

ABSTRACT
The HU superfamily of proteins, with a unique DNA-binding mode, has been extensively studied as the primary chromosome-packaging protein of the bacterial superkingdom. Representatives also play a role in DNA-structuring during recombination events and in eukaryotic organellar genome maintenance. However, beyond these well-studied roles, little is understood of the functional diversification of this large superfamily. Using sensitive sequence and structure analysis methods we identify multiple novel clades of the HU superfamily. We present evidence that a novel eukaryotic clade prototyped by the human CCDC81 protein acquired roles beyond DNA-binding, likely in protein-protein interaction in centrosome organization and as a potential cargo-binding protein in conjunction with Dynein-VII. We also show that these eukaryotic versions were acquired via an early lateral transfer from bacteroidetes, where we predict a role in chromosome partition. This likely happened before the last eukaryotic common ancestor, pointing to potential endosymbiont contributions beyond that of the mitochondrial progenitor. Further, we show that the dramatic lineage-specific expansion of this domain in the bacteroidetes lineage primarily is linked to a functional shift related to potential recognition and preemption of genome invasive entities such as mobile elements. Remarkably, the CCDC81 clade has undergone a similar massive lineage-specific expansion within the archosaurian lineage in birds, suggesting a possible use of the HU superfamily in a similar capacity in recognition of non-self molecules even in this case.
KEYWORDS: bacteroidetes, biological conflict, birds, CCDC81, centrosome, HU, IHF
Introduction
Packaging of the chromosomal DNA polymer, which is several orders of magnitude longer than average cell-dimensions when extended, inside of the cell or nucleus is a universal structural challenge faced by all organisms. Interestingly, despite DNA having been present in the last universal common ancestor (LUCA) as a genetic molecule, various independent solutions for its packaging have been invented across the 3 great superkingdoms of life.1-7 In archaea and eukaryotes the dominant packaging proteins are members of the ancient histone fold which multimerize to form tetramers or octamers around which DNA is wound.8-10 However, in several archaea alternative solutions for the DNA-packaging problem have been adopted in the form of proteins with unrelated structural folds, which might exist alongside or in place of histones.11 These include members of the Alba (IF3-C like fold),3,12 MC1 (distinct α+β fold),13 Cren7 (Zinc-ribbon-like)14 and HU superfamilies.
In all bacteria the primary DNA-packaging protein is HU,15,16 although it might be additionally accompanied by other DNA-packaging proteins such as Fis, the chlamydial-type nucleoid protein, H-NS, MC1 or histones.7,17,18 However, these never displace HU in the genome, suggesting that HU plays an irreplaceable role in DNA compaction in the bacterial nucleoid. Additionally, HU has also been horizontally transferred to certain archaea,15 where it might function in place of or alongside the ancestral histones and other chromosomal proteins.11 HU is also widely seen in eukaryotes with plastids, which are of cyanobacterial origin.19,20 Indeed, HU homologs have been demonstrated to play a role in the packaging of circular plastid DNA in a manner comparable to bacterial DNA in red alga Cyanidioschyzon merolae,19 the green alga Chlamydomonas reinhardtii21 and apicomplexans.22,23 The African Swine Fever Virus of the NCLDV clade has also acquired HU as a packaging protein in line with the previous proposal of convergences between organelle DNA replication and cytoplasmic viral DNA replication.6 Interestingly, in one eukaryotic lineage, the dinoflagellates, members of the HU have also been recruited for nuclear chromosomal DNA packaging alongside the ancestral eukaryotic histones.24,25
Several bacteria possess multiple paralogs of the HU superfamily.26 In proteobacteria these fall into 3 major clades prototyped by the Escherichia coli HU, integration host factor (IHF) α and IHFβ.27,28 While a HU ortholog is widespread, there are several other clades of paralogs specific to certain bacterial lineages. Both HU and IHFs have been shown to be nucleoid structural proteins; however, while HU is a non-specific DNA-binding protein, IHFs have a significantly greater preference for certain sequences.29-31 Beyond their structural roles, binding of DNA by HU/IHF, by virtue of their contacts with DNA, also affects the dynamics of replication, recombination and repair. Specifically, both HU and IHF also participate in DNA-protein complexes formed during integration (IHF: e.g. phage lambda integration,32,33 and CRISPR spacer acquisition by Cas1-Cas234) and inversion of DNA (HU: e.g., Hin recombinase35). Additionally, they have transcription regulatory roles by direct binding of particular sites, thereby affecting DNA supercoiling and facilitating interactions between distantly bound transcription factors via DNA-bending.36-38
The HU superfamily possesses a distinct core fold with a N-terminal bihelical ‘stalk’ followed by a β-sheet with an extended β-hairpin and a further α-helix at the C-terminus (Fig. 1A-B).39,40 This monomeric unit exists as an obligate dimer: the N-terminal stalk plays a key role in dimerization and the extended β-hairpin forms a ‘clasp’ which positions itself deeply within the double-helical groove of the DNA resulting in a bend in the double helical axis (Fig. 1A).16,41 This primary mode of DNA-binding appears to be conserved across the HU superfamily. Sequence diversity in the HU superfamily at this primary DNA interface accounts for the differences in specificity of different clades. In several bacteria (e.g., several actinobacteria, proteobacteria and Deinococcus) positively charged low-complexity extensions comparable to histone tails at either terminus also play a role in DNA-binding, thereby altering the specificity of the core fold.15,42
Figure 1.
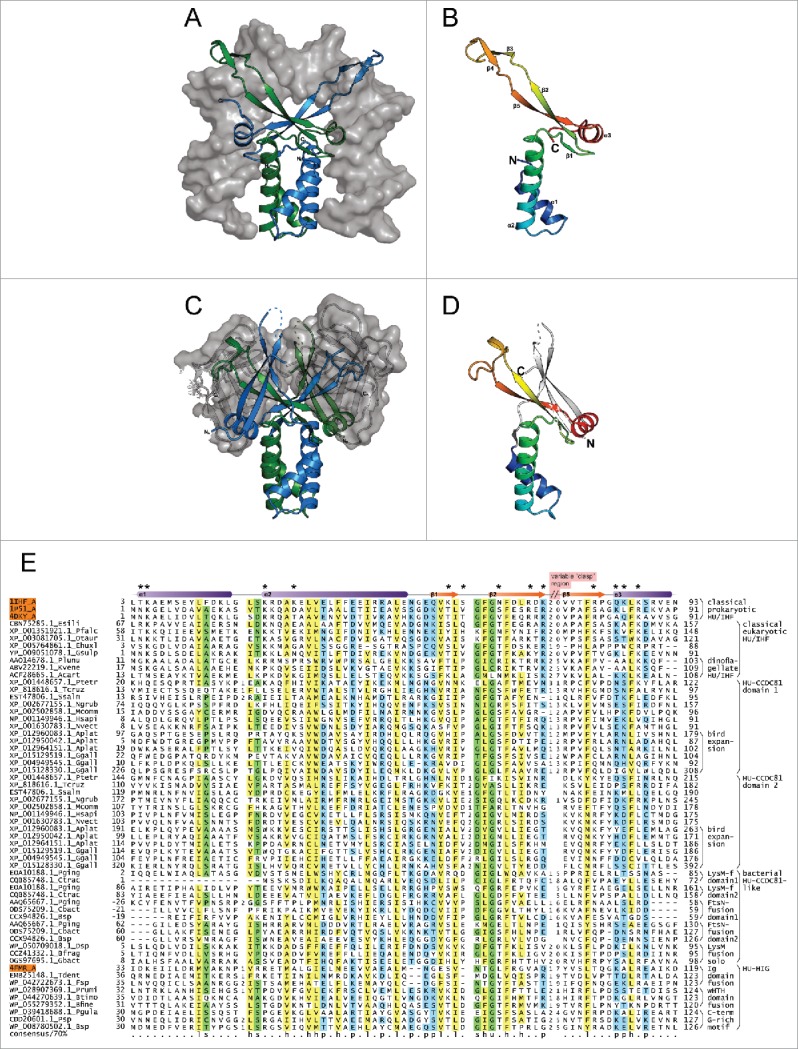
Structural and sequence overview of the HU superfamily. (A-D) Cartoon renderings of HU superfamily members. (A) Integration host factor (IHF) α and β in complex with DNA (PDB: 1IHF). IHFα and IHFβ are represented as ribbons and colored green and blue, respectively. DNA is shown as a surface trace in gray. (B) IHFα (PDB: 1IHF_A). (C) HU-HIG clade homodimer from Bacteroides vulgatus with a C-terminal Ig-like domain fusion (PDB: 4FMR). Coloring as in (A) above. The region corresponding to the Ig-like domain is shown as a superimposed ribbon with surface representation colored in gray. (D) HU domain from chain A of Bacteroides vulgatus HU homolog (PDB: 4FMR_A). The domains are colored and labeled as in (B), with additional secondary structure elements colored white. (E) Multiple sequence alignment of the HU superfamily. Secondary structure provided in top line, with elements labeled to correspond with (B). Positions shown to interact with DNA are denoted by asterisks. Sequences are labeled to left with NCBI accession number and organism abbreviation separated by rightmost underscore; HU family/clade names are given to the right. Negative numbers at left indicate extension of predicted protein start sites in GenBank. The alignment is colored as follows: h, hydrophobic and yellow; l, aliphatic and yellow; s, small and green; p, polar and blue; u, tiny and green. Organism abbreviations: Esili, Ectocarpus siliculosus; Pfalc, Plasmodium falciparum; Otaur, Ostreococcus tauri; Ehuxl, Emiliania huxleyi; Gsulp, Galdieria sulphuraria; Plunu, Pyrocystis lunula; Kvene, Karlodinium veneficum; Acart, Amphidinium carterae; Ptetr, Paramecium tetraurelia; Tcruz, Trypanosoma cruzi; Ssalm, Spironucleus salmonicida; Ngrub, Naegleria gruberi; Mcomm, Micromonas commoda; Hsapi, Homo sapiens; Nvect, Nematostella vectensis; Aplat, Anas platyrhynchos; Ggall, Gallus gallus; Pging, Porphyromonas gingivalis; Ctrac, Chlamydia trachomatis; Cbact, Cytophagaceae bacterium; Bsp, Bacteroides sp, Dsp, Dysgonomonas sp; Bfrag, Bacteroides fragilis; Gbact, Gallionellales bacterium; Tdent, Treponema denticola; Fsp, Flavobacterium sp; Prumi, Prevotella ruminicola; Btimo, Bacteroides timonensis; Bfine, Bacteroides finegoldii; Pgula, Porphyromonas gulae; Psp, Prevotella sp.
While the evolutionary history and sequence features of the major bacterial clades of the HU family, HU, IHFα and IHFβ have been extensively studied,15,26 our preliminary analysis revealed the presence of several additional, poorly-understood clades. Further, given this unique mode of DNA-binding, which is unparalleled in other DNA-binding proteins, we wondered if there are additional members of the HU superfamily that might be deployed beyond bacteria. Hence, to better understand the obscure clades and potentially detect new members of this superfamily we performed a comprehensive analysis of the HU superfamily using sensitive sequence and structure comparison techniques. Consequently, we have unearthed a novel clade of eukaryotic HUs with bacterial cognates, which might play a key role in eukaryotic centrosome function. Our analysis also throws new light on the evolution and functions of poorly understood clades of the HU superfamily in bacteria.
Results
Detection of novel members and revised phyletic overview of the HU superfamily
Given that HU is present in every bacterial genome, iterative sequence profile searches against the NR database tend to ‘saturate’ rapidly before detecting divergent members. Accordingly, we initiated iterative PSI-BLAST43 and JACKHMMER44 searches against a curated database of 6430 complete proteomes drawn from across the tree of life. In addition to the major HU and IHF clades, these searches detected with significant e-values several divergent versions in bacteria, especially from the bacteroidetes-chlorobi lineage, which is known to possess multiple paralogs of this superfamily.26 While these have not been comprehensively sampled in previous studies, our searches enabled us to obtain a comprehensive collection of all members of the superfamily from this lineage, including several divergent ones.
In addition to the previously-known eukaryotic versions from plastid-containing eukaryotes and dinoflagellates, we also detected a distinct eukaryotic version of the HU superfamily prototyped by CCDC81, a recently-identified human centrosomal protein.45 These were by far the most divergent and have not been previously recognized as members of the HU superfamily. The globular region corresponding to the potential HU domains in these proteins partly overlaps with the Pfam model Domain of Unknown Function (DUF)4496.46 We then conducted profile-profile searches with the HHpred program47 to confirm this relationship. Duly, these searches initiated with the newly detected domains in CCDC81 recovered the HU/IHF profiles with significant probability (P-value = 3.5E-17, probability 99%). Similarly, reverse searches with the HU/IHF profiles significantly recovered the CCDC81 profiles suggesting that they were indeed members of the HU superfamily. Accordingly, we named this clade of HU domains the HU-CCDC81 clade and recommend replacement of the PFAM model with our alignment which precisely defines the HU domain boundaries.
In light of these findings we performed a revised sequence-similarity based clustering and phyletic pattern and phylogenetic analysis of the HU superfamily. Beyond being present in all bacterial lineages, members of the core HU/IHF family have been transferred to representatives of the Eury- Thaum- Loki and Thor- archaeota lineages on multiple occasions, suggesting that it is more widely used as an alternative or auxiliary chromosomal protein in the archaeal superkingdom than previously appreciated (see Supplemental Material).48 The bacteroidetes-chlorobi lineage contains distinct, massive lineage-specific expansions (e.g., 22 copies in just Bacteroides helcogenes), which forms a specific clade of HUs that have been occasionally transferred to the spirochaetes (see below, Fig. 1C-E). Most of the previously-known eukaryotic members of the HU superfamily are closely related to the bacterial versions and belong to a single clade that is primarily present in eukaryotes with plastids except land plants. These are often characterized by a N-terminal signal peptide, which allows their import into the plastid where they typically function (see Supplemental Material). These observations suggest that this version was first acquired in the common ancestor of the plant lineage alongside the plastid derived from the primary cyanobacterial endosymbiosis (as seen in the chlorophyte and rhodophyte algae) and subsequently disseminated to other lineages that acquired the plastid via secondary endosymbiosis (as in alveolates and stramenopiles). Beyond these, we also found evidence supporting several possible cases of late, independent lateral transfer of classical HU/IHF proteins into eukaryotes, including in the amoebozoan Acanthamoeba and the basal chordate Oikopleura (see Supplemental Material).
In contrast to the above, the HU-CCDC81 clade represents a distinct clade that is widespread throughout eukaryotes including early-branching eukaryotic lineages such as diplomonads (Giardia), kinetoplastids, and heteroloboseans (Naegleria) (Fig. 1E). However, HU-CCDC81 has been lost in several eukaryotic lineages, such as land plants, amoebozoans, all fungi except chytridiomycota and within Metazoa in nematodes. Curiously, it has undergone expansion within the archosauromorph lineage. Whereas the basal extant branch of this lineage, the turtles, contain only a single copy like other animals, in crocodiles we see up to 4 copies. Birds display an even greater expansion with at least 20 identified copies in the chicken. Further searches initiated with eukaryotic HU-CCDC81 domains against prokaryotic genomes also recovered specific bacterial members of this clade, which are primarily found in the bacteroidetes lineage, although a few related forms appear to have been sporadically disseminated to certain proteobacteria and others (Fig. 1E, Supplemental Material). These observations suggest that it emerged from a single ancient lateral transfer event from the bacteroidetes lineage that happened before the last eukaryotic common ancestor.
Structural features of the HU-CCDC81 clade
To better understand the structure and function of the novel HU-CCDC81 clade, we created a multiple sequence alignment of their HU domain along with representatives of all the clades detected in our searches (see Materials and Methods, Fig. 1E). The eukaryotic HU-CCDC81 domains are on an average shorter (∼74 residues, s.d. 11) than the classical HU domains (∼93 residues, s.d. 2). Further, all eukaryotic proteins of this clade display a tandem duplication of the HU domain within the same polypeptide suggesting that the 2 copies together form a dimeric unit in a single molecule. Mapping the alignment of the HU-CCDC81 clade onto known structures of the HU superfamily (Fig. 1E) reveals that the shorter length of these HU domains relative to the classic versions is due to a truncation of the tip of the extended β-hairpin feature characteristic of the latter domains. The N-terminal copy of the eukaryotic HU-CCDC81 domain is less truncated (average domain length of 85 residues, s.d. 7) while the C-terminal copy is always more drastically truncated (average domain length of 76 residues, s.d 3). The bacterial members of the HU-CCDC81 clade might occur as either a single copy of the HU domain per polypeptide or as 2 tandem copies just as in the eukaryotes. The latter bacterial versions tend to show a pattern of truncation of the tip of the β-hairpin in each repeat comparable to the equivalent eukaryotic versions. This suggests that the duplication and modification of the domains had happened before their transfer to eukaryotes.
Along with the truncation, the sequence composition of the C-terminal DNA-binding region is also dramatically different in the HU-CCDC81 clade. Analysis of the clasp and its immediate flanking regions, which house the greatest concentration of binding residues (Fig. 1E, Supplemental Material), in the classical HU/IHF clades reveals an enrichment in large basic amino acids (K, R) with an average of 8.1 basic residues in that segment per sequence, or 2.6 out of every 10 residues. In contrast, the corresponding segment in the HU-CCDC81 clade has on an average only 2.8 basic residues per sequence, or 1.3 out of every 10 residues. Further, examination of the positions corresponding to the DNA-contacting basic residues in the classical HU/IHF clades reveals that they are either entirely absent or poorly conserved (< 60% per column). However, both groups possess a similar average density of acidic residues in this segment (2.6 and 2.7, respectively), suggesting a specific loss of positively charged positions in the HU-CCDC81 clade. In contrast, the predominantly hydrophobic residues which stabilize the core helices of the fold and the GXG motif which defines the turn between the first strand of the sheet and downstream β-hairpin are well-conserved between the classical HU/IHF clades and HU-CCDC81 clade (Fig. 1E). These observations indicate that the HU-CCDC81 domains, while adopting the same fold, are likely to possess a short, less positively-charged clasp.
Domain architectural features of the HU-CCDC81 clade
Whereas the classical HU/IHF proteins are generally short proteins with at best low-complexity basic extensions at the termini,15 both eukaryotic and bacterial members of the HU-CCDC81 tend to be larger proteins with several distinct architectures. The most common architecture, which is widely represented across eukaryotes, is one where the 2 N-terminal HU domains are fused to a C-terminal coiled coil (CC) that can be over 400 residues in length with a central kink (Fig. 2A, Supplemental Material). In the animal versions there are 2 pairs of CXXC (where C is cysteine and X any residue) motifs bounding the CC suggesting that they are likely to chelate Zn2+ with the 2 sub-regions of the CC folding back and internally dimerizing (Supplemental Material). This “CC segment” is abbreviated or absent in basal eukaryotes, such as Giardia and kinetoplastids, though these versions might have their own lineage-specific C-terminal extensions (Supplemental Material). Notably, certain alveolates (e.g., ciliates) and stramenopiles (oomycetes) might possess multiple paralogs of the HU-CCDC81 clade beyond a conventional version with the C-terminal CC region. One set of these paralogs is fused to multiple C-terminal EF-hand domains and in some cases IQ motifs (Fig. 2A). The other set of paralogs in these organisms is fused at the C-terminus to the Dynein-VII protein, which comprises a core of 6 AAA+ NTPase domain along with dynein heavy chain-specific modules such as DHC-N2 and DH-C (Fig. 2A).
Figure 2.

Phylogenetic relationships and genome associations in HU-CCDC81 and HU-HIG families. (A) Phylogenetic tree depicting higher-level relationships between HU families/clades described in this study. Branches are collapsed at levels containing clearly-delineated monophyletic groups, labeled to the right. Nodes with greater than 65% bootstrap support are marked with yellow circle. Representative conserved domain architectures and gene neighborhoods in a given clade provided to the right. (For complete list see Supplemental Material). Phyletic patterns of a given architecture/neighborhood are found provided to the right in green lettering. Phylogeny abbreviations: b, bacteroidetes; C, Chlamydia; P, Porphyromonas; s, spirochaetes; β, β-proteobacterial; γ, γ-proteobacteria; δ, δ-proteobacteria; v, verrucomicrobia; a, animals; k, kinetoplastids; api, apicomplexa; diplo, diplomonads; N, Naegleria; cili, ciliates; chloro, chlorophytes; stram, stramenopiles; SAR, stramenopile-alveolate-rhizarian group; Phy, Phytopthora; o, oomycetes; G, Guillardia. (B) Phylogenetic tree depicting the multiple paralogs identified in avian expansion of HU-CCDC81 domains. Monophyletic clades, as determined by phyletic distribution conservation patterns, are collapsed and then labeled and colored according to evolutionary depth. Nodes with greater than 70% bootstrap support are marked with yellow circle. Potential lineage-specific expansions within a clade are labeled with total number of non-redundant protein copies and phyletic patterns. (C) Phylogenetic tree depicting rampant LSEs, gene loss, and incomplete lineage-sorting in HU-HIG family based on a set of all HU-HIG sequences retrieved from the 10 bacteroidetes genomes, listed in key to the right, with the highest number of identifiable HU-HIG sequences. Branch coloring in tree corresponds to genome name colors in key. Domain architectures typical of sequences in clustered branches ring the tree, see (A) for explanation of architecture depictions. Complete trees provided in Newick format in Supplemental Material.
Like the eukaryotic versions, the bacterial versions of the HU-CCDC81 clade are also fused to C-terminal domains: all of them display a disordered segment followed by a TM region. The majority of them are further fused to C-terminal extracellular FtsN (Pfam model: SPOR) or LysM domains (Fig. 2A). A still smaller subset are instead fused to a conserved yet small and largely disordered C-terminal extension (Fig. 2A, Supplemental Material). Both the FtsN and LysM extracellular domains are peptidoglycan-binding domains,49,50 suggesting the small disordered extension functions similarly. This architecture suggests that the HU-CCDC81 domain(s) at the N-terminus are located inside the cell with the FtsN and LysM domains in the periplasmic space anchored to the cell wall via interactions with peptidoglycan. Versions with the LysM domain might come with either a single or 2 HU domains, while those with the FtsN domain always come with 2 N-terminal HU domains. Notably, the versions with a single HU-CCDC81 domain are typically encoded as part of a predicted 2 gene operon, wherein the second gene codes for a stand-alone HU domain protein which is similar in size and sequence conservation to the classical HU/IHF clades (Fig. 2A, Supplemental Material). This suggests that the 2 domain version originally arose in the context of the linked LysM domain via fusion with the adjacent gene coding for the stand-alone HU domain protein.
HU-CCDC81 clade is a possible cargo-binding domain in eukaryotic centrosome-linked trafficking
Keeping with the detection of CCDC81 as a centrosomal protein in animals,45 the phyletic pattern of the CCDC81 clade in eukaryotes is congruent with those organisms which possess centrosomes and the primary cilium51,52: 1) while it is present in chytridiomycota and chlorophyte algae, it is lost in other fungi and land plants mirroring the loss of the centrosome/primary cilium. 2) While it is strongly conserved in animals, it is lost in nematodes alone, which lack a centrosome and are characterized by amoeboid sperms. 3) Amoebozoans, which have entirely lost the cilium, lack CCDC81, while it is present in amoeboflagellates like Naegleria, which have the cilium in one stage of their life cycle. Further, the fusion of one of the paralogs of CCDC81 in stramenopiles and alveolates to Dynein-VII (Fig. 2A) links it to the centriole-derived structure, the axoneme, since the dynein-VII clade of dyneins are specifically axonemal dyneins.53 Fusions of the second paralog in these organisms to the EF-hands and IQ motif also links them to the centrosome, as these domains are key players in centrosomal interactions (e.g., EF-hand protein Centrin54 and IQCE55). Together these observations strongly suggest that members of the CCDC81 clade function throughout eukaryotes primarily as a centrosomal protein with potential links to the ciliary compartment.
This conclusion, at first sight, is at odds with the presence of HU domains in CCDC81, as all previously-studied representatives in both bacteria and eukaryotes are known to be DNA-binding proteins and CCDC81 is localized to an organelle distinct from those containing DNA (nucleus, mitochondrion or plastids). However, recent research has shown the nuclear and the ciliary compartments share a similar localization apparatus (based on the RAN GTPase56,57). Moreover, several nuclear DNA-repair signaling proteins are known to localize to the centrosome and shuttle between the 2 compartments. Hence, in principle, it is possible that CCDC81 shows a similar behavior and accesses DNA in the nucleus at specific points in the cell-cycle or under specific stimuli. However, our analysis of the C-terminal clasp structure of HU-CCDC81 points in a different direction: most residues from the β-hairpin which are implicated in DNA binding in the classical HU/IHF proteins are either lost or not conserved in HU-CCDC81 along with a drastic reduction of the positive charge in this region (Fig. 1E, Supplemental Material). Hence, it is likely that the derived HU domains in CCDC81 do not bind nucleic acids but rather interact with other proteins. This, together with its fusion to Dynein-VII,53 raises the possibility that the clasp of HU-CCDC81, rather than binding DNA or RNA, serves to bind protein cargo during microtubule-dependent translocation of proteins.
This leaves us with the enigma of the lineage-specific expansion of HU-CCDC81 in archosaurs, particularly birds (Fig. 2B). Such an expansion is unusual for an ancient eukaryotic protein, which is typically found in a single or a few copies and is predicted to function in a conserved organellar process. While centrosomes of birds have been noted to have some distinct ultrastructural features relative to other vertebrates,58 none of these are dramatic or even indicative of qualitative functional difference vis-à-vis other vertebrates. Phylogenetic trees reveal that the bird sequences for distinct clades are faster evolving compared with other vertebrate CCDC81 proteins (Fig. 2B). A direct measure of this divergence using position-specific entropy comparisons reveal an evenly-distributed and strong diversification propensity across the HU-CCDC81 archosaur expansion relative to mammalian counterpart sequences (Fig. 3A-B). These 2 observations suggest that the multiple observed copies of the CCDC81-HU domains in bird proteins are under rapid pressure to diverge, possibly acquiring novel functional properties. Birds are unusual in possessing numerous gene-rich microchromosomes that are distinct from the gene-poor macrochromosomes.59, 60 The former tend to cluster toward the nuclear center and during cell-division there is a need for a greater number of microtubular filaments radiating from the spindle poles for chromosome segregation. Hence, given their rapid divergence, one possibility is that the expansion of HU-CCDC81 in birds might relate to the expansion of microchromosomes and the organization of the spindle during their segregation. However, another distinct possibility is their role as binding proteins which are recognizing a fast-evolving target, like from a pathogen. Such a possibility is consistent with comparable observation we made for bacterial expansions of HU proteins (see below).
Figure 3.

Positional entropy and sequence diversity comparisons. (A) Positional entropy comparison between Gallus and Meleagris HU-CCDC81 domains (galliform birds) and primate and rodent HU-CCDC81 domains. Entropy values calculated as described in Materials and Methods. (B) Entropy values from (A) plotted along linear sequence of HU-CCDC81 domain, secondary structure provided below and labeled in concordance with Fig. 1(B). (C-F) Sequence diversity plots comparing pairwise sequence evolutionary distances (see Materials and Methods) within representatives of labeled HU families, y-axes set to log scale. Differences in boxplots (A, C-F) are significant (p < 2.2e-16) by Wilcoxon rank sum test.
Bacterial HU-CCDC81 proteins potentially function in tethering the nucleoid to the cell-envelope
Interestingly, architectures of bacterial members of the HU-CCDC81 imply that they are anchored to the membrane and cell wall with the HU domains free to participate in cytoplasmic contacts (Fig. 2A). At least in the case of the versions with a single HU domain, their operonic partner, which is closer in sequence and structure to the classical HU/IHF domains, might be able to support interactions with DNA in the heterodimer. In light of this, we propose that these bacterial versions of this clade might play a role in anchoring the nucleoid to the cell-envelope via direct interactions with DNA. This might be important during cell-division where chromosomes need to be segregated. Indeed, parallel roles have been proposed in other bacteria like Bacillus for proteins implicated in tethering DNA directly (Noc)61 or indirectly (RacA)62 to the cell-envelope. In this context, it is interesting to note that archaeal HUs from the halobacterial lineage are also fused to N-terminal TM helices (Supplemental Material), suggesting that they might have convergently evolved a similar role in tethering the nucleoid to the membrane.
The bacterial HU-CCDC81 proteins with 2 HU domains, however, show sequence features comparable to their eukaryotic counterparts, which suggests that they, too, might not bind DNA. Of particular note, at least the second domain in these versions is evolving rapidly relative to classical HU/IHF counterparts when comparing sequences observed in the same complement of genomes (Fig. 3C-D). Thus, it is possible that they play a comparable role in nucleoid tethering as the above-discussed versions via protein-protein interactions rather than by directly binding DNA as has been proposed for the RacA-DivIVA complex in Bacillus.63 Hence, the shift from DNA-binding to an alternative binding interface appears to have happened within the bacterial HU-CCDC81 proteins in the bacteroidetes-chlorobi lineage. Hence, such a version was likely acquired early in eukaryotic evolution specifically from a bacteroidetes-like source independently of the α-proteobacterial mitochondrial symbiont and recruited for comparable interactions in microtubular trafficking.
The bacteroidetes radiation of HU and its functional implications
The dramatic but poorly understood lineage-specific expansion of HU domains26 and the unusual structural modification of the HU-CCDC81 domain, which emerged in the bacteroidetes lineage, prompted us to more closely examine the evolutionary radiation and functional diversification of the HU superfamily in this lineage. Our phylogenetic analysis revealed that in bacteroidetes, the HU superfamily shows 2 distinct clades beyond the HU-CCDC81 clade (Fig. 2). The first of these typically has only 1–3 members and is close in sequence and size to the classical HU/IHF proteins. A member of this clade from Bacteroides thetaiotaomicron has recently been shown to be a chromosomal protein that functions as a host factor for the integration of the Integrative Conjugative Element CTnDOT64 - a function comparable to the E.coli IHFs and HU. The second clade comprises larger proteins, which are distinguished by a modified HU domain that possesses an additional N-terminal β-hairpin and a C-terminal strand. Together, these form a 3-stranded β-sheet that buttresses the core C-terminal β-sheet while contributing few residues to the binding-pocket of the clasp (Fig. 1C-D, Supplemental Material). This clade has undergone the lineage-specific expansions (LSEs) characteristic of bacteroidetes.26
In-depth phylogenetic analysis of this second clade reveals that it contains 3 distinct subclades with representatives across bacteroidetes, suggesting that they had diverged from each other early in the evolution of the lineage (Fig. 2C). Each sub-clade is marked by distinct domain architectures with C-terminal fusions respectively to a winged HTH (wHTH) domain, an immunoglobulin fold (Ig) domain and a glycine-rich motif with a predicted β-strand-like extended region (Fig. 2A,C). The Ig domain fused subclade can additionally house the glycine-rich motif at the extreme C-terminus (Fig. 2C, Supplemental Material). Accordingly, we hereafter refer to the clade as HU-HIG (wHTH, Ig, Glycine-rich motif). However, within each HU-HIG subclade we observed multiple LSEs and gene-loss events along with incomplete lineage-sorting even between closely related species within a given genus (Fig. 2C). Additionally, we observed that unlike the classical HU/IHF proteins, these are rapidly diverging even within members of a LSE in a single bacteroidetes species (Fig. 2C, Supplemental Material). Further, HU-HIG domains are diverging more rapidly than counterpart classical HU/IHF versions, and at rates comparable to the second domain of the bacterial HU-CCDC81 2-domain architectural configuration (Fig. 3E-F).
Several distinct evolutionary pressures affecting the HU-HIG clade can be identified from these observations. First, the different domain architectures point to a degree of functional divergence. The structural genomics program has determined a structure of one of the proteins with fusion to a C-terminal Ig domain (PDB: 4FMR). Examination of this structure reveals that the Ig domain partly occupies the space that is occupied by DNA in the classical HU/IHF domains (Fig. 1A,C). The interaction between this Ig domain and the clasp of the HU domain also greatly narrows the binding cleft formed by the clasp. Hence, the clasp is unlikely to bind DNA. On the other hand, those fused to the wHTH domain are likely to possess an augmented DNA interface, where that domain makes additional contacts with the adjacent major and minor grooves. Those versions with a glycine-rich tail possess a well-conserved short extended C-terminal region, which might mediate specific interaction with a binding partner. Second, despite these differences, the overall pattern of evolutionary radiation points in a similar direction across the clade: there is clear selection both for maintaining multiple copies as well as their sequence diversification (Fig. 2C and Fig. 3E-F, Supplemental Material). Third, genes for members of this clade almost entirely lack any conserved operonic linkages to any other genes, although sometimes they are observed adjacent to each other in pairs along the genome pointing to recent duplications (Fig. 2A, Supplemental Material).
Together these observations have clear functional implications for these enigmatic HU-HIG proteins. The above features indicate that they are unlikely to function as sequence-specific transcription factors. However, like other proteins, which tend to show rampant lineage-specific expansions coupled with sequence diversification, they are likely to be at the interface of a biologic conflict, for instance in the recognition of similarly diverse non-self-molecules or in evasion of targeting by effectors of invasive elements.65-67 Hence, we propose that the bacteroidetes expansion is indeed related to one such process. It is well-known that across bacteria members of the HU/IHF family are used by diverse invasive DNA elements for insertion into genomes (including the CTnDOT element in Bacteroidetes64). It has also been seen to play a comparable role in the insertion of CRISPR spacer sequences from invasive elements.34 Combining these observations, one attractive possibility that emerges is a subset of HU-HIG members directly intercept the DNA of parasitic elements and prevent their incorporation into genomes. This scenario is likely to have set off an “arms-race” like situation to allow the observed diversification. Furthermore, it also probably selected for versions that interacted with proteins or RNA from invasive elements rather than the DNA itself, potentially accounting for the unusual versions fused to Ig domains.
Discussion
One of the enigmas pertaining to the evolution of DNA-packaging is the use of apparently distinct folds for the packaging of DNA in the bacterial and the archaeo-eukaryotic lineage. Based on their phyletic patterns, it is likely that the ancestral DNA packaging protein in the archaeo-eukaryotic lineage were the histones,8 whereas in the bacterial lineage it was HU.15,68 While their structures and modes of DNA-interaction appear different, they still share some noteworthy features. The core DNA-binding unit in both cases is a dimer and both possess a structurally comparable bihelical element that is the basis of their dimerization. Hence, it is possible that the 2 shared a common ancestor, which underwent drastic divergence in their DNA-binding mode after the divergence of bacteria and archaea from the LUCA.
While this could account for the origin of the unique fold of the widely distributed HU superfamily, it has so far been only characterized as playing a role in a DNA-binding capacity. Our current studies present evidence that the HU superfamily encompasses greater structure and functional diversity than has been previously appreciated. A conspicuous aspect of this diversity is the inclusion of biochemical roles beyond DNA-binding while preserving the core fold. A major part of this functional diversification appears to have happened within the bacteroidetes lineage to encompass forms that are predicted to play roles as diverse as chromosome tethering and countering invasion of the genome by DNA elements. The transfer of one representative clade of this bacteroidetes radiation to give rise to the eukaryotic HU-CCDC81 proteins provides evidence for important contributions from bacterial sources other than the mitochondrial progenitor in eukaryogenesis. This might point to existence of other early endosymbionts, especially given that several extant members of the bacteroidetes lineage are endosymbionts in modern eukaryotes.69 Dramatically, in just the archosaurian lineage there appears to have been a re-utilization of the HU-CCDC81 in a capacity similar to their counterparts in bacteroidetes.
We hope that this investigation of the diversity of the HU superfamily might help in future investigation of its broader functional scope.
Materials and methods
Protein sequences belonging to the bona fide bacterial HU/IHF family were retrieved from the Genbank at the National Center for Biotechnology Information (NCBI). These were used to seed iterative PSI-BLAST43 and JackHMMER44 searches against the above-described, locally maintained protein sequence databases and, as appropriate, the non-redundant (nr) database using an expect (e)-value threshold of 0.01. New sequences retrieved in the process were used to re-iterate searches and collect divergent members. The HHpred program47 was used to detect remote homologs in the PDB and PFAM databases. Clustering of protein sequences was done using the BLASTCLUST program (ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html), adjusting the length of aligned regions and bit-score density threshold empirically. Multiple sequence alignments (MSAs) were generated using the Kalign70 and Muscle71 programs with default parameters. These MSAs were adjusted manually, guided by structure superimpositions, profile-profile alignments and secondary structure prediction. All structures were visualized and compared using the molecular visualization program PyMOL (https://www.pymol.org/). The JPred72 program was used to predict secondary structures.
An approximately maximum-likelihood method as implemented in the FastTree73 program with other default parameters was used to assess the phylogenetic relationships. The FigTree program (http://tree.bio.ed.ac.uk/software/figtree/) was used to render phylogenetic trees. Co-occurring domains and gene neighborhoods were retrieved through custom scripts and using tools of the TASS software package (Anantharaman, V., Balaji, S., Aravind, L., unpublished results).
Position-wise Shannon entropy (H) analysis for MSAs was performed using the equation:
Where M is the number of amino acid types and P is the fraction of residues of amino acid type i. The Shannon entropy for any given position in the MSA ranges from 0 (absolutely conserved one amino acid at that position) to 4.32 (all 20 amino acid residues equally represented at that position). Sequence diversity comparisons were performed across 2 families by extracting all sequences found in the set of bacterial genomes with at least one member in both families. Diversity values were calculated as all-against-all pairwise distance scores between all sequence pairs of the same family, under the Jones-Taylor-Thornton (JTT) substitution model using a Gamma distribution with parameter 1, as implemented by the MEGA5 software package.74 Rstudio (http://www.rstudio.com/) was used for analysis and visualization of the entropy and diversity values thus obtained.
Disclosure of potential conflicts of interest
The authors declare that research was conducted in absence of commercial or financial relationship that could be construed as a conflict of interest.
Funding
This research was supported by the Intramural Research Program of the NIH, National Library of Medicine.
References
- [1].Dillon SC, Dorman CJ. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat Rev Microbiol 2010; 8:185-95; PMID:20140026; https://doi.org/ 10.1038/nrmicro2261 [DOI] [PubMed] [Google Scholar]
- [2].Reeve JN, Bailey KA, Li WT, Marc F, Sandman K, Soares DJ. Archaeal histones: structures, stability and DNA binding. Biochem Soc Trans 2004; 32:227-30; PMID:15046577; https://doi.org/ 10.1042/bst0320227 [DOI] [PubMed] [Google Scholar]
- [3].Sandman K, Reeve JN. Archaeal chromatin proteins: different structures but common function? Curr Opin Microbiol 2005; 8:656-61; PMID:16256418; https://doi.org/ 10.1016/j.mib.2005.10.007 [DOI] [PubMed] [Google Scholar]
- [4].Jones DO, Cowell IG, Singh PB. Mammalian chromodomain proteins: their role in genome organisation and expression. Bioessays 2000; 22:124-37; PMID:10655032; https://doi.org/ 10.1002/(SICI)1521-1878(200002)22:2%3c124::AID-BIES4%3e3.0.CO;2-E [DOI] [PubMed] [Google Scholar]
- [5].Luijsterburg MS, White MF, van Driel R, Dame RT. The major architects of chromatin: architectural proteins in bacteria, archaea and eukaryotes. Crit Rev Biochem Mol Biol 2008; 43:393-418; PMID:19037758; https://doi.org/ 10.1080/10409230802528488 [DOI] [PubMed] [Google Scholar]
- [6].de Souza RF, Iyer LM, Aravind L. Diversity and evolution of chromatin proteins encoded by DNA viruses. Biochim Biophys Acta 2010; 1799:302-18; PMID:19878744; https://doi.org/ 10.1016/j.bbagrm.2009.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sandman K, Pereira SL, Reeve JN. Diversity of prokaryotic chromosomal proteins and the origin of the nucleosome. Cell Mol Life Sci 1998; 54:1350-64; PMID:9893710; https://doi.org/ 10.1007/s000180050259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Arents G, Moudrianakis EN. The histone fold: a ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc Natl Acad Sci U S A 1995; 92:11170-4; PMID:7479959; https://doi.org/ 10.1073/pnas.92.24.11170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997; 389:251-60; PMID:9305837; https://doi.org/ 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- [10].Sandman K, Reeve JN. Chromosome packaging by archaeal histones. Adv Appl Microbiol 2001; 50:75-99; PMID:11677690 [DOI] [PubMed] [Google Scholar]
- [11].White MF, Bell SD. Holding it together: chromatin in the Archaea. Trends Genet 2002; 18:621-6; PMID:12446147; https://doi.org/ 10.1016/S0168-9525(02)02808-1 [DOI] [PubMed] [Google Scholar]
- [12].Wardleworth BN, Russell RJ, Bell SD, Taylor GL, White MF. Structure of Alba: an archaeal chromatin protein modulated by acetylation. EMBO J 2002; 21:4654-62; PMID:12198167; https://doi.org/ 10.1093/emboj/cdf465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Paquet F, Culard F, Barbault F, Maurizot JC, Lancelot G. NMR solution structure of the archaebacterial chromosomal protein MC1 reveals a new protein fold. Biochemistry 2004; 43:14971-8; PMID:15554704; https://doi.org/ 10.1021/bi048382z [DOI] [PubMed] [Google Scholar]
- [14].Guo L, Feng Y, Zhang Z, Yao H, Luo Y, Wang J, Huang L. Biochemical and structural characterization of Cren7, a novel chromatin protein conserved among Crenarchaea. Nucleic Acids Res 2008; 36:1129-37; PMID:18096617; https://doi.org/ 10.1093/nar/gkm1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Grove A. Functional evolution of bacterial histone-like HU proteins. Curr Issues Mol Biol 2011; 13:1-12; PMID:20484776 [PubMed] [Google Scholar]
- [16].Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol 2004; 14:28-35; PMID:15102446; https://doi.org/ 10.1016/j.sbi.2003.12.003 [DOI] [PubMed] [Google Scholar]
- [17].Luijsterburg MS, Noom MC, Wuite GJ, Dame RT. The architectural role of nucleoid-associated proteins in the organization of bacterial chromatin: a molecular perspective. J Struct Biol 2006; 156:262-72; PMID:16879983; https://doi.org/ 10.1016/j.jsb.2006.05.006 [DOI] [PubMed] [Google Scholar]
- [18].Dame RT. The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin. Mol Microbiol 2005; 56:858-70; PMID:15853876; https://doi.org/ 10.1111/j.1365-2958.2005.04598.x [DOI] [PubMed] [Google Scholar]
- [19].Kobayashi T, Takahara M, Miyagishima SY, Kuroiwa H, Sasaki N, Ohta N, Matsuzaki M, Kuroiwa T. Detection and localization of a chloroplast-encoded HU-like protein that organizes chloroplast nucleoids. Plant Cell 2002; 14:1579-89; PMID:12119376; https://doi.org/ 10.1105/tpc.002717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sato N. Was the evolution of plastid genetic machinery discontinuous? Trends Plant Sci 2001; 6:151-5; PMID:11286919; https://doi.org/ 10.1016/S1360-1385(01)01888-X [DOI] [PubMed] [Google Scholar]
- [21].Karcher D, Koster D, Schadach A, Klevesath A, Bock R. The Chlamydomonas chloroplast HLP protein is required for nucleoid organization and genome maintenance. Mol Plant 2009; 2:1223-32; PMID:19995727; https://doi.org/ 10.1093/mp/ssp083 [DOI] [PubMed] [Google Scholar]
- [22].Ram EV, Naik R, Ganguli M, Habib S. DNA organization by the apicoplast-targeted bacterial histone-like protein of Plasmodium falciparum. Nucleic Acids Res 2008; 36:5061-73; PMID:18663012; https://doi.org/ 10.1093/nar/gkn483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Reiff SB, Vaishnava S, Striepen B. The HU protein is important for apicoplast genome maintenance and inheritance in Toxoplasma gondii. Eukaryot Cell 2012; 11:905-15; PMID:22611021; https://doi.org/ 10.1128/EC.00029-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wong JT, New DC, Wong JC, Hung VK. Histone-like proteins of the dinoflagellate Crypthecodinium cohnii have homologies to bacterial DNA-binding proteins. Eukaryot Cell 2003; 2:646-50; PMID:12796310; https://doi.org/ 10.1128/EC.2.3.646-650.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shoguchi E, Shinzato C, Kawashima T, Gyoja F, Mungpakdee S, Koyanagi R, Takeuchi T, Hisata K, Tanaka M, Fujiwara M, et al.. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr Biol 2013; 23:1399-408; PMID:23850284; https://doi.org/ 10.1016/j.cub.2013.05.062 [DOI] [PubMed] [Google Scholar]
- [26].Dey D, Nagaraja V, Ramakumar S. Structural and evolutionary analyses reveal determinants of DNA binding specificities of nucleoid-associated proteins HU and IHF. Mol Phylogenet Evol 2017; 107:356-66; PMID:27894997; https://doi.org/ 10.1016/j.ympev.2016.11.014 [DOI] [PubMed] [Google Scholar]
- [27].Bonnefoy E, Rouviere-Yaniv J. HU and IHF, two homologous histone-like proteins of Escherichia coli, form different protein-DNA complexes with short DNA fragments. EMBO J 1991; 10:687-96; PMID:2001682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Drlica K, Rouviere-Yaniv J. Histonelike proteins of bacteria. Microbiol Rev 1987; 51:301-19; PMID:3118156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Balandina A, Kamashev D, Rouviere-Yaniv J. The bacterial histone-like protein HU specifically recognizes similar structures in all nucleic acids. DNA, RNA, and their hybrids. J Biol Chem 2002; 277:27622-8; PMID:12006568; https://doi.org/ 10.1074/jbc.M201978200 [DOI] [PubMed] [Google Scholar]
- [30].Craig NL, Nash HA. E. coli integration host factor binds to specific sites in DNA. Cell 1984; 39:707-16; PMID:6096022; https://doi.org/ 10.1016/0092-8674(84)90478-1 [DOI] [PubMed] [Google Scholar]
- [31].Benevides JM, Danahy J, Kawakami J, Thomas GJ Jr.. Mechanisms of specific and nonspecific binding of architectural proteins in prokaryotic gene regulation. Biochemistry 2008; 47:3855-62; PMID:18302340; https://doi.org/ 10.1021/bi7009426 [DOI] [PubMed] [Google Scholar]
- [32].Nash HA, Robertson CA. Purification and properties of the Escherichia coli protein factor required for lambda integrative recombination. J Biol Chem 1981; 256:9246-53; PMID:6267068 [PubMed] [Google Scholar]
- [33].Yu A, Haggard-Ljungquist E. Characterization of the binding sites of two proteins involved in the bacteriophage P2 site-specific recombination system. J Bacteriol 1993; 175:1239-49; PMID:8444786; https://doi.org/ 10.1128/jb.175.5.1239-1249.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nunez JK, Bai L, Harrington LB, Hinder TL, Doudna JA. CRISPR Immunological Memory Requires a Host Factor for Specificity. Mol Cell 2016; 62:824-33; PMID:27211867; https://doi.org/ 10.1016/j.molcel.2016.04.027 [DOI] [PubMed] [Google Scholar]
- [35].Johnson RC, Bruist MF, Simon MI. Host protein requirements for in vitro site-specific DNA inversion. Cell 1986; 46:531-9; PMID:3524854; https://doi.org/ 10.1016/0092-8674(86)90878-0 [DOI] [PubMed] [Google Scholar]
- [36].Aki T, Adhya S. Repressor induced site-specific binding of HU for transcriptional regulation. EMBO J 1997; 16:3666-74; PMID:9218807; https://doi.org/ 10.1093/emboj/16.12.3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lewis DE, Geanacopoulos M, Adhya S. Role of HU and DNA supercoiling in transcription repression: specialized nucleoprotein repression complex at gal promoters in Escherichia coli. Mol Microbiol 1999; 31:451-61; PMID:10027963; https://doi.org/ 10.1046/j.1365-2958.1999.01186.x [DOI] [PubMed] [Google Scholar]
- [38].Wei J, Czapla L, Grosner MA, Swigon D, Olson WK. DNA topology confers sequence specificity to nonspecific architectural proteins. Proc Natl Acad Sci U S A 2014; 111:16742-7; PMID:25385626; https://doi.org/ 10.1073/pnas.1405016111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].White SW, Appelt K, Wilson KS, Tanaka I. A protein structural motif that bends DNA. Proteins 1989; 5:281-8; PMID:2508086; https://doi.org/ 10.1002/prot.340050405 [DOI] [PubMed] [Google Scholar]
- [40].Rice PA, Yang S, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell 1996; 87:1295-306; PMID:8980235; https://doi.org/ 10.1016/S0092-8674(00)81824-3 [DOI] [PubMed] [Google Scholar]
- [41].Swinger KK, Lemberg KM, Zhang Y, Rice PA. Flexible DNA bending in HU-DNA cocrystal structures. EMBO J 2003; 22:3749-60; PMID:12853489; https://doi.org/ 10.1093/emboj/cdg351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yee B, Sagulenko E, Fuerst JA. Making heads or tails of the HU proteins in the planctomycete Gemmata obscuriglobus. Microbiology 2011; 157:2012-21; PMID:21511768; https://doi.org/ 10.1099/mic.0.047605-0 [DOI] [PubMed] [Google Scholar]
- [43].Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25:3389-402; PMID:9254694; https://doi.org/ 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR. HMMER web server: 2015 update. Nucleic Acids Res 2015; 43:W30-8; https://doi.org/ 10.1093/nar/gkv397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Firat-Karalar EN, Sante J, Elliott S, Stearns T. Proteomic analysis of mammalian sperm cells identifies new components of the centrosome. J Cell Sci 2014; 127:4128-33; PMID:25074808; https://doi.org/ 10.1242/jcs.157008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bateman A, Coggill P, Finn RD. DUFs: families in search of function. Acta Crystallogr Sect F Struct Biol Cryst Commun 2010; 66:1148-52; PMID:20944204; https://doi.org/ 10.1107/S1744309110001685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 2005; 33:W244-8; PMID:15980461; https://doi.org/ 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Orfaniotou F, Tzamalis P, Thanassoulas A, Stefanidi E, Zees A, Boutou E, Vlassi M, Nounesis G, Vorgias CE. The stability of the archaeal HU histone-like DNA-binding protein from Thermoplasma volcanium. Extremophiles 2009; 13:1-10; PMID:18818867; https://doi.org/ 10.1007/s00792-008-0190-6 [DOI] [PubMed] [Google Scholar]
- [49].Yang JC, Van Den Ent F, Neuhaus D, Brevier J, Lowe J. Solution structure and domain architecture of the divisome protein FtsN. Mol Microbiol 2004; 52:651-60; PMID:15101973; https://doi.org/ 10.1111/j.1365-2958.2004.03991.x [DOI] [PubMed] [Google Scholar]
- [50].Bateman A, Bycroft M. The structure of a LysM domain from E. coli membrane-bound lytic murein transglycosylase D (MltD). J Mol Biol 2000; 299:1113-9; PMID:10843862; https://doi.org/ 10.1006/jmbi.2000.3778 [DOI] [PubMed] [Google Scholar]
- [51].Carvalho-Santos Z, Machado P, Branco P, Tavares-Cadete F, Rodrigues-Martins A, Pereira-Leal JB, Bettencourt-Dias M. Stepwise evolution of the centriole-assembly pathway. J Cell Sci 2010; 123:1414-26; PMID:20392737; https://doi.org/ 10.1242/jcs.064931 [DOI] [PubMed] [Google Scholar]
- [52].Zhang D, Aravind L. Novel transglutaminase-like peptidase and C2 domains elucidate the structure, biogenesis and evolution of the ciliary compartment. Cell Cycle 2012; 11:3861-75; PMID:22983010; https://doi.org/ 10.4161/cc.22068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hook P, Vallee RB. The dynein family at a glance. J Cell Sci 2006; 119:4369-71; PMID:17074830; https://doi.org/ 10.1242/jcs.03176 [DOI] [PubMed] [Google Scholar]
- [54].Dantas TJ, Daly OM, Morrison CG. Such small hands: the roles of centrins/caltractins in the centriole and in genome maintenance. Cell Mol Life Sci 2012; 69:2979-97; PMID:22460578; https://doi.org/ 10.1007/s00018-012-0961-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pusapati GV, Hughes CE, Dorn KV, Zhang D, Sugianto P, Aravind L, Rohatgi R. EFCAB7 and IQCE regulate hedgehog signaling by tethering the EVC-EVC2 complex to the base of primary cilia. Dev Cell 2014; 28:483-96; PMID:24582806; https://doi.org/ 10.1016/j.devcel.2014.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fan S, Whiteman EL, Hurd TW, McIntyre JC, Dishinger JF, Liu CJ, Martens JR, Verhey KJ, Sajjan U, Margolis B. Induction of Ran GTP drives ciliogenesis. Mol Biol Cell 2011; 22:4539-48; PMID:21998203; https://doi.org/ 10.1091/mbc.E11-03-0267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jekely G. Origin of the nucleus and Ran-dependent transport to safeguard ribosome biogenesis in a chimeric cell. Biol Direct 2008; 3:31; PMID:18652645; https://doi.org/ 10.1186/1745-6150-3-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Soley JT. A comparative overview of the sperm centriolar complex in mammals and birds: Variations on a theme. Anim Reprod Sci 2016; 169:14-23; PMID:26907939; https://doi.org/ 10.1016/j.anireprosci.2016.02.006 [DOI] [PubMed] [Google Scholar]
- [59].Habermann FA, Cremer M, Walter J, Kreth G, von Hase J, Bauer K, Wienberg J, Cremer C, Cremer T, Solovei I. Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res 2001; 9:569-84; PMID:11721954; https://doi.org/ 10.1023/A:1012447318535 [DOI] [PubMed] [Google Scholar]
- [60].Ellegren H. The avian genome uncovered. Trends Ecol Evol 2005; 20:180-6; https://doi.org/ 10.1016/j.tree.2005.01.015 [DOI] [PubMed] [Google Scholar]
- [61].Adams DW, Wu LJ, Errington J. Nucleoid occlusion protein Noc recruits DNA to the bacterial cell membrane. EMBO J 2015; 34:491-501; PMID:25568309; https://doi.org/ 10.15252/embj.201490177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wu LJ, Errington J. RacA and the Soj-Spo0J system combine to effect polar chromosome segregation in sporulating Bacillus subtilis. Mol Microbiol 2003; 49:1463-75; PMID:12950914; https://doi.org/ 10.1046/j.1365-2958.2003.03643.x [DOI] [PubMed] [Google Scholar]
- [63].van Baarle S, Celik IN, Kaval KG, Bramkamp M, Hamoen LW, Halbedel S. Protein-protein interaction domains of Bacillus subtilis DivIVA. J Bacteriol 2013; 195:1012-21; PMID:23264578; https://doi.org/ 10.1128/JB.02171-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ringwald K, Gardner J. The Bacteroides thetaiotaomicron protein Bacteroides host factor A participates in integration of the integrative conjugative element CTnDOT into the chromosome. J Bacteriol 2015; 197:1339-49; PMID:25645562; https://doi.org/ 10.1128/JB.02198-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hurst LD, Atlan A, Bengtsson BO. Genetic conflicts. Q Rev Biol 1996; 71:317-64; PMID:8828237; https://doi.org/ 10.1086/419442 [DOI] [PubMed] [Google Scholar]
- [66].Werren JH. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc Natl Acad Sci U S A 2011; 108 Suppl 2:10863-70; PMID:21690392; https://doi.org/ 10.1073/pnas.1102343108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Aravind L, Anantharaman V, Zhang D, de Souza RF, Iyer LM. Gene flow and biological conflict systems in the origin and evolution of eukaryotes. Front Cell Infect Microbiol 2012; 2:89; PMID:22919680; https://doi.org/ 10.3389/fcimb.2012.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Oberto J, Drlica K, Rouviere-Yaniv J. Histones, HMG, HU, IHF: Meme combat. Biochimie 1994; 76:901-8; PMID:7748933; https://doi.org/ 10.1016/0300-9084(94)90014-0 [DOI] [PubMed] [Google Scholar]
- [69].Toft C, Andersson SG. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet 2010; 11:465-75; PMID:20517341; https://doi.org/ 10.1038/nrg2798 [DOI] [PubMed] [Google Scholar]
- [70].Lassmann T, Sonnhammer EL. Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics 2005; 6:298; PMID:16343337; https://doi.org/ 10.1186/1471-2105-6-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792-7; PMID:15034147; https://doi.org/ 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: a protein secondary structure prediction server. Nucleic Acids Res 2015; 43:W389-94; PMID:25883141; https://doi.org/ 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 2010; 5:e9490; PMID:20224823; https://doi.org/ 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evolution 2011; 28:2731-9; PMID:21546353; https://doi.org/ 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]