methylation sum’. We used our bisulfite data to characterize the methylation sum and other descriptive information across all possible fragment-sized loci (Figure 2 and Supplementary Table S3) as described in the ‘Materials and Methods’ section. Because bisulfite estimated methylation can differ across regions depending on number of sites and absolute methylation level, we get a different methylation sum for each region. The genome-wide distribution of the regional methylation sums are plotted in Figure 2D. The median of all methylation sums was 2.4 for mCG and 0.9 for hmCG. Although only a small proportion of cytosines were methylated outside the CG context, the median CH methylation sum was comparable at 1.9 due of the large numbers of cytosines per locus.
To evaluate our enrichment panel we calculated sensitivity (proportion of methylated loci correctly detected), specificity (proportion of non-methylated loci correctly detected), and overall agreement (proportion of loci correctly classified as methylated or non-methylated). For this purpose we classified loci as methylated/non-methylated based on the presence/absence of methylated sites in the fragment-sizes loci as described in the ‘Materials and Methods’ section. Next, we calculated the sensitivity/specificity/agreement of the enrichment methods by comparing methylation status called from the bisulfite data versus methylation status as estimated from the enrichment data. To avoid choosing a single arbitrary threshold for classifying methylation status in the enrichment data, we considered a range of enrichment coverage cut-offs. For example, a coverage threshold of one means that we need at least one read to call a locus methylated. There are multiple ways to motivate the choice of the threshold. For each evaluation metric, we took the mean of the four possible duplicate combinations (i.e. enrichment assay 1 or 2 versus bisulfite assay 1 or 2). This method will underestimate the sensitivity/specificity/agreement of the enrichment methods because we do not know the true methylation status since the bisulfite data will incorrectly classify a subset of the loci. For this reason we also calculated the sensitivity/specificity between bisulfite duplicates (see horizontal dashed lines in Figure 3). These bisulfite duplicates provide and a benchmark against which to compare the observed correspondence between bisulfite and enrichment results.
Figure 3.

Methylome-wide coverage of enrichment methods. To study sensitivity and specificity (methylome-wide coverage) of our enrichment panel, we first used our bisulfite data to classify loci as methylated or non-methylated. (A) mCG results (MBD) showed that sensitivity decreases and specificity increases, as the coverage threshold is heightened. As methylated loci were much more abundant than non-methylated loci, the overall agreement between bisulfite and MBD closely followed sensitivity. The sensitivity of MBD approached that of the bisulfite methods at a coverage cut-off of 0.5, while retaining higher specificity. The MBD sensitivity and specificity approached equivalency at an enrichment coverage threshold of about 0.9. (B) The sensitivity and specificity of hmCG capture by hMe-Seal was very similar to TAB-seq at a threshold of ∼2. At a threshold of ∼5, sensitivity and specificity were approximately equal for the enrichment method. (C) To avoid any possible bias due to imperfect depletion for mCG, indices for mCH were calculated using only cytosines (∼80 million) located more than the maximum fragment size away from CG dinucleotides. Sensitivity was generally lower but specificity higher for MBD-DIP compared to the bisulfite method. Like mCG, the pattern of results for mCH indicates a coverage threshold of 0.5 achieves relatively good sensitivity while retaining acceptable specificity, with sensitivity and specificity equivalent at a threshold of about 0.9.
Results (Figure 3) showed that the sensitivity of enrichment approximated the sensitivity of the bisulfite methods, and that specificity was generally better for enrichment compared to bisulfite methods. There are multiple ways to choose a specific coverage threshold. One option is to use the point where sensitivity equals specificity and those values are reported in the legend of Figure 3. However, in the main text we will use the coverage threshold where the enrichment methods approximate the sensitivity/specific of the bisulfite method. For mCG, at a coverage cut-off of 0.5, the ratio of the specificity/sensitivity of MBD relative to the bisulfite method was 0.91/1.09. Relative to the bisulfite method, the overall agreement between MBD and bisulfite was 0.92. Agreement counts the number of times the enrichment and bisulfite method classified methylation status identically. As there were substantially more methylated than non-methylated loci, the number of correctly identified methylated sites (i.e. sensitivity) has a larger impact on overall agreement. The hMe-Seal enrichment method for assaying hmC displayed very high sensitivity. At an enrichment coverage cut-off of two, its performance was very comparable to the bisulfite data, achieving a relative sensitivity of 0.98, relative specificity of 0.97 and relative agreement of 0.98. For mCH (MBD-DIP) a coverage threshold of 0.5 achieved a relative sensitivity of 0.76, relative specificity of 2.27 and relative agreement of 0.77. Further, the rank correlation between estimates of mCG from MBD-DIP and bisulfite was −0.07, confirming that MBD-DIP samples were successfully pre-depleted of mCG and predominantly captured mCH.
We also studied the rank correlations between the bisulfite estimated methylation sum versus fragment coverage estimated from the enrichment data (Supplementary Table S4). To obtain a benchmark against which to compare these results, we first calculated correlations between the bisulfite duplicates that were 0.97, 0.81 and 0.62 for mCG, hmCG and mCH, respectively. Relative to these bisulfite duplicate correlations, correlations between bisulfite and enrichment data were 0.80, 0.70 and 0.77 for mCG, hmCG and mCH. The implications of these correlations provides critical support for an enrichment methods in MWAS, as it means that differences in methylation levels caused by phenotype can be detected as differences in enrichment coverage estimates.
For assaying mCG, profiles across genomic features were very similar for WGB and MBD (Figure 4A). This suggested that both technologies captured comparable information on mCG. For hmCG, the patterns between TAB and hMe-Seal were very similar as well (Figure 4B). However, the effect sizes were systematically larger for hMe-Seal than TAB. This is unlikely due to sampling variation because the effects (i) were in the same direction, and (ii) showed a pattern comparable to that as seen for mCG (Figure 4A). Instead, it suggested that hMe-Seal outperformed TAB in terms of more accurate classification of methylation status. In contrast, for mCH, the effect sizes were diminished for the enrichment method (Figure 4C). Although, patterns were still similar to WGB, effect sizes for MBD-DIP were smaller, suggesting it may be less accurate.
Figure 4.

Methylation profiles across genomic features. We classified loci as methylated versus non-methylated, and genomic features as present versus absent. Using these 2 by 2 tables as input, we calculated odds ratios that indicated whether sites in the studied featured were more likely to be methylated compared to sites not in this feature. For the purpose of plotting we took the log10 of the odds ratio. Thus, a value of zero, log10(1) = 0, indicated by the dashed lines in the figures, means no enrichment of methylated sites. Odds ratios for mCH were calculated using only cytosines located more than the maximum fragment size away from CG dinucleotides. This explains the missing values in panel C for CG islands, as those regions contain no such cytosines. The full list of tested features is provided in Supplementary Table S5. For assaying mCG, profiles across genomic features were very similar for WGB and MBD (A). This suggested that both technologies covered similar features. For hmCG, the two patterns were also very comparable but the odds ratios were systematically larger for hMe-Seal (B). This is unlikely the result of sampling variation because the effects (i) were in the same direction, and (ii) displayed a pattern comparable to that observed for mCG (A). Instead, it suggested that hMe-Seal outperformed TAB in terms of more accurately classifying methylation status. In contrast, enrichment for mCH had an opposite relationship to WGB (C). Although, patterns were still similar, odds ratios were smaller for MBD-DIP suggesting it may be less accurate.
There is the possibility that mapping or library prep may have introduced differences between bisulfite and enrichment data. For this reason we read coverage profiles across a variety of genomic features for input and bisulfite samples (Supplemental Figure S2). The input sample involved unmodified DNA from the same subject for which we obtained whole genome sequencing data. An unbiased process should result in uniformly distributed read coverage across the genome. However, results show that read coverage is not perfectly uniform where bisulfite methods are more severely affected. Moreover, profiles sometimes deviated for bisulfite and input controls for enrichment methods, suggesting that this bias could have lowered the agreement between bisulfite and enrichment data.
Noting possible a confound of cell-type heterogeneity within cortical tissue, we sought to repeat our evaluation with genomic material from a less complex source. Using protocols described elsewhere (38,39) we isolated the nuclei of neurons from other cells present in brain tissue from the same subject and repeated all assays. The overall pattern of results was very comparable to those obtained in bulk tissue, providing support for the robustness of our findings (Supplementary Figure S3).
A recent comparison (48) found that the antibody based MeDIP enrichment approach outperformed a variety of commercially available alternatives that yielded meager CG coverage ranging from merely 2% for commonly used methylation arrays to 19.6% for the most comprehensive bisulfite targeted capture sequencing method. However, for our panel we used the protein based MBD enrichment. To justify this choice, we also performed standard MeDIP assays on the same sample. A direct comparison (Supplementary Figure S4) showed that for assaying mCG, sensitivity/specific/agreement was consistently higher for MBD versus MeDIP enrichment. At a coverage threshold of 0.5, sensitivity was 0.91 for MBD versus 0.86 for MeDIP; specificity was 1.09 for MBD versus 0.94 for MeDIP. In addition, the correlation with the bisulfite data was 0.80 for MBD versus 0.51 for MeDIP (Supplementary Table S4). We did use a modified version of MeDIP, labeled MBD-DIP, to assay mCH. The modification consisted of first depleting samples of mCG, giving MBD-DIP the advantage of being able to discriminate between mCG and mCH. Furthermore, compared to standard MeDIP, MBD-DIP had better sensitivity/specificity (Supplementary Figure S4) to detect mCH, and showed higher correlations with the bisulfite estimates of mCH methylation sum (0.77 for MBD-DIP versus 0.67 for standard MeDIP).
Enrichment methods do not provide methylation information about the individual sites in the fragment-sized loci. However, for the purpose of MWAS the resolution of our panel is still quite high, as association signals will implicate fragment sized loci (e.g. ∼140 bp windows as estimated from the sequencing data). Furthermore, the bisulfite data showed that the variance in methylation between sites within a locus was generally low (Figure 2C), with over 90% of loci showing a variance <0.05 (Figure 5). This implies that in many instances the single base resolution offered by bisulfite methods may not substantially improve resolution or help fine-map MWAS results to specific sites. Thus, because methylation statuses are highly spatially correlated within our fragment-sized loci, the sites in these loci will tend to produce association signals of the same magnitude. This interpretation may not apply to mCH as the small variance in non-CG methylation likely resulted from the very small number of methylated sites (∼2) among the many possible methylation sites (∼73). Here, single base resolution could potentially help prune the list of methylation sites responsible for the association signal.
Figure 5.
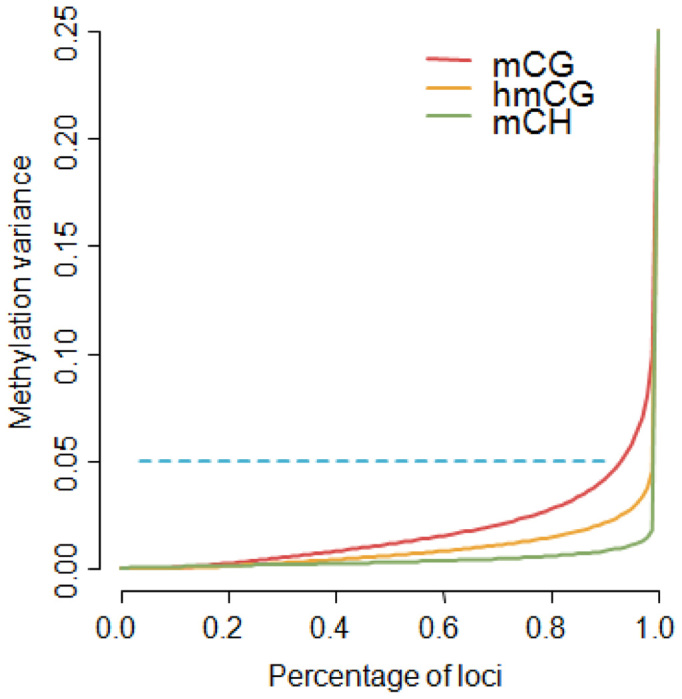
Methylation variance at fragment sized loci. The variance was low for all three types of bisulfite methylation estimates (Figure 3C), suggesting sites tend to have similar methylation levels within our fragment sized loci. To further quantify this phenomenon, we plotted the percentiles of the methylation variances. Over 90% of the loci had a variance <0.05. There were slight differences among the three types, with mCG showing relatively more variability than hmCG and mCH being the most invariant.
DISCUSSION
The goal of MWAS is to identify links between methylation sites and outcomes of interest. MWAS is typically performed using microarray technologies that assay only a very small fraction of the CG methylome and may entirely miss additional forms of methylation as are present in tissues such as brain. The alternative is the use of WGB sequencing but this approach is not yet practically feasible with the sample sizes required for adequate statistical power in MWAS (21). In this article we revisited methylation enrichment approaches, an ‘older’ group of methods to assay the methylome. After selecting the best enrichment methods among the many options we further optimized the selected protocols. Results showed that the sensitivity (proportion of methylated loci correctly detected) of our panel approximated the sensitivity obtained with the bisulfite methods (relative sensitivity was 0.91, 0.97 and 0.76 for mCG, hmCG and mCH, respectively). By its nature sensitivity is contingent upon and needs to be balanced against specificity (proportion of non-methylated loci correctly detected). For example, by simply calling all loci methylated, sensitivity would be 100% but specificity 0%. In our analysis specificity was generally better for the enrichment methods as compared to bisulfite methods (relative specificity was 1.09, 0.98 and 2.27 for mCG, hmCG and mCH, respectively). Finally, results showed that methylation profiles for enrichment methods (closely) tracked patterns observed in the bisulfite data across a variety of genomic features.
In contrast to bisulfite methods, enrichment methods do not provide estimates of absolute methylation levels at single base resolution. However, this is not a major hindrance for MWAS. First, for all three components of our panel we observed high correlations (0.7–0.8 relative to bisulfite) between the estimated fragment coverage and the amount of methylation as assayed by bisulfite methods. This is an important property for MWAS as it means that differences in methylation levels can be detected using enrichment methods. The fact that fragment coverage does not provide a direct estimate of absolute methylation levels (i.e. how much methylation occurs) is irrelevant for MWAS as long as it is sensitive to methylation differences at a locus. Second, because enrichment methods assay the total amount of methylation for loci that are about the size of the sequenced fragments (methylation sum), they cannot pinpoint the specific site that caused the MWAS signal. However, the resolution is still quite high on a genomic scale (e.g. ∼140 bp in this study when estimated from the sequencing data (42)) and MWAS findings obtained with enrichment methods can be readily followed up using a targeted bisulfite approach. Critically, we also found that methylation levels across sites in the fragment-size loci tended to be similar in magnitude (variance <0.05 for 90% of sites). Thus, bisulfite methods may often not be able to substantially improve the resolution of MWAS findings.
To improve performance, we did have to do some optimization to the laboratory protocols of our enrichment panel. This included the use of a low salt elution buffer during MBD enrichment to improve sensitivity to low levels of mCG, the use of mCG depleted samples to improve capture of mCH and fragmenting gDNA to the shortest length compatible with current sequencers. We previously showed that our optimized protocol to capture mCG outperformed (35) the best kit identified in a study comparing five MBD kits (49). In this article, we further showed that our protocol outperformed standard MeDIP, which had previously been shown to outperform other commonly used mCG methylation assays (48). The most challenging task appeared to be probing the mCH status of the ∼1 billion genomic cytosines. This challenge, however, was not unique for our enrichment assay and was true for our bisulfite data and has also been observed by others (50). One reason could be that CH methylation is always strand specific, thereby lowering the reliability of the estimates that are based on half the coverage compared to sites showing ‘symmetric’ CG methylation. Furthermore, because the vast majority of cytosines outside the CG context are not methylated they will be converted by the bisulfite treatment. This lowers the diversity of the regions (i.e. only three bases remain) which is known to negatively affect the quality of the base calls during sequencing. Depleting for mCG means that those samples will also be depleted for any mCH co-occurring on the fragments containing mCG. In this sense, our approach does not provide a complete fractionation. To study the risk of missing such mCH signals in MWAS, we calculated the correlation between mCG and mCH methylation in fragment sized regions containing mCG. This correlation was 0.85, meaning that part of such mCH signal will be captured by studying mCG methylation, thereby mitigating the loss of mCH coverage near CG sites.
While our parallel enrichments can identify genomic regions in which both hmC and mC occur, it does not provide information about whether these two marks co-occur on the same molecules (intrachromosomal) or different molecules from the same region (interchromosomal). This issue is of potential scientific interest as regions in which both methylation and hmC are coincident, may represent loci undergoing epigenetic transitions that are possibly relevant to disease. A double enrichment approach as demonstrated in our MBD-DIP protocol may in principle be adapted to provide this refinement. Thus, we could first enrich for the subset of molecules with mCG and then, from within that same mCG enriched sample, perform a second enrichment to estimate the fraction that of molecules that also have hmC. Even further refinement could entail identifying the specific cells in which this transition is occurring, but currently neither enrichment nor bisulfite methods can feasibly probe all methylation types at single-cell resolution. While single-cell methylation analysis by nanopore sequencing (51,52) and cell lineage tracing in human brain (53,54) are possible, they are not currently viable for large-scale MWAS.
CONCLUSION
Whereas our triplet of enrichment methods approximated the sensitivity/specificity obtained with WGB/TAB, reagent costs of our panel were <5% of those for WGB methods. In practice, the cost benefit is much greater as bisulfite sequencing uses longer and paired-end reads (in our case 300 versus 75 bp) and requires more total runs (in our case 1 sample per run versus 10 samples per run). This will further increase the cost difference (laboratory technician time, sequencer depreciation, etc.) and assuming the coverage/read length used in this article, the time to complete a project by a factor of ∼15. Thus, enrichment methods allow for comprehensive, adequately powered and cost-effective genome-wide investigations of the brain methylome. Being currently the only viable option for comprehensive brain methylome studies, enrichment methods may be critical for moving the field forward.
ACCESSION NUMBER
The data is available from GEO under accession number GSE94866.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Mental Health [R03MH102723 to K.A, R01MH104576, R01MH099110 to E.O.]. Funding for open access charge: National Institute of Mental Health [R03MH102723 to K.A, R01MH104576, R01MH099110 to E.O.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Gage F.H., Temple S.. Neural stem cells: generating and regenerating the brain. Neuron. 2013; 80:588–601. [DOI] [PubMed] [Google Scholar]
- 2. Heyward F.D., Sweatt J.D.. DNA methylation in memory formation: emerging insights. Neuroscientist. 2015; 21:475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aberg K.A., McClay J.L., Nerella S., Clark S., Kumar G., Chen W., Khachane A.N., Xie L., Hudson A., Gao G. et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry. 2014; 71:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mill J., Tang T., Kaminsky Z., Khare T., Yazdanpanah S., Bouchard L., Jia P., Assadzadeh A., Flanagan J., Schumacher A. et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 2008; 82:696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang T., Pan Q., Lin L., Szulwach K.E., Song C.X., He C., Wu H., Warren S.T., Jin P., Duan R. et al. Genome-wide DNA hydroxymethylation changes are associated with neurodevelopmental genes in the developing human cerebellum. Hum. Mol. Genet. 2012; 21:5500–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernstein B.E., Meissner A., Lander E.S.. The mammalian epigenome. Cell. 2007; 128:669–681. [DOI] [PubMed] [Google Scholar]
- 7. Bird A.P. CpG-rich islands and the function of DNA methylation. Nature. 1986; 321:209–213. [DOI] [PubMed] [Google Scholar]
- 8. Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009; 462:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lister R., Mukamel E.A., Nery J.R., Urich M., Puddifoot C.A., Johnson N.D., Lucero J., Huang Y., Dwork A.J., Schultz M.D. et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013; 341:1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xie W., Barr C.L., Kim A., Yue F., Lee A.Y., Eubanks J., Dempster E.L., Ren B.. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012; 148:816–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Junjie U.G., Su Y., Joo H.S., Shin J., Li H., Xie B., Zhong C., Hu S., Le T., Fan G. et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2013; 17:215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Day J.J., Sweatt J.D.. DNA methylation and memory formation. Nat. Neurosci. 2010; 13:1319–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roth E.D., Roth T.L., Money K.M., SenGupta S., Eason D.E., Sweatt J.D.. DNA methylation regulates neurophysiological spatial representation in memory formation. Neuroepigenetics. 2015; 2:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen L., Zhang Y.. 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr. Opin. Cell Biol. 2013; 25:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ito S., D/'Alessio A.C., Taranova O.V., Hong K., Sowers L.C., Zhang Y.. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010; 466:1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Penn N.W., Suwalski R., O’Riley C., Bojanowski K., Yura R.. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem. J. 1972; 126:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dyrvig M., Gøtzsche C.R., Woldbye D.P.D., Lichota J.. Epigenetic regulation of Dnmt3a and Arc gene expression after electroconvulsive stimulation in the rat. Mol. Cell. Neurosci. 2015; 67:137–143. [DOI] [PubMed] [Google Scholar]
- 18. LaPlant Q., Vialou V., Covington H.E., Dumitriu D., Feng J., Warren B., Maze I., Dietz D.M., Watts E.L., Iñiguez S.D. et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010; 13:1137–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li X., Wei W., Zhao Q.Y., Widagdo J., Baker-Andresen D., Flavell C.R., D’Alessio A., Zhang Y., Bredy T.W.. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:7120–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rudenko A., Dawlaty M.M., Seo J., Cheng A.W., Meng J., Le T., Faull K.F., Jaenisch R., Tsai L.H.. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013; 79:1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rakyan V.K., Down T.A., Balding D.J., Beck S.. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011; 12:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laird P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010; 11:191–203. [DOI] [PubMed] [Google Scholar]
- 23. Beck S., Rakyan V.K.. The methylome: approaches for global DNA methylation profiling. Trends Genet. 2008; 24:231–237. [DOI] [PubMed] [Google Scholar]
- 24. Metzker M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 2010; 11:31–46. [DOI] [PubMed] [Google Scholar]
- 25. Frommer M., McDonald L.E., Millar D.S., Collis C.M., Watt F., Grigg G.W., Molloy P.L., Paul C.L.. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu M., Hon G.C., Szulwach K.E., Song C.X., Jin P., Ren B., He C.. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat. Protoc. 2012; 7:2159–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mohn F., Weber M., Schubeler D., Roloff T.C.. Methylated DNA immunoprecipitation (MeDIP). Methods Mol. Biol. 2009; 507:55–64. [DOI] [PubMed] [Google Scholar]
- 28. Serre D., Lee B.H., Ting A.H.. MBD-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010; 38:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brinkman A.B., Simmer F., Ma K., Kaan A., Zhu J., Stunnenberg H.G.. Whole-genome DNA methylation profiling using methylcap-seq. Methods. 2010; 52:232–236. [DOI] [PubMed] [Google Scholar]
- 30. Li N., Ye M., Li Y., Yan Z., Butcher L.M., Sun J., Han X., Chen Q., Zhang X., Wang J.. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods. 2010; 52:203–212. [DOI] [PubMed] [Google Scholar]
- 31. Harris R.A., Wang T., Coarfa C., Nagarajan R.P., Hong C., Downey S.L., Johnson B.E., Fouse S.D., Delaney A., Zhao Y. et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010; 28:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ziller M.J., Gu H., Muller F., Donaghey J., Tsai L.T., Kohlbacher O., De Jager P.L., Rosen E.D., Bennett D.A., Bernstein B.E. et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013; 500:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stevens M., Cheng J.B., Li D., Xie M., Hong C., Maire C.L., Ligon K.L., Hirst M., Marra M.A., Costello J.F. et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res. 2013; 23:1541–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bock C., Tomazou E.M., Brinkman A.B., Muller F., Simmer F., Gu H., Jager N., Gnirke A., Stunnenberg H.G., Meissner A.. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat. Biotechnol. 2010; 28:1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aberg K.A., Xie L., Chan R.F., Zhao M., Pandey A.K., Kumar G., Clark S.L., van den Oord E.J.. Evaluation of methyl-binding domain based enrichment approaches revisited. PLoS One. 2015; 10:e0132205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Song C.X., Szulwach K.E., Fu Y., Dai Q., Yi C., Li X., Li Y., Chen C.H., Zhang W., Jian X. et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011; 29:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thomson J.P., Hunter J.M., Nestor C.E., Dunican D.S., Terranova R., Moggs J.G., Meehan R.R.. Comparative analysis of affinity-based 5-hydroxymethylation enrichment techniques. Nucleic Acids Res. 2013; 41:e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spalding K.L., Bergmann O., Alkass K., Bernard S., Salehpour M., Huttner H.B., Bostrom E., Westerlund I., Vial C., Buchholz B.A. et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013; 153:1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matevossian A., Akbarian S.. Neuronal nuclei isolation from human postmortem brain tissue. J. Vis. Exp. 2008; 20:e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aberg K.A., McClay J.L., Nerella S., Xie L.Y., Clark S.L., Hudson A.D., Bukszar J., Adkins D., Consortium S.S., Hultman C.M. et al. MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case–control samples. Epigenomics. 2012; 4:605–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van den Oord E.J., Bukszar J., Rudolf G., Nerella S., McClay J.L., Xie L.Y., Aberg K.A.. Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinformatics. 2013; 14:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu M., Hon G.C., Szulwach K.E., Song C.X., Zhang L., Kim A., Li X., Dai Q., Shen Y., Park B. et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012; 149:1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu M., Hon G.C., Szulwach K.E., Song C.-X., Jin P., Ren B., He C.. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat. Protoc. 2012; 7:2159–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu M., Hon Gary C., Szulwach Keith E., Song C.-X., Zhang L., Kim A., Li X., Dai Q., Shen Y., Park B. et al. Base-resolution analysis of 5- hydroxymethylcytosine in the mammalian genome. Cell. 2012; 149:1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo W., Fiziev P., Yan W., Cokus S., Sun X., Zhang M.Q., Chen P.Y., Pellegrini M.. BS-seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics. 2013; 14:774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wen L., Li X., Yan L., Tan Y., Li R., Zhao Y., Wang Y., Xie J., Zhang Y., Song C. et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014; 15:R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walker D.L., Bhagwate A.V., Baheti S., Smalley R.L., Hilker C.A., Sun Z., Cunningham J.M.. DNA methylation profiling: comparison of genome-wide sequencing methods and the infinium human methylation 450 Bead Chip. Epigenomics. 2015; 7:1287–1302. [DOI] [PubMed] [Google Scholar]
- 49. De Meyer T., Mampaey E., Vlemmix M., Denil S., Trooskens G., Renard J.P., De Keulenaer S., Dehan P., Menschaert G., Van Criekinge W.. Quality evaluation of methyl binding domain based kits for enrichment DNA-methylation sequencing. PLoS One. 2013; 8:e59068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sun Z., Dai N., Borgaro J.G., Quimby A., Sun D., Corrêa I.R. Jr, Zheng Y., Zhu Z., Guan S.. A sensitive approach to map genome-wide 5-hydroxymethylcytosine and 5-formylcytosine at single-base resolution. Mol. Cell. 2015; 57:750–761. [DOI] [PubMed] [Google Scholar]
- 51. Zahid O.K., Zhao B.S., He C., Hall A.R.. Quantifying mammalian genomic DNA hydroxymethylcytosine content using solid-state nanopores. Sci. Rep. 2016; 6:29565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laszlo A.H., Derrington I.M., Brinkerhoff H., Langford K.W., Nova I.C., Samson J.M., Bartlett J.J., Pavlenok M., Gundlach J.H.. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:18904–18909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Evrony G.D., Cai X., Lee E., Hills L.B., Elhosary P.C., Lehmann H.S., Parker J.J., Atabay K.D., Gilmore E.C., Poduri A. et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012; 151:483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Evrony G.D., Lee E., Mehta B.K., Benjamini Y., Johnson R.M., Cai X., Yang L., Haseley P., Lehmann H.S., Park P.J. et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron. 2015; 85:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.