


Abstract
The virulon of Staphyloccocus aureus is controlled by intricate connections between transcriptional and post-transcriptional regulators including proteins and small non-coding RNAs (sRNAs). Many of the sRNAs regulate gene expression through base-pairings with mRNAs. However, characterization of the direct sRNA targets in Gram-positive bacteria remained a difficult challenge. Here, we have applied and adapted the MS2-affinity purification approach coupled to RNA sequencing (MAPS) to determine the targetome of RsaA sRNA of S. aureus, known to repress the synthesis of the transcriptional regulator MgrA. Several mRNAs were enriched with RsaA expanding its regulatory network. Besides mgrA, several of these mRNAs encode a family of SsaA-like enzymes involved in peptidoglycan metabolism and the secreted anti-inflammatory FLIPr protein. Using a combination of in vivo and in vitro approaches, these mRNAs were validated as direct RsaA targets. Quantitative differential proteomics of wild-type and mutant strains corroborated the MAPS results. Additionally, it revealed that RsaA indirectly activated the synthesis of surface proteins supporting previous data that RsaA stimulated biofilm formation and favoured chronic infections. All together, this study shows that MAPS could also be easily applied in Gram-positive bacteria for identification of sRNA targetome.
INTRODUCTION
The importance of post-transcriptional regulation in bacteria is now widely recognized for several adaptive cellular processes such as iron regulation, biofilm formation, quorum sensing and virulence (1). These regulations are essentially achieved through a combination of proteins (RNA binding proteins, ribonucleases) and small non-coding RNAs (sRNAs). Pioneering studies performed in Escherichia coli and closely related bacteria have revealed that most of the sRNAs regulate gene expression through direct mRNA binding (2,3). Because many sRNAs regulate transcriptional/post-transcriptional regulatory proteins, they induce downstream effects rendering challenging the identification of their primary targets. In addition, computational target predictions are delicate because basepairing interactions can be rather short and non-contiguous despite the fact that they often involved conserved and unpaired regions of the sRNAs (reviewed in 4,5). Hence, several approaches have been developed over the years to identify primary sRNA targets in order to characterize their regulatory networks and their functional impact on cell physiology (6,3,7). In E. coli and closely related bacteria, analysis of the rapid changes in mRNA levels after pulse induction of the analysed sRNA has been for a long time one of the most successful methods (6). Besides, other approaches were used to assess targets of sRNAs independent of mRNA levels such as differential proteomic approaches (8), ribosome profiling (9) or purification of regulatory complexes using protein factors as baits (10,11). More recently, affinity purification of in vivo regulatory complexes coupled with high-throughput RNA sequencing methodology or MAPS standing for ‘MS2 affinity purification coupled to RNA sequencing’ (7,12,13) has proven to be powerful to access to the whole targetome of any type of RNAs in E. coli. In this work, we have adapted the MAPS strategy to Staphylococcus aureus sRNAs, and in particular to RsaA.
S. aureus is a Gram-positive opportunistic pathogen that is well adapted to diverse hosts and ecological niches. The bacterium is present in a third of the population on skin and in nostrils as commensal bacteria, but is also responsible for a wide range of infections from minor skin abscesses to life-threatening diseases such as bacteremia, infective endocarditis or toxic shock syndrome. Mainly recognized as extracellular pathogen, it can nevertheless invade professional and non-professional phagocytes leading to chronic infections that are difficult to treat (14). S. aureus produces a plethora of virulence factors including secreted toxins, exopolysaccharides, the major component of the biofilm matrix, capsule and several surface proteins contributing to host adhesion. Transcriptional factors, two components systems and RNA regulators fine-tune the virulence expression programs (15). The discovery of RNAIII, the major RNA regulator of virulence in S. aureus (16,17), has opened new ways of research. Today, more than 100 sRNAs have been described in S. aureus although the functions of many of them are still unknown (reviewed in 18). Several studies revealed that RNAIII and other sRNAs contain conserved UCCC seed sequences directed against the ribosome-binding sites (RBS) of target mRNAs (i.e. 19,20,21). This mode of pairings is conserved among several Gram-positive bacteria (e.g. 22,23). This interaction hinders ribosome binding to inhibit translation initiation but in contrast to Gram-negative bacteria, does not necessarily lead to rapid depletion of the target mRNAs. This was the case with the Sigma B-dependent RsaA sRNA, which was first identified in S. aureus genome using bioinformatics and expression studies (24). By comparing the cytosolic protein contents of the wild-type (WT) and the mutant ΔrsaA strains by a 2D-gel approach, the synthesis of proteins belonging to the regulon of the global transcriptional regulator MgrA was altered and the effect of RsaA was opposite to that of MgrA (25). In agreement with these observations, RsaA was shown to repress the translation of mgrA mRNA by forming imperfect duplexes between its C-rich motif and RBS of mgrA mRNA further stabilized by a loop–loop interaction within the coding region of mgrA. Consequently, RsaA causes enhanced production of biofilm, decreased synthesis of capsule formation, and decreased protection of S. aureus against opsonophagocytic killing by polymorphonuclear leukocytes (25).
In order to extend the repertoire of RsaA targets, we adapted for the first time the MAPS technology to S. aureus. RsaA was tagged at its 5΄ end with two hairpin motifs recognized by the MS2 coat protein and expressed in vivo. After cell lysis, affinity chromatography purification followed by RNA-seq, we have confirmed that mgrA mRNA was the main mRNA target of RsaA and have revealed other mRNAs encoding cell wall hydrolases and FLIPr (also called FlR), a secreted immunomodulatory molecule that interferes with effective opsonization by the complement and/or IgG antibodies (26). The MAPS approach appears to be a powerful methodology to extend RsaA regulon of S. aureus. This strategy was further corroborated to a complete differential proteomic analysis of the WT and ΔrsaA strains including cytosolic and secreted proteins.
MATERIALS AND METHODS
Strains, plasmids and growth conditions
S. aureus strains and plasmids used in this study are listed in Supplementary Table S1. E. coli strain DC10B (27) was used as a host strain for plasmid construction. Plasmids extracted from E. coli DC10B can be used directly for S. aureus electroporation. E. coli strain was cultivated in Luria-Bertani (LB) medium (1% peptone, 0.5% yeast extract, 1% NaCl) supplemented with ampicillin (100 μg/ml) when necessary. LB-agar plates (with or without ampicillin) were also used for growth on solid medium. S. aureus strains were grown in Brain-Heart Infusion (BHI) medium (Sigma-Aldrich) supplemented with erythromycin (10 μg/ml) when necessary. Blood-agar (VWR Chemicals) and BHI plates (with or without erythromycin) were used for growth on solid medium. Plasmids were prepared from transformed E. coli pellets using the Nucleospin Plasmid kit (Macherey-Nagel). Transformation of both E. coli and S. aureus strains was performed by electroporation (Bio-Rad Gene Pulser).
All plasmids were prepared using pCN51 as template vector (28). Synthesis of polymerase chain reaction (PCR) products was performed using Phusion Polymerase (Thermoscientific). To remove the cadmium inducible promoter, pCN51 was digested by SphI/PstI. The P3 promoter region was then amplified by PCR and cloned into pCN51 following SphI/PstI digestion, forming pCN51::P3. PCR products containing either the sole MS2 tag, or MS2 tag fused to the 5΄ end of RsaA were cloned into pCN51::P3 following digestion of products and of the plasmid by PstI/BamHI (see Supplementary Table S2 for primers containing a double MS2 tag sequence). Construction of both pCN51::P3 and its derivatives (Supplementary Table S1) was conducted in DC10B. Plasmids from positive clones were sequenced (GATC Biotech) before being electroporated into S. aureus strains.
Preparation of crude bacterial extracts
For crude bacterial extract preparation, S. aureus cells were grown in 50 ml BHI medium to an OD600nm of 5 (∼6 h of culture at 37°C), immediately chilled on ice, and then pelleted by centrifugation (3750 rpm, 15 min, 4°C). Supernatants were carefully removed and the pellets were resuspended in 4 ml ice-cold Buffer A (20 mM Tris–HCl pH 8, 150 mM KCl, 1 mM MgCl2, 1 mM DTT). Cells were then transferred into Lysis Matrix B Tubes (MP Biomedicals) and lysis was performed with FastPrep apparatus (MP Biomedicals). The tubes were then centrifuged 15 min (13 000 rpm, 4°C). The supernatants (around 4 ml) were carefully removed and placed in new ice-cold tubes that were maintained at 4°C until subsequent use for the affinity chromatography.
Affinity purification of regulatory complexes containing MS2-tagged RsaA
The MS2 coat protein fused to maltose binding protein (MS2-MBP) was purified as previously described (29). All steps for affinity chromatography purification were performed at 4°C. For each sample, 300 μl of amylose resin (New England BioLabs) per column was used. The columns containing the amylose resin were washed three times with 10 ml Buffer A, and 3600 pmoles of MS2-MBP (diluted in 6 ml of Buffer A) were then immobilized in each column. The crude bacterial extracts (4 ml) were directly loaded on each column, followed by three washes with 10 ml Buffer A. Finally, RNAs and proteins were eluted from the column with 1 ml of Buffer E (20 mM Tris–HCl pH 8, 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 0.1% Triton X-100, 12 mM maltose). Eluted RNAs were extracted with phenol/chloroform:isoamylalcohol (25 :24 :1 (v/v), Roth) and precipitated with three volumes of cold absolute ethanol in the presence of sodium acetate 0.3 M. The RNA samples were either used for Northern blot or treated with DNase I prior to RNA-seq analysis. Isolation of tagged sRNAs and the co-purified RNAs was performed in duplicates.
Northern blot
Total RNAs were prepared from different volumes of S. aureus cultures taken at 2, 4 and 6 h of culture. After centrifugation, bacterial pellets were resuspended in RNA Pro Solution (MP Biomedicals). Lysis was performed with FastPrep and the RNA purification followed strictly the procedure described for the FastRNA Pro Blue Kit (MP Biomedicals).
Electrophoresis of either total RNA (10 μg) or MS2-eluted RNA (1 μg) was performed on 1% agarose gel containing 25 mM guanidium thiocyanate. After migration, RNAs were vacuum transferred on nitrocellulose membrane. Hybridization with specific digoxygenin (DIG)-labelled probes complementary to RsaA, mgrA, hu or the MS2-tag sequences followed by luminescent detection were carried out as described previously (21).
RNA-seq analysis
For both total RNA extracts and MS2-eluted RNAs, DNase I (0.1 U/μl) treatment was performed 1 h at 37°C. The reaction mixtures were then purified by phenol/chloroform/isoamylalcohol and subsequent ethanol precipitation as described above. RNA pellets were re-suspended in sterile milliQ water. RNA quality and quantity assessments were performed on Agilent Nano Chip on the Bioanalyzer 2100. The RNAs for total transcriptomics were then treated to deplete abundant rRNAs, and the cDNA libraries were performed using the Random Hexamer approach and sequenced by Fasteris (Switzerland). The standard protocol used is the ‘TruSeq Stranded mRNA’ which is based on the TruSeq Illumina kit and which preserves the information about the orientation of the transcripts and produces reads of 125 nts, which map on the complementary strand. The libraries were sequenced using Illumina Hi-Seq 2500 with High-Output (HO) mode using a V4 chemistry sequencing kit (Illumina). Each RNA-seq was performed in duplicates. The reads were then processed to remove adapter sequences and poor quality reads by Trimmomatic (30), converted to the FASTQ format with FASTQ Groomer (31), aligned on the HG001 genome closely related to HG003 genome (32) using BOWTIE2 (33). Finally, the number of reads mapping to each annotated feature has been counted with HTSeq (34) using the interception non-empty protocol. All processing steps were performed using the Galaxy platform (35). To estimate the enrichment values for the MAPS experiment or the differential expression analysis for the transcriptomic experiment, we used DEseq2 (36). The statistical analysis process includes data normalization, graphical exploration of raw and normalized data, test for differential expression for each feature between the conditions, raw P-value adjustment and export of lists of features having a significant differential expression (threshold P-value = 0.05; fold change threshold = 2) between the conditions.
Preparation of RNAs for in vitro experiments
Transcription of RsaA and RsaAmutC was performed using linearized pUC18 vectors (25). PCR fragments containing the 5΄UTR and the 120 first nucleotides of selected mRNA targets were directly used as templates for in vitro transcription using T7 RNA polymerase. The RNAs were then purified using a 6% polyacrylamide-8 M urea gel electrophoresis. After elution with 0.5 M ammonium acetate pH 6.5 containing 1 mM ethylenediaminetetraacetic acid (EDTA), the RNAs were precipitated in cold absolute ethanol, washed with 85% ethanol and vacuum-dried. The labelling of the 5΄ end of dephosphorylated RNAs (RsaA/RsaAmutC) and DNA oligonucleotides were performed with T4 polynucleotide kinase (Fermentas) and [γ32P] adenosine triphosphate as previously described (21).
Before use, cold or labelled RNAs were renaturated by incubation at 90°C for 1 min in 20 mM Tris–HCl pH 7.5, cooled 1 min on ice and incubated 10 min at 20°C in ToeP+ buffer (20 mM Tris–HCl pH 7.5, 10 mM MgCl2, 60 mM KCl, 1 mM DTT).
In vitro translation assays
The in vitro translation assays were performed using the full-length mgrA mRNA and the PURESYSTEMII Classic Kit (Cosmo Bio Co, Japan). The construct carries an additional sequence corresponding to the FLAG peptide, which was inserted at the 3΄ end of mgrA (25). The reactions were performed at 37°C for 1 h in the presence of 25 μl of the commercial solution A and 10 μl of the commercial solution B in the presence of 10 pmoles of mgrA mRNA. Experiments were also carried out in the presence of increasing concentrations of RsaA or of MS2-RsaA transcript (5–30 pmoles). Reactions were stopped by adding an equal volume of Laemmli buffer. The proteins were detected by western blot with an antibody against the FLAG tag (Sigma).
Gel retardation assays
Radiolabelled purified RsaA or RsaAmutC (50 000 cps/sample, concentration < 1 pM) and cold mRNAs were renaturated separately as described above. For each experiment, increasing concentrations of cold mRNAs (20–240 nM for ssaA2_3, 50–900 nM for the other mRNAs) were added to the 5΄ end labelled RsaA or RsaAmutC in a total volume of 10 μl containing the ToeP+ buffer. Complex formation was performed at 37°C during 15 min. After incubation, 10 μl of glycerol blue was added and the samples were loaded on a 6% polyacrylamide gel under non-denaturing conditions (1 h, 300 V, 4°C). Quantification of the data corresponding to free RsaA or RsaAmutC and to RsaA/mRNA complex present on the autoradiography was done with ImageQuant TL software (GE Healthcare Life Sciences). Under these conditions where the concentration of the labelled RNA is negligible, the KD dissociation constant can be evaluated as the concentration of the cold RNA that showed 50% of binding.
Toe-printing assays
The preparation of E. coli 30S subunits, the formation of a simplified translational initiation complex with mRNA, and the extension inhibition conditions were performed as previously described (37). Increasing concentrations of either RsaA or RsaAmutC concentrations were used to monitor their effects on the formation of the initiation complex with mgrA. Experimental details are given in the Supplementary Data.
Mapping RsaA–mRNA interactions using footprinting
RsaA-flr mRNA formation was carried out at 37°C for 15 min in ToeP+ buffer. Enzymatic hydrolysis was performed on the unlabelled mRNA (1 pmole) free or bound to RsaA in the presence of 1 μg carrier tRNA at 37°C for 5 min in 20 μl of ToeP+ buffer. The reactions were performed with RNase T1 (0.0025 units), RNase V1 (0.5 units) or RNase T2 (0.05 U). The reactions were stopped by phenol extraction followed by RNA precipitation. The enzymatic cleavages in flr mRNA were detected by primer extension with reverse transcriptase according to (38).
Relative quantification of RNAs by RT-qPCR
Fresh BHI broth was inoculated with an overnight culture to an initial OD600 nm of 0.05 and grown 6 h at 37°C with shaking (200 rpm). The bacterial cells were harvested, washed in 10 mM Tris–HCl pH 8 and diluted to a standard OD600 of 1.0. An aliquot of the diluted bacterial suspension (1 ml) was treated with 20 μg lysostaphin. The RNA isolation was performed using the RNeasy Plus mini kit (QIAGEN) according to the manufacturer's instructions. Then, 1 μg of total RNA was transcribed into cDNA using Reverse Transcriptase System (Promega) and random primer according to the manufacturer's instructions. Finally, 2 μl of 1/5 diluted cDNA was used as a template for the real-time PCR amplification using light cycler Fast Start DNA Syber Green I kit (Roche) and a LightCycler instrument (Roche) following the manufacturer's instructions. Specific primers are shown in Supplementary Table S2. The amplification products were detected using SYBR Green. The relative amounts of amplicons for each gene were determined using quantitative PCR relative to an internal standard (gyrB). The expression levels of the RNA were expressed as n-fold differences relative to the calibrator. The RT-qPCR experiments were processed in triplicates.
In vivo ß-galactosidase assays
Translation fusions were constructed with plasmid pLUG220, a derivative of pTCV-lac, a low-copy-number promoter-less lacZ vector (Supplementary Table S1), containing the constitutive rpoB promoter. The whole leader region of ssaA2_3 (nts −83 to +87) and glpF (nts −215 to +114) mRNAs (primers in Supplementary Table S2) were cloned downstream the PrpoB in frame with lacZ. ß-galactosidase activity was measured four times as described (25).
Differential proteomics for cytoplasmic and secreted proteins
Triplicate protein extracts from supernatant or cytoplasm of HG001 (WT), ΔrsaA mutant strain (LUG1630) and the same strain complemented with a plasmid expressing RsaA, were analysed in separate LC/MS experiments. MS/MS spectra numbers were compared for each protein. Total protein extracts were prepared as follows: 1.5 ml of a S. aureus culture (OD600nm = 5) was centrifuged and the pellet resuspended in 150 μl of lysis buffer (10 mM Tris pH 7.5, 20 mM NaCl, 1 mM EDTA, 5 mM MgCl2) in the presence of 50 μg/ml lysostaphin, 15 μl of protease inhibitor cocktail (Thermo Fischer Scientific), 2 μl DNase 10 U/μl (Roche), 2 μl RNase 500 μg/ml (Roche) and incubated for 30 min at 37°C. Then, 1 ml Trizol Reagent (Life Technologies) was used according to the manufacturer's protocol. The final proteins phases were then precipitated in ice-cold acetone at least for 2 h at −20°C. Secreted proteins were prepared as follows: supernatants of cultures were filtered through a 0.22 μM membrane and precipitated with five volumes of 0.1 M ammonium acetate in methanol. To quantify protein extracts by Bradford assay, air-dried protein pellets were resuspended in 2D buffer (7 M Urea, 2 M Thiourea, 4% Chaps, 25 mM Tris pH 8) for total extracts or Triton buffer (1% triton × 100, 50 mM NaCl, 50 mM Tris pH 8) for secreted proteins. Proteins (5 μg) were precipitated with methanol/0.1 M ammonium acetate, reduced and alkylated (5 mM DTT, 10 mM iodoacetamide) and digested overnight with 1/25 (W/W) of trypsin. The peptide mixtures (1 μg /sample) were analysed using a NanoLC-2DPlus system coupled to a TripleTOF 5600 mass spectrometer (ABSciex) as previously described (22).
Protein identifications were assigned using Mascot algorithm (version 2.5, Matrix Science, London, UK) through ProlineStudio 1.2 package (http://proline.profiproteomic.fr/). Data were searched against the S. aureus HG001 genome. Peptide modifications allowed during the search were: N-acetyl (protein), carbamidomethylation (C) and oxidation (M). Mass tolerances in MS and MS/MS were set to 20 ppm and 0.5 Da, respectively. Two trypsin missed cleavages sites were allowed. After the import of the Mascot data files, proteins were validated on Mascot pretty rank equal to 1.1% false discovery rate (FDR), on peptide spectrum matches (PSM) based on PSM score, and 1% FDR on protein sets on protein set score. A spectral counting quantitative strategy was applied on the Mascot identification summaries. To evaluate the reproducibility, a statistical Student t-test was applied to this experiment.
RESULTS
MS2-RsaA is functional in vivo
The MS2 tagged version of RsaA was expressed from a plasmid under the control of the agr-dependent P3 promoter allowing an accumulation of the RNA at the stationary phase of growth in the mutant ΔrsaA strain (LUG1630; Supplementary Table S1). RsaA was detected by Northern blot using total RNAs extracted at 2, 4 and 6 h of growth. Using a DIG-labelled RsaA probe, we show that MS2-RsaA is efficiently transcribed from the P3 promoter (Figure 1A) in a manner highly similar to RsaA expressed from its endogenous promoter in the WT strain (Figure 1A). We then measured the half-lives of MS2-RsaA and of RsaA transcribed from the chromosome in BHI medium at 37°C after rifampicin treatment at 4 h of growth (Supplementary Figure S1). Under these conditions, MS2-RsaA, even if slightly overexpressed, is as stable as RsaA, with a half-life > 60 min (24). These data show that the steady state level and the stability of RsaA were not affected by the presence of the MS2 tag.
Figure 1.

The MS2-RsaA variant is normally expressed, correctly retained by affinity chromatography and functional. (A) Northern blots showing the expression of RsaA and the MS2-RsaA variant in HG001 WT and HG001-ΔrsaA strains. Total RNAs were prepared after 2, 4 and 6 h of culture in BHI medium at 37°C. Hybridization against 5S RNA was used as loading control. (B) Northern blot targeting RsaA, MS2-RsaA, mgrA or hu performed on RNAs purified after MS2 chromatography affinity; 1 μg of total RNA was loaded on a 2% agarose gel. CE: crude extract; FT : Flow-through ; W : Washing ; EL : Elution. mgrA* denotes a short fragment of the mgrA mRNA (below 274 nts) that was specifically detected by the mgrA probe in the elution fraction. This RNA fragment most likely represented a degradation product of mgrA mRNA containing the sequences interacting with RsaA. (C) In vitro translation assay performed with PURESYSTEM. The reactions were performed with 10 pmol of wild-type (WT) mgrA mRNA and in the presence of increasing quantities of WT RsaA or MS2-RsaA. The proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (10%) and were revealed using a FLAG-specific antibody. *unspecific protein revealed with the FLAG-antibody, this protein was used as an internal loading control.
Affinity chromatography was then performed to verify the specific retention of MS2-RsaA by the MS2-MBP fused protein. The RNAs extracted from the different fractions (Crude extract, Flowthrough, Wash and Elution) were analysed by Northern blot and revealed with a DIG-probe against RsaA. The data showed that MS2-RsaA was specifically enriched in the elution fraction (Figure 1B). As we have previously shown that RsaA is a translational repressor of mgrA mRNA, we have tested whether the mRNA was co-purified together with MS2-RsaA. We also used hu mRNA, which was not enriched together with RsaA. The Northern blot performed with a DIG-RNA probe revealed that mgrA mRNA was strongly enriched in the eluted fraction while hu mRNA was primarily detected in the Flowthrough and Wash fractions (Figure 1B). We have further verified that the MS2-tagged derivative of RsaA is functional. In vitro translation assays (PURE system) were performed with the whole mgrA mRNA whose translation is known to be repressed by RsaA (Romilly et al. (25)). A FLAG tag was added at the C-terminus part of the protein MgrA to monitor its synthesis using Western blot analysis. Addition of increasing concentrations of RsaA or MS2-RsaA led to a strong inhibition of MgrA synthesis (Figure 1C).
All in all, addition of the MS2 tag to RsaA had no significant effect on its expression, stability and its regulatory activities. Therefore, the MAPS approach was performed to characterize the targetome of RsaA.
The MAPS approach reveals novel mRNAs as potential targets of RsaA
In the following experiments, we have prepared crude extracts from the ΔrsaA mutant strain expressing MS2-RsaA and the WT strain expressing the MS2 tag alone from a plasmid (Supplementary Table S1). Bacterial growth was performed in BHI medium for 6h where RsaA should be sufficiently abundant for the identification of the targets. After affinity chromatography, RNA was extracted from the eluted fraction and prepared for sequencing. In parallel, we have performed from total RNA extracts prepared from the WT strain and the mutant strain expressing MS2-RsaA, a transcriptomic analysis to gain some indication on the expression levels of the mRNAs (Supplementary Table S3). The differential expression analysis revealed no major changes in the corresponding transcriptomes and also confirmed that the RsaA level was only slightly more abundant when MS2-RsaA was expressed from the plasmid (1.7-fold). We could also estimate the cellular abundance of each of the transcripts of the RsaA targetome, which has been used in combination with the enrichment values of the MAPS analysis to estimate in vivo affinities (designated as affinity rank in Supplementary Table S5) between RsaA and its targets. The data were analysed and visualized using Galaxy (35) and the Integrative Genomics Viewer (IGV) browser, respectively (39) (Table 1, and Supplementary Table S4). A detailed protocol for the bioinformatic analysis is provided in ‘Materials and Methods’ section. Briefly, we aligned the sequencing reads onto our reference genome HG001, counted per feature and normalized, we have estimated the enrichment of putative targets by comparing the number of reads obtained from the tagged MS2-RsaA purification and the MS2 alone as control. Reads were found to map all along the co-purified RNAs and RsaA showing that the RNAs were not degraded during the treatment (Supplementary Figure S2). The data were reproduced by two independent experiments. Interestingly, the best enrichment was obtained for mgrA mRNA, which was previously experimentally validated (25). Due to the number of total reads, we decided to fix the cut-off at around 2.5-fold using the DeSeq2 method. Using these criteria of selection, we obtained a restricted list of candidates (Table 1). Interestingly, several of the mRNAs code for proteins involved in common pathways. They include ssaA2_3, ssaA2_4, ssaA2_2 and ssaA_2, all belonging to the family of staphylococcal secretory antigen A (SsaA). These proteins contain a CHAP domain corresponding to an amidase function involved in peptidoglycan metabolism (reviewed in (40)). In addition, we identified another mRNA encoding a protein (orf HG001_01977) containing a SH3 domain, which is present in glycyl-glycine endopeptidases like lysostaphine. We also found flr mRNA, which encodes FLIPr, a secreted immunomodulatory molecule that interferes with effective opsonization by complement and/or IgG antibodies (41). Other mRNAs encode the ribosomal protein L19, a drug efflux pump NorB that confers quinolone resistance and a D-serine/D-alanine/glycine transporter. Transcriptomic analysis revealed that among the co-purified mRNAs, the mRNAs ssaA_2 and flr were weakly expressed suggesting that both mRNAs are most probably specifically recognized by RsaA (Supplementary Table S3). Therefore, no direct correlation was found between mRNA abundance and their enrichment after the affinity chromatography.
Table 1. List of RNA sequenced by MAPS using RsaA as bait.
| Id | Gene | Product | RNA fold change* | P-value* |
|---|---|---|---|---|
| HG001_00625 | mgrA | HTH-type transcriptional regulator MgrA | 14.641 | 7.15E-33 |
| HG001_00516 | RsaA | 13.901 | 3.64E-31 | |
| HG001_02338 | ssaA2_3 | Staphylococcal secretory antigen ssaA2 precursor | 5.162 | 1.14E-12 |
| HG001_01977 | Bacterial SH3 domain protein | 4.024 | 1.63E-09 | |
| HG001_02583 | ssaA2_4 | Staphylococcal secretory antigen ssaA2 precursor | 3.868 | 4.16E-09 |
| HG001_02334 | ssaA2_2 | Staphylococcal secretory antigen ssaA2 precursor | 3.455 | 8.21E-09 |
| HG001_01615 | cycA_1 | D-serine/D-alanine/glycine transporter | 3.262 | 9.99E-08 |
| HG001_01092 | rplS | 50S ribosomal protein L19 | 2.882 | 1.59E-06 |
| HG001_01204 | hypothetical protein | 2.889 | 1.93E-06 | |
| HG001_01150 | glpF | Glycerol uptake facilitator protein | 2.739 | 5.91E-06 |
| HG001_01863 | hypothetical protein | 2.654 | 1.84E-05 | |
| HG001_01003 | flr | FPRL1 inhibitory protein precursor | 2.605 | 2.89E-05 |
| HG001_01295 | norB_4 | Quinolone resistance protein NorB | 2.581 | 3.96E-05 |
| HG001_02606 | ssaA_2 | Staphylococcal secretory antigen SsaA precursor | 2.413 | 0.0001149 |
*Fold change (MS2-RsaA/MS2 control) and P-values were calculated by DESeq2 using shrinkage estimation for dispersions and fold changes .
We then searched for possible intermolecular basepairing interactions between RsaA and its potential mRNA targets using RNACoFold and IntaRNA (Table 1 and Supplementary Table S5). Again, the best hit was mgrA mRNA, but stable pairings were also predicted with the four ssaA mRNAs and flr mRNA. Less stable interactions were detected for norB, cycA, glpF and rplS. Interestingly, most of the predictions involved the conserved UCCCA motif of RsaA and the Shine and Dalgarno (SD) sequence of the mRNAs. Only for HG001_01977 mRNA, stable pairings were between the 5΄UTR of the mRNA and the first apical loop of RsaA (Supplementary Table S5). In many cases such as mgrA, ssaA and flr mRNAs, a second distant region of interactions was predicted in their coding sequences (Supplementary Table S5).
These data suggest that RsaA would bind directly to many of these mRNAs, and would repress the translation through sequestration of the SD sequence.
RsaA forms stable complexes with various mRNAs
Based on the MAPS data, we first analysed whether RsaA directly binds to the mRNA candidates using gel retardation assays (Figure 2 and Supplementary Figure S3). In vitro 5΄ end-labelled RsaA was incubated with increasing concentrations of the various mRNAs including the four ssaA species, flr, norB-4, cycA, HG001_01977, rplS and glpF. All the mRNA fragments contain the whole 5΄UTR and at least 120 nts of the coding region (Supplementary Table S2). The experiments were performed in a buffer containing magnesium and salt, without prior denaturation of the mRNA targets. The data showed that RsaA formed complexes with high affinity (between 20 and 100 nM, see Supplementary Table S5) with ssaA_2, ssaA2_3 and flr. For flr mRNA, two distinct complexes were observed, indicating that two RsaA molecules are able to bind to the mRNA. Affinity of complexes between RsaA and HG001_01977 or ssaA2_2 was weaker (>250 nM), but the deletion of the 5΄UTR of ssaA2_2 significantly enhanced duplex formation (Supplementary Figure S3A). Finally, no gel retardation was observed when complex formation was performed with RsaA and increasing concentrations of ssaA2_4, glpF, rplS, norB-4 and cycA mRNAs (up to 1 μM, Supplementary Figure S3B).
Figure 2.

Gel retardation assays to monitor RsaA binding to several mRNAs. Experiments were performed on four different mRNA targets of RsaA, which are ssaA2_3 (nucleotides −83 to +130), flr (nucleotides −47 to +120), ssaA_2 (nucleotides −128 to +125), HG001_01977 (nucleotides −390 to +122). The 5΄ end-labelled WT RsaA (A) or the mutant RsaAmutC (B) were incubated with increasing concentrations (nM) of mRNAs. In the case of flr mRNA, two complexes could be detected with WT RsaA. In the gel shift assays with RsaAmutC, residual complexes are indicated with a star. (C) Representation of RsaA secondary structure. The C-rich residues (with the mutation introduced in the RsaAmutC variant) and the second region of interaction with mgrA mRNA are shown in red.
Because the C-rich motif of RsaA was predicted to basepair with the SD of mRNA targets, we analysed whether mutations at these positions would affect RsaA binding as it was demonstrated for mgrA (25). Complex formation was carried out under strictly identical conditions for the WT and a version of RsaA in which the GUUCUCCC sequence had been replaced by CAAGACCC (RsaAmutC). The data showed that RsaAmutC failed to bind to ssaA2_3 and flr mRNAs (Figure 2B). Surprisingly, the second complex with flr also disappeared when using RsaAmutC, which would indicate cooperation between both sites. In these cases, the C-rich motif of RsaA might act as a seed sequence to initiate mRNA binding. In contrast, mutations in RsaAmutC slightly altered the stability of the complex formed with ssaA_2 mRNA while they had no significant effect on HG001_01977 mRNA binding as expected, since the major site for basepairing interactions involved the first hairpin loop of RsaA (Figure 2 and Supplementary Figure S5).
Because the gel retardation assays suggested that two RsaA molecules could bind to flr mRNA, we have performed footprinting experiments using the double-strand specific RNase V1, the single-strand specific RNase T2 (preference for unpaired adenines) and RNase T1 (specific for unpaired guanine) (Figure 5B). The data revealed that the addition of RsaA induced significant changes in the region encompassing the SD sequence and part of the coding sequence from A − 10 to C + 38. Enhanced RNase V1 cuts were located close to the SD sequence (at adenines −7, −8 and −10) and in the coding sequence at C + 35/U + 36. Concomitantly, binding of RsaA reduced RNase T2 hydrolysis at positions U + 24, A + 25 and U + 27 while enhanced cleavages were observed at A + 4 to A + 9. These changes are well correlated with the positioning of two RsaA molecules. In this model, the CCCU motif would interact with the SD sequence.
Figure 5.

Characterization of the inhibitory RsaA–flr mRNA complex. (A) Genetic organization of the pathogenic island containing the flr gene and effect of RsaA on the formation of initiation ribosomal complex on flr mRNA (50 nM). Lane 1: incubation control of mRNA; lane 2: formation of the ribosomal initiation complex containing mRNA, the 30S subunits and tRNAfMet (tRNAi); lane 3: incubation control of mRNA with WT RsaA ; lane 4–6: formation of the initiation complex in presence of increasing concentrations of WT RsaA : 100, 200, 400 nM. lane 7: incubation control of mRNA with RsaAmutC; lanes 8–10: formation of the initiation complex in the presence of increasing concentrations of RsaAmutC: 100, 200, 400 nM. Lanes U, A, G, C: sequencing ladders. The SD sequence, the start site of translation (A +1 of the AUG initiation codon) and the toe-printing signals (N +16) are indicated. * indicates the RT stop induced by RsaA binding on flr mRNA. Graph showing the quantification of the toe-print signals. The toe-print signals with flr mRNA in the presence of increasing concentrations of RsaA or RsaAmutC were normalized according to the total amount of radioactivity (full-length extension and +16 product bands) using the ImageQuanTL software (GE Healthcare). (B) Footprinting assays to map RsaA–flr mRNA interactions. Enzymatic reactions performed on flr mRNA free or bound to either RsaA or RsaAmutC (25 and 50 nM). The first three lanes represent incubation controls of free mRNA, bound to RsaA WT or RsaAmutC, respectively. Lanes T, G, A: dideoxy sequencing reactions. The experiments were performed with RNase V1 (V1), RNase T1 (T1) and RNase T2 (T2). On the sequence of flr mRNA are shown the RNase cleavages, which have been reproducibly found in two independent experiments: triangles denote RNase V1 cuts, filled arrows denote RNase T2 cuts. Effect of RsaA binding: protections are given by circles and enhanced or new RNase V1/T2 cleavages are labelled by stars.
Taken together, the gel retardation assays correlate well with the prediction of the basepairing interactions and suggest that the C-rich motif of RsaA is one of its major regulatory determinant sites.
RsaA hinders ribosome binding site of several mRNA targets
Because sRNA–mRNA pairings can alter mRNA decay (stabilization/degradation), we have probed whether RsaA affects the steady state levels of the target mRNAs. We therefore performed RT-qPCR in triplicate on selected target mRNAs using total RNAs extracted from the WT HG001 strain, the isogenic HG001ΔrsaA mutant strain and HG001ΔrsaA complemented with pCN51::PrsaA (Supplementary Table S1). The levels of each mRNA were quantified relative to gyrB (Figure 3). The data showed that the deletion of rsaA did not significantly affect the steady state levels of HG001_01977, flr, ssaA2_2 and ssaA 2_3 mRNAs. Only for ssaA_2 mRNA, the deletion of rsaA led to a reproducibly weak increase in mRNA yield, although the statistics of the data cannot be considered relevant. Such a mild RsaA-dependent effect was also observed for mgrA mRNA (25).
Figure 3.

The expression of target mRNAs was quantified by RT-qPCR. The data were normalized to the level of gyrB mRNA expression from total RNA extracts prepared from in vitro culture to the late-exponential phase (6 h) of HG001 (WT), the ΔrsaA mutant strain and the complemented strain with a plasmid expressing RsaA under its own promoter.
Toe-printing assays were then used to decipher the effect of RsaA and of RsaAmutC on the formation of the ternary initiation complex formed in the presence of mRNA, the initiator tRNA and the 30S subunit. The experiments performed with ssaA_2, ssaA2_2, ssaA2_3 and flr mRNAs, showed that the ribosome efficiently recognized the mRNAs as illustrated by the presence of the toe-print signal at position +16 (+1 being the initiation codon). The addition of RsaA together with the 30S strongly decreased the toe-print signal showing that RsaA competes efficiently with the ribosome for mRNA binding (Figures 4 and 5, Supplementary Figure S4). The RsaA-dependent inhibition was albeit less efficient for ssaA2_2 (Supplementary Figure S4). Toe-printing experiment was also performed for HG001_01977 mRNA (Supplementary Figure S5). However in this case, we did not observe any toe-print signal correlated with the absence of a SD sequence. The addition of RsaA caused a specific pause of the reverse transcriptase that corresponds to the predicted basepairing interactions (Supplementary Figure S5 and Table S5).
Figure 4.

Toe-printing assays to monitor the effect of RsaA on the initiation ribosomal complex formation. (A) Effect of RsaA on the formation of initiation ribosomal complex on ssaA_2 and ssaA2_3 mRNAs (50 nM). Lane 1 : incubation control of mRNA ; lane 2: incubation control of mRNA with 30S subunits; lane 3 : formation of the ribosomal initiation complex containing mRNA, 30S and the initiator tRNAfMet (tRNAi); lane 4: incubation control of mRNA with WT RsaA; lane 5–7: formation of the initiation complex in presence of increasing concentrations of WT RsaA : 100, 200, 400 nM. Lane 8: incubation control of mRNA with RsaAmutC; lane 9–11: formation of the initiation complex in presence of increasing concentrations of RsaAmutC: 100, 200, 400 nM. Lanes U, A, G, C: sequencing ladders. The Shine and Dalgarno (SD) sequence, the start site of translation (A +1 of the AUG initiation codon) and the toe-printing signals (N + 16) are indicated. * indicates the RT stop induced by RsaA binding on the two mRNAs, the RsaA-dependent RT pause on ssaA_2 is weaker than on ssaA2_3. (B) β-galactosidase activity (Miller Units) measured from PrpoB-ssaA2_3 (−83/+87)::lacZ and PrpoB-glpF (−215/+114)::lacZ expressed in HG001-ΔrsaA complemented either with the plasmid pCN38 without insert, or containing the WT RsaA (pRsaA) or RsaAmutC (pMutC). β-galactosidase activity was normalized for total density and the results represented the mean of four independent experiments. *P < 0.05, **P < 0.01 and ns = not significant.
For flr, ssaA2_2 and ssaA2_3 mRNAs, the formation of the initiation complex was completely restored when RsaAmutC was added in the reaction (Figures 4, 5A and Supplementary Figure S4). This result correlates well with the gel retardation assays, which showed that the C-rich motif of RsaA is essential for stable duplex formation. In the case of ssaA_2 mRNA, addition of RsaAmutC to the reaction did not alter the formation of the initiation ribosomal complex as evidenced by the presence of a toe-printing signal comparable to the reaction performed in the presence of RsaA, supporting the Electrophoretic Mobility Shift Assay results. In this case, the mutations are silent because the second binding site occurring in the coding sequence of the mRNA contribute significantly to the stability of the duplex.
To further validate the in vivo relevance of RsaA-dependent repression of the mRNA target ssaA2_3, we analysed the expression of a reporter construct carrying the regulatory region of ssaA2_3 fused to lacZ in S. aureus HG001-ΔrsaA strain expressing the WT RsaA ou RsaAmutC under its own promoter. We also analysed the expression of glpF-lacZ mRNA. The glpF mRNA was pulled out in the MS2 purification but did not interact with RsaA in the EMSA experiment (Supplementary Figure S3). In these constructs, the entire leader regulatory region of ssaA2_3 and glpF genes and around 100 nts from their coding sequences were cloned in-frame with the lacZ gene into the pLUG220::PrpoB vector (Supplementary Table S2). The ß-galactosidase activity was respectively 3- and 5-fold lower in the ΔrsaA mutant strain expressing WT RsaA than in the same strain transformed with the control vector or with the RsaAmutC vector (Figure 4). Thus, disruption of the predicted basepairings alleviated the repression of the reporter ssaA2_3-lacZ gene. In contrast, the glpF fusion was poorly expressed and did not show any RsaA-dependent regulation.
In conclusion, these data strongly suggest that RsaA would primarily regulate translation initiation of several of the newly identified target mRNAs by sequestrating their ribosome binding sites.
Quantitative proteomics suggest that RsaA indirectly regulates cell surface proteins
Cytoplasmic and secreted proteins extracts from the WT strain (HG001), the mutant HG001ΔrsaA (LUG1630) strain and the same strain complemented with a plasmid expressing RsaA under its own promoter were analysed in triplicates by LC/MSMS coupled to spectral count analysis, which ended with more than 1000 identified proteins (Supplementary Table S6). We considered a P-value lower than 0.02 and a ratio of spC (number of spectra identified by protein) of the deleted strain compared to the complemented strain, which is smaller than 1/6 or bigger than 6. Using these criteria, a list of 46 putative proteins whose translation might be regulated directly or indirectly by RsaA was obtained (Table 2). Among them, the synthesis of two proteins, MgrA and SsaA2_3, for which the mRNAs have been demonstrated as primary targets of RsaA, were found enhanced in the mutant strains (Table 2). In the remaining protein set, we also found FLIPr, but the protein was poorly detected in the culture supernatant, represented by only one MSMS spectrum in the analysis. However it was reproducibly found enriched by a factor of almost two in the ΔrsaA strain. The protein SsaA2_2 was well expressed both in the total proteins and supernatant extracts, but enriched only by a factor below two in the mutant strain (Supplementary Table S6). In these experiments, the proteins SsaA_2, the SH3 domain-containing protein (HG001_01977), CycA and NorB_4 were not detected while the synthesis of L19, and GlpF were found unchanged in the various strains. Interestingly, several surface proteins were found significantly activated by RsaA. These proteins include the extracellular matrix proteins EbH_1 and 3, the immunoglobulin G binding protein A, the surface protein G SasG, the Serine-aspartate repeat-containing proteins C and D StrC/StrD, the amidase LytN and the clumping factor ClfB. Because the corresponding mRNAs were not detected in the MAPS approach and that no significant basepairings were predicted, we postulated that these effects are most likely indirect and linked to the RsaA-dependent repression of mgrA (54).
Table 2. Differential proteomic analysis.
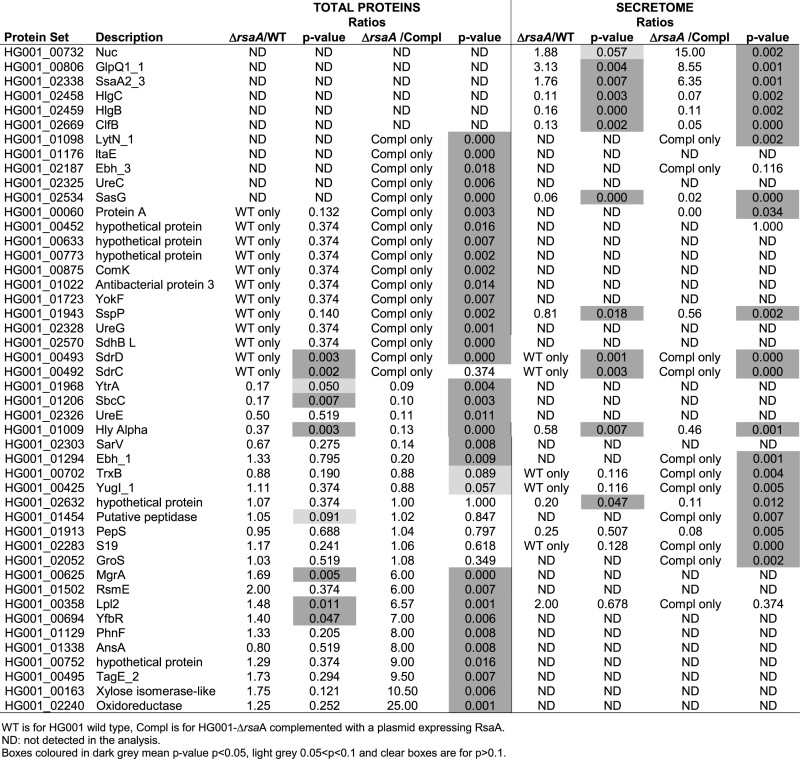
|
DISCUSSION
Many bacterial sRNAs act as antisense RNAs and form short and non-contiguous basepairing interactions with a group of mRNAs often functionally related to adapt cell growth in response to specific stress, environmental changes and various hosts. Although S. aureus genome codes for numerous sRNA genes, the functions of only few of them have been identified. In this organism, transcriptomic analysis was poorly adapted to monitor the rapid depletion of mRNAs specifically repressed by a specific sRNA. A recent approach, the so-called MAPS, has been recently developed to detect in vivo the whole sets of mRNAs interacting with a defined sRNA. This powerful approach has been applied on two E. coli sRNAs, RyhB and RybB (12). For the first time, this study provided a whole sRNA targetome and besides the previously identified mRNA targets, has revealed the unexpected existence of sponge RNAs issued from precursor tRNAs. Here, we demonstrated that this technology could also be applied to S. aureus sRNAs independently of their regulatory mechanisms, and in a WT strain context expressing all ribonucleases.
As a proof of concept, we adapted the protocol to one small non-coding sRNA, RsaA, which functionally links two master regulatory factors, Sigma B and MgrA proteins (25). We first showed that the tagged RsaA behaved as the WT sRNA and was able to efficiently regulate the translation of mgrA mRNA (Figure 1). Analysis of the enriched RNAs that co-purified with RsaA revealed that the first most abundant mRNA (15-fold) was mgrA. A second set of mRNAs (>3-fold) encodes several amidases that are involved in peptidoglycan metabolism and remodelling (reviewed in (40)). These enzymes play also a role in biofilm-associated infections (42). A third group of mRNAs (below 3-fold) codes for various proteins that are associated to the membrane (transporters, efflux pump), and the secreted FLIPr protein. Our analysis revealed that most of the mRNAs could potentially form stable duplexes with RsaA involving either its interhelical unpaired domain encompassing the C-rich motif or the first hairpin loop motif. It is of interest that a potential correlation exists between the order of the enriched mRNAs and the strength of the predicted basepairings (Table 1 and Supplementary Table S5). However, gel retardation assays revealed that only several mRNAs (flr, mgrA, ssaA_2, ssaA2_2, ssaA2_3, HG001_01977) were competent to form stable complexes with RsaA. For the other mRNAs (glpF, cycA-1, norB-4, rplS, ssaA2_4), one cannot exclude that a trans-acting RNA-binding protein would help to facilitate the formation of complexes, and/or alternatively that RsaA would bind to the mRNAs during the transcription process to avoid secondary structure elements masking the regulatory elements. However, we also cannot exclude the possibility that these RNAs were retained together with another partner, through an indirect manner. For instance, we observed enrichment of the quorum sensing induced regulatory RNAIII (enrichment just below 2,5), which has been recently shown to stabilize mgrA mRNA through direct binding with the 5΄UTR of one of the mgrA transcripts (43). Therefore, it is highly possible that RNAIII was co-purified together with mgrA. Toe-printing assays further demonstrated that RsaA primarily represses translation initiation of flr, ssaA_2, ssaA2_2 and ssaA2_3 mRNAs. As established for mgrA, the C-rich motif of RsaA binds to the strong SD sequences of the mRNAs but we also observed that complementarities extend to the coding sequences of the target mRNAs. The regulatory regions present in the four ssaA mRNAs are highly conserved. Based on our data, we propose that for ssaA mRNAs, the two interaction sites involve one molecule of RsaA while for flr mRNA, two RsaA molecules would cover almost 40 nts of the mRNA (Figures 2 and 5). Interestingly enough, mutations in the C-rich motif of RsaA hindered duplex formation and restored ribosome binding for flr mRNA, ssaA_2, ssaA2_3 mRNAs arguing that initial pairings occur at the SD sequence. Proteomic analysis is well correlated with the MAPS data because RsaA significantly repressed the synthesis of these proteins (MgrA, FLIPr, SsaA2_3, SsaA_2). All these data strongly support that these mRNAs are direct targets of RsaA.
The first 5΄ hairpin loop of RsaA is a second regulatory site, which has been involved in the formation of a loop–loop interaction stabilizing mgrA–RsaA complex (25). Here our results demonstrate that this domain of RsaA might also bind to the large 5΄UTR of the hypothetical mRNA HG001_01977 encoding a SH3 domain-containing protein (Supplementary Figure S5). Indeed, a strong RsaA-dependent RT pause was found at the 3΄ boundary of the mRNA bound to RsaA, and mutation of the C-rich motif of RsaA did not alter the formation of the stable complex (Figure 2). It is intriguing that the steady state level of this RNA did not change in the WT and mutant strains (Figure 3). We also did not observe the formation of an active initiation ribosomal complex questioning on the coding capacity of this hypothetical mRNA. The gene is located on a pathogenicity island (phiSA3, which carry the immune evasion cluster), just downstream sak mRNA encoding staphylokinase (Supplementary Figure S5). This exotoxin activates plasminogen to digest fibrin clots, but also inhibits phagocytosis by cleaving IgG and complement component. In the same area, several non-coding RNAs have been identified; one of them called SprE is expressed from the same strand between the sak gene and HG001_01977 (44). We need to address more specifically the functional consequences of the mRNA–RsaA complex, and to decipher if HG001_01977 mRNA is translated. Alternatively, we cannot rule out that this hypothetical mRNA might regulate the activity of RsaA. Recent reports have indeed described novel RNA functions modulating the action of sRNAs; some of them act as RNA decoys (45) or as RNA sponges (46) to regulate the stability of the target sRNA and consequently affect the expression of their regulons. More recently, Lalaouna et al. (12) have demonstrated that the tRNA spacers can serve as RNA sponges to prevent transcriptional noise of several sRNAs in E. coli. In this latter case, the decay of the regulated sRNAs is not affected. It was proposed that such sRNA sponge would set up a concentration threshold that sRNAs must exceed to induce target regulation under specific conditions (12). Finally, a bacteriophage encoded RNA called AgvB was able to counteract the action of the enterohemorrhagic E. coli GcvB sRNA through sequence mimicry (11). Its interaction does not alter the stability of GcvB although the synthesis of its targets was deregulated. Interestingly, HG001_01977 gene locus is also located on a pathogenicity island, which arose from bacteriophage (Supplementary Figure S5). Whether HG001_01977 regulates the RsaA action remains to be studied.
Analysis of the functions of the RsaA targets reveals several interconnected links. Four of the ssaA mRNAs encode proteins that belong to the CHAP amidase family endowed with D-alanyl-glycyl endopeptidase activity that cleave between the crossbridge and the stem peptide. A SH3 domain found in glycyl-glycine endopeptidases, that cut the pentaglycine bridge specific to staphylococcal cell wall, was predicted in the protein encoded by mRNA HG001_01977. Cell wall metabolism is vital for bacteria and endopeptidases play multiple functions in cell shape, division, resistance to stresses, protein secretion, biofilm formation and in host–pathogen interactions. These enzymes should also be tightly regulated to avoid perturbation of the cell wall particularly at the stationary phase of growth. Indeed, they are under the control of several transcriptional regulatory proteins, among which the essential two-component system WalKR (40,47,48). Inactivation of WalKR at the stationary phase of growth caused transcription arrest of these enzymes (40), and interestingly, the expression of SsaA2_2 proteins and LytM were found sufficient to by pass WalKR essentiality (47). An additional post-transcriptional level of regulation was recently described in which S. aureus RNAIII contributed to the repression of lytM translation at high cell density (49). Hence, RsaA would be the second RNA coordinating the translation repression of SsaA family enzymes at the stationary phase of growth in a Sigma B-dependent manner. Among the new mRNA targets of RsaA, one of them encodes a secreted virulence factor, the FLIPr protein (26,50). Interestingly, the gene is located on an immune evasion cluster (pathogenicity island 4; (51) and Figure 5) that includes staphylococcal complement inhibitor and several fibrinogen-binding proteins (Scn-2, Fib-1, Fib-2). FLIPr protein is a secreted staphylococcal anti-inflammatory protein that acts as an antagonist of the receptor protein FPRL1. This protein is expressed in various cells such as neutrophils, macrophages and microglials (52), and triggers many cellular functions including chemotaxis, exocytosis and superoxide generation. Hence, by counteracting FPRL1, FLIPr contributes to the evasion of bacteria from the human defense system (26). This novel function of RsaA is somehow related to the translation repression of mgrA mRNA, a master regulator of S. aureus (53,54). MgrA is a major activator of capsule formation and regulates autolytic activities, efflux pumps, several transporters and virulence factors. Particularly, the two-component system ArlRS and MgrA formed a regulatory pathway that led to the repression of several cell surface proteins (SraP, SasG, FmtB, SdrD and protein A) (53,54). This repression confers to S. aureus the ability to form clumps in the presence of fibrinogen, which facilitates adhesion to the host tissues but also protect the bacteria. Interestingly, mgrA mutants are unable to form clumping due to the synthesis of the cell surface proteins Ebh, SraP and SasG. In addition, the mgrA mutant has enhanced biofilm production due to the expression of SasG protein, which is known to favour intercellular contacts (54). In agreement with this study, quantitative differential proteomic analysis revealed that the deletion of RsaA causes a strong decrease in the synthesis of the cell surface proteins EbH, protein A, SdrD, SdrC, SasG, LytN and the clumping factor ClfB (Table 2 and Supplementary Table S6). These effects are most likely indirect and linked to the direct repression of mgrA. Indeed, our previous study has also shown that the induction of RsaA inhibits capsule formation and enhances biofilm formation through the repression of mgrA (25). The expression of RsaA was also accompanied by a decreased protection of S. aureus against opsonophagocytic killing by neutrophils, and acute systemic infection in mice was attenuated while chronic catheter infection was enhanced. Clearly, the RsaA-dependent translation repression of both MgrA and FLIPr supports these phenotypes while the repression of the SsaA family enzymes helps to maintain the integrity of the cell wall, a property that is certainly essential to maintain the correct presentation of the cell surface proteins. Hence, RsaA is part of a regulatory network that modifies the complex interactions with the host immune system (Figure 6). The MAPS approach also revealed that RsaA mediates crosstalk between the core genome and the mobile elements through the regulation of genes (flr, HG001_01977) located on pathogenicity islands thus integrating horizontally acquired genes into pre-existing post-transcriptional networks (55,56).
Figure 6.

Schematic drawing summarizing the regulatory networks involving RsaA and its mRNA targets in Staphylococcus aureus. RsaA activated by σB binds to mgrA, flr and ssaA mRNAs to inhibit their translation. The role of RsaA interaction with HG001_01977 is still unclear. Arrows are for activation, bars for repression. In blue are the transcriptional protein regulators, in red the regulatory RNA and in grey the virulence factors. Red lines corresponded to post-transcriptional regulation and black lines to transcriptional regulation. Dotted line represented the regulatory events for which direct regulation is not yet demonstrated.
As shown for E. coli (12), the MAPS approach can also be used to gain a better knowledge of the overall functions regulated by a specific sRNA in Gram-positive bacteria. In this work, we utilized an sRNA carrying a C-rich motif that has been shown as an ideal motif for targeting the ribosome binding sites of mRNAs, indicating that RsaA would primarily act as a translational regulator. However, because these experiments have been conducted in a WT strain (in the presence of all ribonucleases), we do not exclude that mRNAs that are rapidly depleted could be underestimated. Two of the ribonucleases, RNase III and RNase J1/J2, generate multiple effects on RNA metabolism and regulation (57,58). Furthermore, RsaA and mgrA mRNA were both co-immunoprecipitated with WT and catalytically inactive RNase III but the other identified mRNAs were not found in this study (57). Further experiments will be required to analyse whether RsaA acts in coordination with RNases to regulate its target mRNAs. MAPS could also be used to identify RNA-binding proteins although a crosslinking step would probably be required to stabilize the interactions that are often transient and dynamic. More recently, novel high-throughput techniques have been developed to map RNA–RNA interactions at global scale in vivo in eukaryotes (59,60) or bacteria (61). These methods, which combine in vivo crosslinks, ligation of interacting RNAs and high-throughput sequencing, revealed an impressive diversity of intra- and intermolecular RNA–RNA pairings. Such approaches applied to S. aureus could certainly explore the complexity of RNA-RNA interactions taking place and to track their dynamics occurring under various stress conditions and during infection.
ACCESSION NUMBERS
Data has been deposited in GEO under accession number GSE96814.
The complete genome sequence for the HG001 strain has been deposited in GenBank under the accession number GCA_001900185.1.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Anne-Catherine Helfer for her help in the footprinting and toe-printing assays, to Jessica Baude for the RT-qPCR experiments and to Lauriane Kuhn and Philippe Hammann for their assistance with mass spectrometry analysis (IBMC proteomic platform). We are thankful to Redmond Smyth for critical reading of the manuscript, to Delphine Bronesky and David Lalaouna for helpful advices and discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the Centre National de la Recherche Scientifique (CNRS) to [P.R.] and has been published under the framework of the LABEX: ANR-10-LABX-0036 NETRNA to [P.R.], a funding from the state managed by the French National Research Agency as part of the investments for the future program. A Tomasini was supported by Fondation pour la Recherche Médicale (FDT20150532433). Funding for open access charge: ANR [ANR-10-LABX-0036].
Conflict of interest statement. None declared.
REFERENCES
- 1. Caldelari I., Chao Y., Romby P., Vogel J.. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb. Perspect. Med. 2013; 3:a010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Storz G., Vogel J., Wassarman K.M.. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011; 43:880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barquist L., Vogel J.. Accelerating discovery and functional analysis of small RNAs with new technologies. Annu. Rev. Genet. 2015; 49:367–394. [DOI] [PubMed] [Google Scholar]
- 4. Wagner E.G.H., Romby P.. Small RNAs in bacteria and archea: whao they are, what they do, and how they do it. Adv. genet. 2015; 90:133–208. [DOI] [PubMed] [Google Scholar]
- 5. Updegrove T.B., Shabalina S.A., Storz G.. How do base-pairing small RNAs evolve. FEMS Microbiol. Rev. 2015; 39:379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vogel J., Wagner E.G.H.. Target identification of small noncoding RNAs in bacteria. Curr. Opin. Microbiol. 2007; 10:262–270. [DOI] [PubMed] [Google Scholar]
- 7. Lalaouna D., Massé E.. Identification of sRNA interacting with a transcript of interest using MS2-affinity purification coupled with RNA sequencing (MAPS) technology. Genomics Data. 2015; 5:136–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Udekwu K.I., Darfeuille F., Vogel J., Reimegård J., Holmqvist E., Wagner E.G.H.. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev. 2005; 19:2355–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J., Rennie W., Liu C., Carmack C.S., Prévost K., Caron M.-P., Massé E., Ding Y., Wade J.T.. Identification of bacterial sRNA regulatory targets using ribosome profiling. Nucleic Acids Res. 2015; 43:10308–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chao Y., Papenfort K., Reinhardt R., Sharma C.M., Vogel J.. An atlas of Hfq-bound transcripts reveals 3΄UTR and regulates the expression of an immune-all RNAs. EMBO J. 2012; 31:4005–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tree J.J., Granneman S., McAteer S.P., Tollervey D., Gally D.L.. Identification of bacteriophage-encoded anti-sRNAs in pathogenic Escherichia coli. Mol. Cell. 2014; 55:199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lalaouna D., Carrier M.-C., Semsey S., Brouard J.S., Wang J., Wade J.T., Massé E.. A 3΄ external transcribed spacer in a tRNA transcript act as a sponge for small RNAs to prevent transcriptional noise. Mol. Cell. 2015; 58:393–405. [DOI] [PubMed] [Google Scholar]
- 13. Carrier M.-C., Lalaouna D., Massé E.. A game of tag: MAPS catches up on RNA interactomes. RNA Biol. 2016; 13:473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Surmann K., Michalik S., Hildebrandt P., Gierok P., Depke M., Brinkmann L., Bernhardt J., Salazar M.G., Sun Z., Shteynberg D. et al. . Comparative proteome analysis reveals conserved and specific adaptation patterns of Staphylococcus aureus after internalization by different types of human non-professional phagocytic host cells. Front. Microbiol. 2014; 5:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tomasini A., François P., Howden B.P., Fechter P., Romby P., Caldelari I.. The importance of regulatory RNAs in Staphylococcus aureus. Infect. Genet. Evol. 2014; 21:616–626. [DOI] [PubMed] [Google Scholar]
- 16. Janzon L., Arvidson S.. The role of the delta-lysin gene (hld) in the regulation of virulence genes by the accessory gene regulator (agr) in Staphylococcus aureus. EMBO J. 1990; 9:1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Novick R.P., Ross H.F., Projan S.J., Kornblum J., Kreiswirth B., Moghazeh S.. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993; 12:3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Felden B., Vandenesch F., Bouloc P., Romby P.. The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog. 2011; 7:e1002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huntzinger E., Boisset S., Saveanu C., Benito Y., Geissmann T., Namane A., Lina G., Etienne J., Ehresmann B., Ehresmann C. et al. . Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J. 2005; 24:824–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geisinger E., Adhikari R.P., Jin R., Ross H.F., Novick R.P.. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol. Microbiol. 2006; 61:1038–1048. [DOI] [PubMed] [Google Scholar]
- 21. Boisset S., Geissmann T., Huntzinger E., Fechter P., Bendridi N., Possedko M., Chevalier C., Helfer A.C., Benito Y., Jacquier A. et al. . Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 2007; 21:1353–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Durand S., Braun F., Lioliou E., Romilly C., Helfer A.C., Kuhn L., Quittot N., Nicolas P., Romby P., Condon C.. A nitric oxide regulated small RNA controls expression of genes involved in redox homeostasis in Bacillus subtilis. PLoS Genet. 2015; 11:e1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sievers S., Sternkopf Lillebæk E.M., Jacobsen K., Lund A., Mollerup M.S., Nielsen P.K., Kallipolitis B.H.. A multicopy sRNA of Listeria monocytogenes regulates expression of the virulence adhesion LapB. Nucleic Acids Res. 2014; 42:9383–9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geissmann T., Chevalier C., Cros M.-J., Boisset S., Fechter P., Noirot C., Schrenzel J., François P., Vandenesch F., Gaspin C. et al. . A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 2009; 37:7239–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Romilly C., Lays C., Tomasini A., Caldelari I., Benito Y., Hammann P., Geissmann T., Boisset S., Romby P., Vandenesch F.. A non-coding RNA promotes bacterial persistence and decreases virulence by regulating a regulator in Staphylococcus aureus. PLoS Pathog. 2014; 10:e1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prat C., Bestebroer J., Haas C.J.C.d., Strijp J.A.G.V, Kessel K.P.M.V. A new staphylococcal anti-inflammatory protein that antagonizes the formyl peptide receptor-Like 1. J. Immunol. 2006; 177:8017–8026. [DOI] [PubMed] [Google Scholar]
- 27. Monk I.R., Shah I.M., Xu M., Tan M.-W., Foster T.J.. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. Mbio. 2012; 3:e00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Charpentier E., Anton A.I., Barry P., Alfonso B., Fang Y., Novick R.P.. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 2004; 70:6076–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Said N., Rieder R., Hurwitz R., Deckert J., Urlaub H., Vogel J.. In vivo expression and purification of aptamer-tagged small RNA regulators. Nucleic Acids Res. 2009; 37:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blankenberg D., Gordon A., Von Kuster G., Coraor N., Taylor J., Nekrutenko A.. Manipulation of FASTQ data with Galaxy. Bioinformatics. 2010; 26:1783–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sassi M., Felden B., Augagneur Y.. Draft genome sequence of Staphylococus aureus subsp. aureus Strain HG003, an NCTC8325 Derivative. Genome Announc. 2014; 2, doi:10.1128/genomeA.00855-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anders S., Pyl P.T., Huber W.. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Eberhard C. et al. . The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Varet H., Brillet-Guéguen L., Coppée J.-Y., Dillies M.-A.. SARTools: a DESeq2- and EdgeR-Based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS One. 2016; 11:e0157022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fechter P., Chevalier C., Yusupova G., Yusupov M., Romby P., Marzi S.. Ribosomal initiation complexes probed by toeprinting and effect of trans-acting translational regulators in bacteria. Methods Mol. Biol. (Clifton, N.J.). 2009; 540:247–263. [DOI] [PubMed] [Google Scholar]
- 38. Chevalier C., Geissmann T., Helfer A.-C., Romby P.. Probing mRNA structure and sRNA-mRNA interactions in bacteria using enzymes and lead (II). Methods Mol. Biol. (Clifton, N.J.). 2009; 540:215–232. [DOI] [PubMed] [Google Scholar]
- 39. Thorvaldsdóttir H., Robinson J.T., Mesirov J.P.. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dubrac S., Bisicchia P., Devine K.M., Msadek T.. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 2008; 70:1307–1322. [DOI] [PubMed] [Google Scholar]
- 41. Stemerding A.M., Köhl J., Pandey M.K., Kuipers A., Leusen J.H., Boross P., Nederend M., Vidarsson G., Weersink A.Y.L., van de Winkel J.G.J. et al. . Staphylococcus aureus formyl peptide receptor-like 1 inhibitor (FLIPr) and its homologue FLIPr-like are potent FcγR antagonists that inhibit IgG-mediated effector functions. J. Immunol. (Baltimore, Md.: 1950). 2013; 191:353–362. [DOI] [PubMed] [Google Scholar]
- 42. Resch A., Rosenstein R., Nerz C., Götz F.. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl. Environ. Microbiol. 2005; 71:2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gupta R.K., Luong T.T., Lee C.Y.. RNAIII of the Staphylococcus aureus agr system activates global regulator MgrA by stabilizing mRNA. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:14036–14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pichon C., Felden B.. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:14249–14254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Figueroa-Bossi N., Valentini M., Malleret L., Bossi L.. Caught at its own game: regulatory small RNA inactivated by an inducible transcript mimicking its target. Genes Dev. 2009; 23:2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miyakoshi M., Chao Y., Vogel J.. Cross talk between ABC transporter mRNAs via a target mRNA-derived sponge of the GcvB small RNA. EMBO J. 2015; 34:1478–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Delaune A., Poupel O., Mallet A., Coic Y.-M., Msadek T., Dubrac S.. Peptidoglycan crosslinking relaxation plays an important role in Staphylococcus aureus WalKR-dependent cell viability. PLoS One. 2011; 6:e17054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Poupel O., Moyat M., Groizeleau J., Antunes L.C.S., Gribaldo S., Msadek T., Dubrac S.. Transcriptional analysis and subcellular protein localization reveal specific features of the essential WalKR system in Staphylococcus aureus. PLoS One. 2016; 11:e0151449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lioliou E., Fechter P., Caldelari I., Jester B.C., Dubrac S., Helfer A.-C., Boisset S., Vandenesch F., Romby P., Geissmann T.. Various checkpoints prevent the synthesis of Staphylococcus aureus peptidoglycan hydrolase LytM in the stationary growth phase. RNA Biol. 2016; 13:427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prat C., Haas P.-J., Bestebroer J., de Haas C.J.C., van Strijp J.A.G., van Kessel K.P.M.. A homolog of formyl peptide receptor-like 1 (FPRL1) inhibitor from Staphylococcus aureus (FPRL1 inhibitory protein) that inhibits FPRL1 and FPR. J. Immunol. 2009; 183:6569–6578. [DOI] [PubMed] [Google Scholar]
- 51. Novick R.P., Christie G.E., Penadés J.R.. The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 2010; 8:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Migeotte I., Communi D., Parmentier M.. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006; 17:501–519. [DOI] [PubMed] [Google Scholar]
- 53. Luong T.T., Lee C.Y.. The arl locus positively regulates Staphylococcus aureus type 5 capsule via an mgrA-dependent pathway. Microbiology. 2006; 152:3123–3131. [DOI] [PubMed] [Google Scholar]
- 54. Crosby H.A., Schlievert P.M., Merriman J.A., King J.M., Salgado-Pabón W., Horswill A.R.. The Staphylococcus aureus global regulator MgrA modulates clumping and virulence by controlling surface protein expression. PLoS pathog. 2016; 12:e1005604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Papenfort K., Vogel J.. Regulatory RNA in bacterial pathogens. Cell Host Microbe. 2010; 8:116–127. [DOI] [PubMed] [Google Scholar]
- 56. Chabelskaya S., Gaillot O., Felden B.. A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLoS Pathog. 2010; 6:e1000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lioliou E., Sharma C.M., Caldelari I., Helfer A.C., Fechter P., Vandenesch F., Vogel J., Romby P.. Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet. 2012; 8:e1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Linder P., Lemeille S., Redder P.. Transcriptome-wide analyses of 5΄-ends in RNase J mutants of a gram-positive pathogen reveal a role in RNA maturation, regulation and degradation. PLoS Genet. 2014; 10:e1004207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Aw J.G.A., Shen Y., Wilm A., Sun M., Lim X.N., Boon K.-L., Tapsin S., Chan Y.-S., Tan C.-P., Sim A.Y.L. et al. . In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell. 2016; 62:603–617. [DOI] [PubMed] [Google Scholar]
- 60. Sharma E., Sterne-Weiler T., O’Hanlon D., Blencowe B.J.. Global mapping of human RNA-RNA interactions. Mol. Cell. 2016; 62:618–626. [DOI] [PubMed] [Google Scholar]
- 61. Melamed S., Peer A., Faigenbaum-Romm R., Gatt Y.E., Reiss N., Bar A., Altuvia Y., Argaman L., Margalit H.. Global mapping of small RNA-target interactions in bacteria. Mol. Cell. 2016; 63:884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.