

ABSTRACT
Despite the fact that temozolomide (TMZ) has been widely accepted as the key chemotherapeutic agent to prolong the survival of patients with glioblastoma, failure and recurrence cases can still be observed in clinics. Glioma stem-like cells (GSCs) are thought to be responsible for the drug resistance. In this study, we investigate whether endothelial monocyte-activating polypeptide-II (EMAP-II), a pro-inflammatory cytokine, can enhance TMZ cytotoxicity on U87MG and GSCs or not. As described in prior research, GSCs have been isolated from U87MG and maintained in the serum-free DMEM/F12 medium containing EGF, b-FGF, and B27. TMZ and/or EMAP-II administration were performed for 72 h, respectively. The results showed that TMZ combined with EMAP-II inhibit the proliferation of U87MG and GSCs by a larger measure than TMZ single treatment by decreasing the IC50. EMAP-II also enhanced TMZ-induced autophagy-mediated cell death and G2/M arrest. Moreover, we found that EMAP-II functioned a targeted suppression on mTOR, which may involve in the anti-neoplasm mechanism. The results suggest that EMAP-II could be considered as a combined chemotherapeutic agent against glioblastoma by sensitizing U87MG and GSCs to TMZ.
KEYWORDS: autophagy, EMAP-II, G2/M arrest, glioma stem cells, temozolomide
Introduction
Glioblastomas (GBMs) are the most common tumors of central nervous system disease, comprising 16% of all primary brain and central nervous system neoplasms.1 Originally, GBMs were thought to be derived solely from glial cells; however, evidence has confirmed the existence of a small subpopulation as glioma stem-like cells (GSCs).2 GSCs are thought to contribute to tumor progression, treatment resistance, and tumor recapitulation post-treatment.3 Current standard therapy includes maximal safe surgical resection, followed by concurrent radiation with temozolomide (TMZ), an oral alkylating chemotherapeutic agent, and then adjuvant chemotherapy with TMZ.4 Cytotoxicity of TMZ is related to DNA methylation and the subsequent formation of O6-MeG (O6-methylguanine),5 followed by an arrest of the cell cycle during the G2/M phase.6 MGMT-mediated DNA repairation is regarded as the most likely reason for TMZ resistance, resulting in uncontrolled tumor growth.7
Endothelial monocyte-activating polypeptide-II (EMAP-II) is a pro-inflammatory cytokine. Data have revealed both its anti-angiogenic properties8 and its regulatory effect on the blood-brain barrier.9 Further evidence suggested EMAP-II as a possible anti-glioma agent, but its effect is transient,10 which provides a new opportunity for combined treatment in glioblastoma chemotherapy.
In the present study, we found that EMAP-II enhanced TMZ decreasing cell viability via the induction of autophagy, which was further shown to be defective. Moreover, EMAP-II was found to enhance TMZ suppressing tumor growth by inducing G2/M arrest. Additionally, our data also showed that TMZ inhibited PI3K/AKT/mTOR signaling pathway and EMAP-II enhanced targeted suppression on mTOR. Our results demonstrated that combined EMAP-II and TMZ treatment induced a larger degree of autophagy and increased cell cytotoxicity.
Materials and methods
Reagents and antibodies
Temozolomide was obtained from Schering-Plough (Merck, United Kingdom), resuspended at 10 mg/ml in 100% DMSO, aliquoted and stored at −20°C in a concentration of 50 mM. EMAP-II, EGF, and b-FGF were obtained from PeproTech (St. Louis, MO, USA), and diluted with 0.9% sodium chloride. Dulbecco's Modified Eagle's Medium (DMEM), DMEM/F12, and fetal bovine serum (FBS) were purchased from Gibco (Carlsbad, CA, USA). Anti-Nestin and anti-CD133 were purchased from Millipore (Temecula, CA, USA). The CCK-8 Kit and Cell Cycle Kit were purchased from Beyotime (Jiangsu, China). Antibodies against GAPDH and the secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies, including LC3B, p62/SQSTM1, PI3K, p-PI3K, AKT, p-AKT, mTOR, and p-mTOR, were purchased from Abcam (Cambridge, MA, USA).
Cell line and cell culture
The human glioblastoma cell line U87MG was obtained from Shanghai Institutes for Biological Sciences and Cell Resource Center. Cells were cultured in DMEM, supplemented with 10% FBS. The generation and identification of GSCs derived from U87MG cells have been described previously.10 GSCs stained with allophycocyanin (APC)-conjugated CD133 antibodies (Miltenyi Biotec, USA) were sorted by fluorescence-activated cell sorting (FACS) on a FACS Calibur instrument. The CD133-positive cells were collected and maintained in serum-free DMEM/F12 stem cell media containing 20 ng/ml EGF, bFGF and B27 (1:50). All the cells were incubated in a 5% CO2 humidified incubator at 37°C.
Treatment
U87MG cells and GSCs were assigned to four groups, and then treated with (1) DMSO (1:1000 dilution), (2) EMAP-II (0.5 nM), (3) TMZ (100μM), and (4) combined EMAP-II (0.5 nM) with TMZ (100μM), respectively, for 72 h.
Cell viability assay
Cell viability was determined by the CCK-8 assay according to the manufacturer's instructions. Briefly, 2000 U87MG cells/well or 10000 GSCs/well were seeded into 96-well plates overnight and were then treated by EMAP-II and/or TMZ administration. Ten μL CCK-8 solutions were added to the cells of each group after the 72-hour treatment. After incubation for another 2 h, the optical density (OD) was measured at 450 nm using a microplate reader (BioTek, USA). The data were from 3 separate experiments, with 5 replications each time.
Cell cycle analysis
After treated with EMAP-II and/or TMZ administration for 72 h, the U87MG cells and GSCs were harvested and centrifuged at 500 g for 5 min. They were then resuspended in phosphate-buffered saline (PBS) and fixed in ice-cold 70% ethanol at −20°C, stored at 4°C overnight. Cells were then washed twice with PBS and incubated in the dark in PBS containing 0.5 mg/ml RNase A and 50μg/ml propidium iodide (PI) (Sigma, USA) at 37°C for 30 min. The cell cycle was analyzed on a flow cytometer (BD Biosciences, CA). Duplicates were performed in all of the experiments, and the experiments were performed on 2 separate occasions.
Immunofluorescence
The cells were collected and fixed with 4% PFA for 20 min at room temperature, washed twice with PBS, and permeabilized with a blocking solution containing 5% bovine serum albumin 0.01% triton X-100, diluted in PBS. Next, the cells were incubated overnight at 4°C with the LC3B primary antibody. Immunoreactivity was visualized with Alexa-488-conjugated secondary antibodies at a dilution 1:1000 for 2 h. The nuclei were counterstained with Hoechst 33242 (blue on images). Glass coverslips were mounted with fluorescence Mounting Medium (Southern Biotech, USA). The images were captured with an Olympus BX61 fluorescence microscope.
Western blotting
Cells of each group lysates were prepared by lysing the cells in a RIPA buffer, which contained 0.01% of a protease and phosphatase inhibitor cocktail. The protein concentration was determined by the bicinchoninic acid (BCA) protein assay; equal amounts (40μg) of denatured proteins were fractionated through SDS-PAGE on a 10% gel, and then transferred to PVDF membranes. The membrane was blocked with 5% milk/Tris-buffered saline plus Tween 20 (TBST) and incubated with the appropriate primary antibodies overnight at 4°C. HRP goat anti-mouse IgG and HRP goat anti-rabbit IgG were used as the secondary antibodies The immunoreactive bands were visualized using ECL detection reagents. Equal loading was assessed after probing the same membrane with the GAPDH antibody.
Statistical analysis
The data were expressed as the mean ± standard deviation (SD). Statistical significance was estimated using the SPSS 13.0 software (SPSS, Chicago, IL) for two-way ANOVA to compare each group. p < 0.05 was considered statistically significant.
Results
EMAP-II enhanced cytotoxic effect of TMZ on U87MG and GSCs
As in our previously protocol, we derived GSCs from U87MG. After the formation of spheres (Fig. 1A), stemness markers were identified by immunofluorescence which showed double positive in CD133 and Nestin (Fig. 1B). To assess the cytotoxicity of TMZ on U87MG and GSCs, cells were treated with TMZ alone or a combination of TMZ and EMAP-II with assigned concentration for 72 h respectively, the dose-inhibition response curves were made according to CCK-8 assay. The IC50 value was defined as the dose needed for a 50% reduction in OD, calculated from the curve. As shown in Figure 1C, TMZ inhibited cell viability in a dose-dependent manner in both cell lines. IC50 of TMZ on U87MG was remarkably decreased from 659.4 ± 1.06μM to 427.0 ± 1.08 μM with the presence of EMAP-II (p < 0.05). Similarly, IC50 on GSCs was significantly decreased from 1059.0 ± 1.04 μM to 635.9 ± 1.07 μM (p < 0.05). The higher IC50 on GSCs indicated its chemoresistant characteristic to TMZ, at the same time EMAP-II enhanced its cytotoxic effect on both U87MG and GSCs. For further investigation, we treated the cells with 100μM TMZ and/or 0.5 nM EMAP-II for 72 h and analyzed the inhibition rates (Fig. 1D). The result revealed that there was no significant inhibition of cell viability on U87MG or GSCs in the single EMAP-II treatment group and the same result was received in the single TMZ treatment group on GSCs. Interestingly, the combination of TMZ and EMAP-II exerted a remarkable anti-tumor effect on GSCs meanwhile this trend was also observed on U87MG. As previously reported that enhanced MGMT expression might contribute to TMZ resistance,11 there was a lower inhibition rate observed on GSCs in either single TMZ treatment group or combination group.
Figure 1.
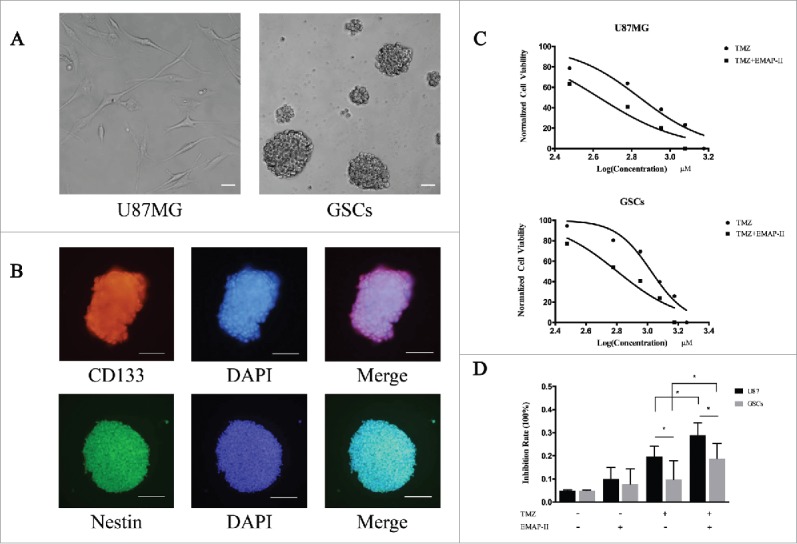
EMAP-II enhanced cytotoxic effect of TMZ on U87MG and GSCs (A) U87MG cells were cultured in the DMEM, containing 10% FBS in flasks. After sorted by FACS, the CD133-positive cells were collected and maintained in the DMEM/F12, containing 2% B27 supplements, 20ng/ml EGF, and bFGF. Suspended cells became spheres after 48 h culture. (B) GSCs expressing the stemness biomarkers of CD133 and Nestin by immunofluorescence. (C) Both U87MG and GSCs were treated with TMZ alone or a combination of TMZ and EMAP-II with assigned concentration for 72 h respectively, IC50 of TMZ calculated from dose-inhibition response curves was remarkably decreased on both U87MG and GSCs. (D) Cells were treated with 100μM TMZ and/or 0.5 nM EMAP-II for 72 h respectively and cell viability was then determined. EMAP-II enhanced the TMZ-induced cytotoxic effect on U87MG and GSCs.
EMAP-II increased TMZ-induced autophagy
Growing evidence have revealed the autophagic cell death induced by novel cancer drugs including temozolomide12 or ionizing radiation.13 The death-promoting and -executing mechanisms involved in the different paradigms of autophagic cell death are very diverse and complex.14 Accepted methods for analyzing macroautophagy in vitro include electron microscopy, Western blotting for LC3B, and green fluorescence protein-LC3B immunofluorescence.15 To investigate whether EMAP-II sensitized U87MG and GSCs to TMZ in an autophagic death-promoting manner or not, immunofluorescent staining of LC3B was performed after separated or combined treatment of 72 h. Compared to the control, cells treated with either EMAP-II or TMZ both showed an obviously higher level of LC3BII expression, and cells treated with both EMAP-II and TMZ showed even higher LC3BII staining, indicating autophagic induction was increased by combination treatment (Fig. 2A). In addition, we used the normalized LC3BII/LC3BI ratio as the quantification of autophagy by western blot. Increased ratio of LC3BII/LC3BI was detected in either EMAP-II or TMZ treatment group on both U87MG and GSCs. Moreover, there was a large-scale increasing ratio in both EMAP-II and TMZ treated cells than that in the either TMZ or EMAP-II treated cells. No significant changes of LC3BII/LC3BI ratio were found between single TMZ or EMAP-II treated GSCs. Differently, on U87MG, a lower amount of LC3BII/LC3BI ratio was observed with single EMAP-II treatment comparing to single TMZ treatment. Recent studies revealed that p62/SQSTM1, which is an autophagosome cargo protein that targets other proteins that bind to it for selective autophagy, has also been shown to interact with LC3B.16 Although there were controversial results pertaining to the changes of p62/SQSTM1 in the autophagic progress,17 it also was regarded as an indicator of autophagy. In the present study, a higher level decline of p62/SQSTM1 expression was found in the combination treatment group on both U87MG and GSCs comparing to that in the single TMZ treatment, but there were no changes found in the single EMAP-II treatment group on either U87MG or GSCs, which was different from LC3B induced by induction by single EMAP-II treatment (Fig. 2B-C).
Figure 2.
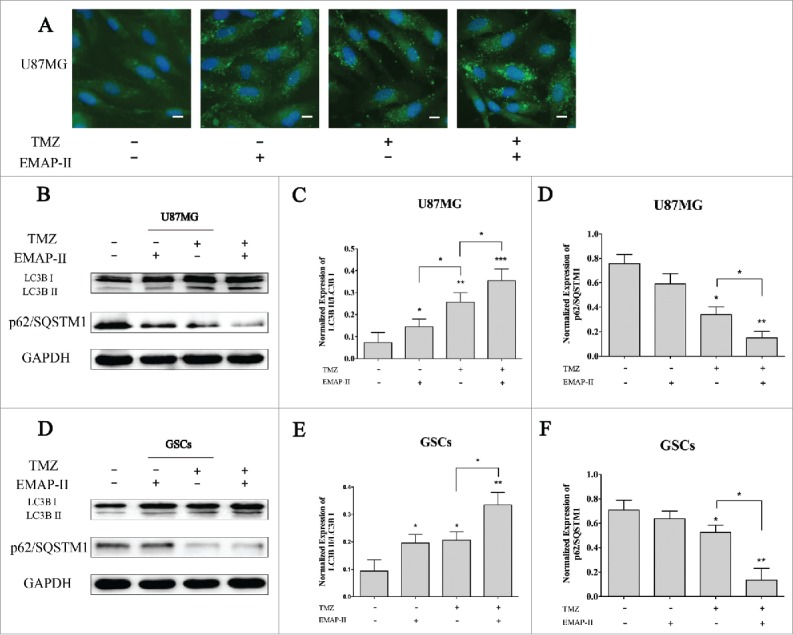
EMAP-II increased TMZ-induced autophagy (A) U87MG was treated with either TMZ or EMAP-II and both TMZ and EMAP-II for 72 h respectively. Expression and distribution of LC3B II were determined by immunofluorescence staining, all images were taken with exactly the same settings. Compared to the control, cells treated with either EMAP-II or TMZ both showed an obvious higher level of LC3BII expression, and cells treated with both EMAP-II and TMZ showed even higher LC3BII staining. (scale bar = 20μM). (B-D) U87MG was treated with either TMZ or EMAP-II and both TMZ and EMAP-II for 72 h. TMZ induced a higher LC3BII/LC3BIlevel than EMAP-II; Combination of TMZ and EMAP-II showed a significant elevation on LC3BII/LC3BIthan either TMZ or EMAP-II; P62/SQSTM1 was obviously inhibited by combination of TMZ and EMAP-II;(E-G) GSCs was treated with either TMZ or EMAP-II and both TMZ and EMAP-II for 72 h. The combination of TMZ and EMAP-II showed a significant elevation on LC3BII/LC3BIthan either TMZ or EMAP-II; P62/SQSTM1 was obviously inhibited by the combination of TMZ and EMAP-II; Values present means ± SD (n = 3, each). *p < 0.05, **p < 0.01, or ***p < 0.001.
Effect on the cell progression
In most cells, TMZ produces cell cycle arrest at G2/M through the activation of ATM/ATR-Chk1/2.18 Chk1/2 can activate Wee1, the kinase that phosphorylates meanwhile inhibits CDC25A, the phosphatase responsible for dephosphorylating this site,19 thus leading to an arrest before mitosis. Activation of the G2 checkpoint acts primarily as a pro-survival mechanism that gives time to the cells to repair their DNA. Some cancer types, such as GBM, are intrinsically resistant to apoptosis and may be more sensitive to other mechanisms of cell death, such as autophagy, senescence, and mitotic catastrophe.20,21 After U87MG and GSCs were treated with either TMZ or EMAP-II and combination of TMZ with EMAP-II for 72 h, the cell population in different phase were determined by flow cytometry. As shown in Figure 3, compared to the control, EMAP-II slightly promoted accumulation of G2/M population on both U87MG and GSCs but showed no significance. Meanwhile, U87MG treated with TMZ showed an increased proportion of the G2/M population but GSCs treated with TMZ was not found a G2/M accumulation. Moreover, combined TMZ and EMAP-II induced a higher G2/M accumulation than TMZ on both U87MG and GSCs.
Figure 3.
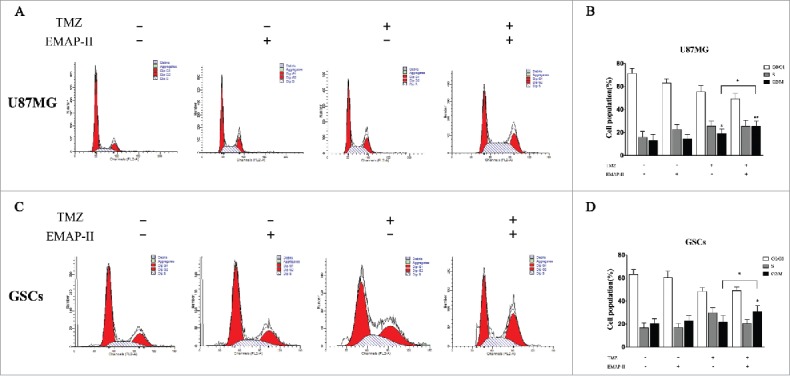
Effect on the cell progression. After U87MG and GSCs were treated with either TMZ or EMAP-II and combination of TMZ with EMAP-II for 72 h, the cell population in different phase were determined by flow cytometry. (A-B) U87MG treated with TMZ showed an increased proportion of the G2/M population, combined TMZ and EMAP-II induced a higher G2/M accumulation than TMZ. (C-D) GSCs treated TMZ did not show an increased proportion of the G2/M population. only combined TMZ and EMAP-II induced a higher G2/M accumulation. The values present means ± SD (n = 4, each). *p < 0.05
Effect on the PI3K/AKT/mTOR signaling pathway
The PI3K/AKT/mTOR signaling pathway is a cell survival pathway that is important for normal cell growth and proliferation.22 On one hand, mTOR might inhibit autophagy predominantly by activating a downstream molecule, p70S6 kinase (p70S6K).23 On the other hand, PI3K/AKT as well as their downstream molecule CHK1 could regulate cell cycle.24 To investigate the effect on the PI3K/AKT/mTOR signaling pathway, western blot analyses were performed after U87MG and GSCs were treated with either TMZ or EMAP-II and combination of TMZ with EMAP-II for 72 h. As Figure 4 shows, Total PI3K, AKT, and mTOR did not change in both U87MG and GSCs. Compared to the control, p-PI3K and p-AKT were not affected by EMAP-II, only TMZ and combination of TMZ with EMAP-II decreased expressions of p-PI3K, p-AKT, and p-mTOR. Moreover, there was a significant decline of p-mTOR/mTOR with the combination of TMZ with EMAP-II than that treated with TMZ in both U87MG and GSCs.
Figure 4.
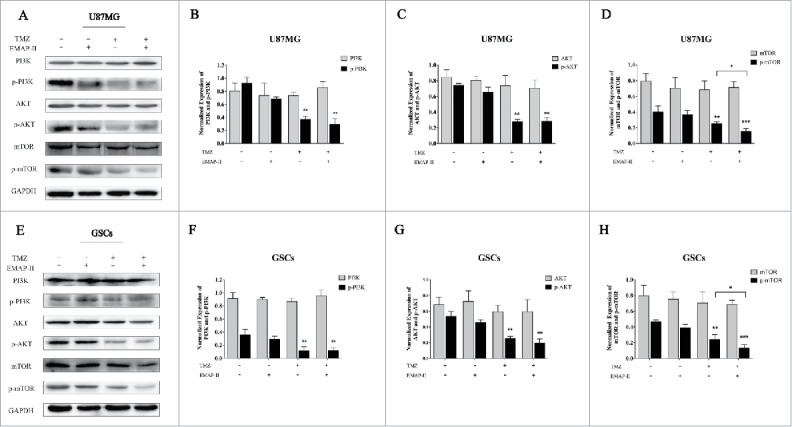
Effect on PI3K/AKT/mTOR pathway. U87MG and GSCs were treated with either TMZ or EMAP-II and combination of TMZ with EMAP-II for 72 h. (A-D) Western Blot showed relative expressions of PI3K, p-PI3K, AKT, p-AKT, mTOR and p-mTOR, combination of TMZ with EMAP-II enhanced inhibition of p-mTOR/mTOR on U87MG; (E-H) Western Blot showed relative expressions of PI3K, p-PI3K, AKT, p-AKT, mTOR and p-mTOR, combination of TMZ with EMAP-II enhanced inhibition of p-mTOR/mTOR on GSCs. The values present means ± SD (n = 4, each). *p < 0.05, **p < 0.01, or ***p < 0.001.
Discussion
EMAP-II was first discovered in 1990 as a pro-inflammatory cytokine with anti-angiogenic properties. 25 Our previous work has revealed that a low-dose EMAP- II could increase the permeability of the blood-tumor barrier26 and inhibit the proliferation of glioblastoma cells in the first hour of treatment.10 As EMAP-II showed an anti-tumor characteristic as TMZ, it was promising that EMAP-II acts as an adjuvant chemotherapeutic agent of TMZ.
In the present study, we found that the cells viability of U87MG cells and GSCs were decreased to around 60% after the 72 h combination treatment period. TMZ showed a remarkable lower IC50 value (427.0 ± 1.08μM on U87MG, and 635.9 ± 1.07μM on GSC, respectively, p < 0.05) with the combination of EMAP-II. Some researchers have reported evidence that TMZ has a wide range of IC50 on different cell lines,27,28 especially on the MGMT-positive GSCs.11 A lower IC50 suggested that EMAP-II could sensitize U87MG and GSCs to TMZ. The enhancement of autophagy and G2/M arrest by the combination of TMZ and EMAP-II were observed, which contributed to the anti-glioma mechanism. Moreover, the inhibition of PI3K/AKT/mTOR signaling pathway was found by TMZ and EMAP-II showed a higher inhibition ability targeting p-mTOR which might involve in the cytotoxicity of TMZ and EMAP-II combined treatment.
GSCs, also known as glioma initial cells, performed a unique biologic behavior in the cell population. The key properties that distinguish GSCs from other tumor cells include their ability to (1) self-renew, (2) differentiate into heterogeneous types of tumor cells, and (3) sustain tumor growth in vivo.29 There is increasing evidence that they are responsible for tumor initiation, the propagation of the disease, the resistance to current therapies, and tumor recurrence.2,30-32 Our data showed that U87MG was induced more autophagy-mediated cell death compared with GSCs, which means that GSCs are more resistant to the combined treatment of TMZ and EMAP-II. Although there is no consensus regarding whether GSCs express more or less MGMT, the evidence indicates that CpG methylation of the MGMT promoter in the tumor cells derived from the patients increased dramatically.33 This suggests that MGMT methylation, which could reduce MGMT activity, was enriched with the GSC culture in vitro. It increases the drug resistance of GSCs up to 10 times,34 and its role in mediating chemoresistance has repeatedly been confirmed.34-37
Autophagy, a lysosome-dependent degradation progress, has been thought to act as a pro-survival or pro-death response to several stressors, especially chemotherapy and radiotherapy, at the cellular and organic levels.14 The mechanism by which autophagy could perform these seemingly opposite roles remained elusive until recently. Under moderate stimulus conditions, the autophagic pathway operates to supply cells with a metabolic substrate, contributing to the maintenance of cell survival.38 However, a considerable body of literature has reported that uncontrolled autophagy is also a cell death mechanism that can occur either in the absence of detectable signs of apoptosis or concomitantly with apoptosis.39 Autophagy has been demonstrated to be involved in DNA damage.40-42 In the treatment of glioblastomas, chemotherapeutic drugs, including arsenic trioxide and TMZ,43 can trigger autophagy-associated cell death and further improve their therapeutic effects. Atg8/LC3B is the most widely mentioned autophagy-related protein, and the ratio of LC3BII/LC3BI is used as the quantification of autophagy.44 In the present study, increased ratio of LC3BII/LC3BI induced EMAP-II was detected, but cell viability was not decreased. We assume that autophagy induced by EMAP-II was not severe enough for the cell death, but if it was added to the autophagy induced by TMZ, the uncontrolled autophagy-mediated cell death would significantly decrease the cell viability. In addition to LC3B, p62/SQSTM1 was found decreased, accompanied with an increased ratio of LC3BII/LC3BI in U87MG and GSCs. Despite the fact that there is not always a clear correlation between increases in LC3BII and decreases in p62/SQSTM1, it can still be used as a protein marker, at least in certain settings.45,46 For example, p62/SQSTM1 can be detected as puncta by IHC in cancer cells, similar to LC3B.47 The p62/SQSTM1 protein serves as a link between LC3B and ubiquitinated substrates.48 p62/SQSTM1 and p62/SQSTM1-bound polyubiquitinated proteins become incorporated into completed autophagosomes and are degraded in autolysosomes, thus serving as an index of autophagic degradation. Inhibition of autophagy correlates with increased levels of p62/SQSTM1 in mammals and Drosophila, suggesting that steady-state levels of this protein reflect the autophagic status.49-53 Similarly, decreased p62/SQSTM1 levels are associated with autophagy activation.54,55
It is widely known that mTOR, a downstream effector of AKT, plays a critical role in regulating autophagy in cells, from yeast to mammalian cells.40,56 Our data showed p-PI3K and p-AKT were not affected by EMAP-II compared with the control. EMAP-II might function a targeted suppression on p-mTOR followed by the induction of autophagy, which is similar to the results of other studies.57 CHK1, a cell cycle controlling molecule, is another downstream target regulated by the PI3K/AKT pathway. When DNA damage occurred, cell cycle checkpoints were activated in the eukaryotic cells—complex kinase signaling networks that prevented further progression through the cell cycle. Parallel to implementing a cell cycle arrest followed by apoptosis, checkpoint signaling also mediates the recruitment of DNA-repair pathways. DNA-damage checkpoints usually cause G1/S arrest to prevent the replication of damaged DNA, G2/M arrest to prevent the segregation of damaged chromosomes during mitosis, or S-phase responses activated by genotoxic insults.58-60 Previous studies investigating TMZ showed that TMZ induced G2 phase arrest, followed by apoptosis in tumor cells.61 In the present study, we found that TMZ alone did not induce G2/M arrest in GSCs, but it did induce G2/M arrest with the presence of EMAP-II. It has now been confirmed that G2/M arrest is largely dependent on the CHK1-mediated signaling pathway, leading to the inhibition of cyclin B1/CDC2 activity. CHK1 is activated by phosphorylation on at least 2 residues (Ser345 and Ser317), located in a Ser/Thr-Gln-rich domain by DNA damage-activated ATR and ATM kinases.58-60,62,63 The inhibition of CHK1 represents a more effective cytotoxicity of TMZ to reduce the growth of human GBM.64 The knockdown of CHK1 could induce the increasing radiosensitivity of GSCs. In addition, our observations regarding decreased p-PI3K/p-AKT/p-mTOR expression are consistent with previous studies that reported that DNA-damage induced activation of CHK1 is downregulated by the inhibition of the PI3K/AKT pathway.24
In conclusion, we found that the cytotoxicity of TMZ was amplified with the presence of EMAP-II on U87MG and GSCs via the induction of autophagy-mediated cell death and G2/M arrest. Moreover, U87MG was more sensitive to the combination chemotherapy compared with GSCs due to the unique characteristics of GSCs. More investigation should be conducted in this area, such as regulation of MGMT and CHK1-mediated G2/M arrest, to clarify how GSCs behave in a chemoresistant manner. We suggest that EMAP-II could be regarded as a novel combined chemotherapy agent against glioblastoma in the future.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work is supported by grants from the Natural Science Foundation of China (81672511, 81372484 and 81573010), Liaoning Science and Technology Plan Project (No. 2015225007), Shenyang Science and Technology Plan Projects (Nos. F15–199–1–30 and F15–199–1–57) and outstanding scientific fund of Shengjing hospital (No. 201304).
References
- [1].Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, Villano JL. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev 2014; 23(10):1985-96; PMID:25053711; https://doi.org/ 10.1158/1055-9965.EPI-14-0275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Filatova A, Acker T, Garvalov BK. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim Biophys Acta 2013; 1830(2):2496-508; PMID:23079585; https://doi.org/ 10.1016/j.bbagen.2012.10.008 [DOI] [PubMed] [Google Scholar]
- [3].Mahon BP, Okoh CO, McKenna R. Targeting aggressive cancers with an artificial sweetener: Could saccharin be a lead compound in anticancer therapy?. Future Oncol 2015; 11(15):2117-9; PMID:26235178; https://doi.org/ 10.2217/fon.15.137 [DOI] [PubMed] [Google Scholar]
- [4].NCCN Clinical Practice Guidelines in Oncoloy Central Nervous System Cancers.Version 1. 2016. [Google Scholar]
- [5].Agarwala SS, Kirkwood JM. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 2000; 5(2):144-51; PMID:10794805; https://doi.org/ 10.1634/theoncologist.5-2-144 [DOI] [PubMed] [Google Scholar]
- [6].Hirose Y, Berger MS, Pieper RO. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res 2001; 61(5):1957-63; PMID:11280752 [PubMed] [Google Scholar]
- [7].Zhang J, Stevens MF, Bradshaw TD. Temozolomide: Mechanisms of action, repair and resistance. Curr Mol Pharmacol 2012; 5(1):102-14; PMID:22122467; https://doi.org/ 10.2174/1874467211205010102 [DOI] [PubMed] [Google Scholar]
- [8].Murray JC, Heng YM, Symonds P, Rice K, Ward W, Huggins M, Todd I, Robins RA. Endothelial monocyte-activating polypeptide-II (EMAP-II): A novel inducer of lymphocyte apoptosis. J Leukoc Biol 2004; 75(5):772-6; PMID:14982944; https://doi.org/ 10.1189/jlb.1003487 [DOI] [PubMed] [Google Scholar]
- [9].Li Z, Liu YH, Xue YX, Liu LB, Xie H. Mechanisms for endothelial monocyte-activating polypeptide-II-induced opening of the blood-tumor barrier. J Mol Neurosci 2012; 47(2):408-17; PMID:21969114; https://doi.org/ 10.1007/s12031-011-9657-5 [DOI] [PubMed] [Google Scholar]
- [10].Liu J, Liu L, Xue Y, Meng F, Li S, Wang P, Liu Y. Anti-neoplastic activity of low-dose endothelial-monocyte activating polypeptide-II results from defective autophagy and G2/M arrest mediated by PI3K/Akt/FoxO1 axis in human glioblastoma stem cells. Biochem Pharmacol 2014; 89(4):477-89; PMID:24792437; https://doi.org/ 10.1016/j.bcp.2014.04.014 [DOI] [PubMed] [Google Scholar]
- [11].Qiu ZK, Shen D, Chen YS, Yang QY, Guo CC, Feng BH, Chen ZP. Enhanced MGMT expression contributes to temozolomide resistance in glioma stem-like cells. Chin J Cancer 2014; 33(2):115-22; PMID:23958055; https://doi.org/ 10.5732/cjc.012.10236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Natsumeda M, Aoki H, Miyahara H, Yajima N, Uzuka T, Toyoshima Y, Kakita A, Takahashi H, Fujii Y. Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 2011; 31(5):486-93; PMID:21269334; https://doi.org/ 10.1111/j.1440-1789.2010.01197.x [DOI] [PubMed] [Google Scholar]
- [13].Sharma K, Le N, Alotaibi M, Gewirtz DA. Cytotoxic autophagy in cancer therapy. Int J Mol Sci 2014; 15(6):10034-51; PMID:24905404; https://doi.org/ 10.3390/ijms150610034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fulda S, Kogel D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015; 34(40):5105-13; PMID:25619832; https://doi.org/ 10.1038/onc.2014.458 [DOI] [PubMed] [Google Scholar]
- [15].Barth S, Glick D, Macleod KF. Autophagy: Assays and artifacts. J Pathol 2010; 221(2):117-24; PMID:20225337; https://doi.org/ 10.1002/path.2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7(3):279-96; PMID:21189453; https://doi.org/ 10.4161/auto.7.3.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res 2012; 2(4):397-413; PMID:22860231 [PMC free article] [PubMed] [Google Scholar]
- [18].Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat Rev 1997; 23(1):35-61; PMID:9189180; https://doi.org/ 10.1016/S0305-7372(97)90019-0 [DOI] [PubMed] [Google Scholar]
- [19].Perry JA, Kornbluth S. Cdc25 and Wee1: Analogous opposites?. Cell Div 2007; 2:12; PMID:17480229; https://doi.org/ 10.1186/1747-1028-2-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lefranc F, Facchini V, Kiss R. Proautophagic drugs: A novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist 2007; 12(12):1395-403; PMID:18165616; https://doi.org/ 10.1634/theoncologist.12-12-1395 [DOI] [PubMed] [Google Scholar]
- [21].Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001; 4(5):303-13; PMID:11991684; https://doi.org/ 10.1054/drup.2001.0213 [DOI] [PubMed] [Google Scholar]
- [22].Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002; 296(5573):1655-7; PMID:12040186; https://doi.org/ 10.1126/science.296.5573.1655 [DOI] [PubMed] [Google Scholar]
- [23].Zhuang W, Qin Z, Liang Z. The role of autophagy in sensitizing malignant glioma cells to radiation therapy. Acta Biochim Biophys Sin (Shanghai) 2009; 41(5):341-51; PMID:19430698; https://doi.org/ 10.1093/abbs/gmp028 [DOI] [PubMed] [Google Scholar]
- [24].Kurosu T, Nagao T, Wu N, Oshikawa G, Miura O. Inhibition of the PI3K/Akt/GSK3 pathway downstream of BCR/ABL, Jak2-V617F, or FLT3-ITD downregulates DNA damage-induced Chk1 activation as well as G2/M arrest and prominently enhances induction of apoptosis. PLoS One 2013; 8(11):e79478; PMID:24260231; https://doi.org/ 10.1371/journal.pone.0079478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].van Horssen R, Eggermont AM, ten Hagen TL. Endothelial monocyte-activating polypeptide-II and its functions in (patho)physiological processes. Cytokine Growth Factor Rev 2006; 17(5):339-48; PMID:16945568; https://doi.org/ 10.1016/j.cytogfr.2006.08.001 [DOI] [PubMed] [Google Scholar]
- [26].Li Z, Liu XB, Liu YH, Xue YX, Wang P, Liu LB, Liu J, Yao YL, Ma J. Roles of Serine/Threonine phosphatases in Low-Dose endothelial Monocyte-Activating Polypeptide-II-Induced opening of Blood-Tumor barrier. J Mol Neurosci 2015; 57(1):11-20; PMID:26087743; https://doi.org/ 10.1007/s12031-015-0604-8 [DOI] [PubMed] [Google Scholar]
- [27].Wang Y, Chen L, Bao Z, Li S, You G, Yan W, Shi Z, Liu Y, Yang P, Zhang W, et al.. Inhibition of STAT3 reverses alkylator resistance through modulation of the AKT and beta-catenin signaling pathways. Oncol Rep 2011; 26(5):1173-80; PMID:21887474 [DOI] [PubMed] [Google Scholar]
- [28].Ananta JS, Paulmurugan R, Massoud TF. Nanoparticle-Delivered antisense MicroRNA-21 Enhances the effects of temozolomide on glioblastoma cells. Mol Pharm 2015; 12(12):4509-17; PMID:26559642; https://doi.org/ 10.1021/acs.molpharmaceut.5b00694 [DOI] [PubMed] [Google Scholar]
- [29].Najbauer J, Kraljik N, Nemeth P. Glioma stem cells: Markers, hallmarks and therapeutic targeting by metformin. Pathol Oncol Res 2014; 20(4):789-97; PMID:25168767; https://doi.org/ 10.1007/s12253-014-9837-z [DOI] [PubMed] [Google Scholar]
- [30].Binello E, Germano IM. Targeting glioma stem cells: A novel framework for brain tumors. Cancer Sci 2011; 102(11):1958-66; PMID:21848914; https://doi.org/ 10.1111/j.1349-7006.2011.02064.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Facchino S, Abdouh M, Bernier G. Brain cancer stem cells: current status on glioblastoma multiforme. Cancers (Basel) 2011; 3(2):1777-97; PMID:24212782; https://doi.org/ 10.3390/cancers3021777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer 2006; 6(6):425-36; PMID:16723989; https://doi.org/ 10.1038/nrc1889 [DOI] [PubMed] [Google Scholar]
- [33].Sciuscio D, Diserens AC, van Dommelen K, Martinet D, Jones G, Janzer RC, Pollo C, Hamou MF, Kaina B, Stupp R, et al.. Extent and patterns of MGMT promoter methylation in glioblastoma- and respective glioblastoma-derived spheres. Clin Cancer Res 2011; 17(2):255-66; PMID:21097691; https://doi.org/ 10.1158/1078-0432.CCR-10-1931 [DOI] [PubMed] [Google Scholar]
- [34].Beier D, Rohrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, Proescholdt M, Brawanski A, Bogdahn U, Trampe-Kieslich A, et al.. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res 2008; 68(14):5706-15; PMID:18632623; https://doi.org/ 10.1158/0008-5472.CAN-07-6878 [DOI] [PubMed] [Google Scholar]
- [35].Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343(19):1350-4; PMID:11070098; https://doi.org/ 10.1056/NEJM200011093431901 [DOI] [PubMed] [Google Scholar]
- [36].Happold C, Roth P, Silginer M, Florea AM, Lamszus K, Frei K, Deenen R, Reifenberger G, Weller M. Interferon-beta induces loss of spherogenicity and overcomes therapy resistance of glioblastoma stem cells. Mol Cancer Ther 2014; 13(4):948-61; PMID:24526161; https://doi.org/ 10.1158/1535-7163.MCT-13-0772 [DOI] [PubMed] [Google Scholar]
- [37].Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al.. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352(10):997-1003; PMID:15758010; https://doi.org/ 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- [38].Galluzzi L, Morselli E, Vicencio JM, Kepp O, Joza N, Tajeddine N, Kroemer G. Life, death and burial: Multifaceted impact of autophagy. Biochem Soc Trans 2008; 36(Pt 5):786-90; PMID:18793137; https://doi.org/ 10.1042/BST0360786 [DOI] [PubMed] [Google Scholar]
- [39].Codogno P, Meijer AJ. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ 2005; 12(Suppl 2):1509-18; PMID:16247498; https://doi.org/ 10.1038/sj.cdd.4401751 [DOI] [PubMed] [Google Scholar]
- [40].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290(5497):1717-21; PMID:11099404; https://doi.org/ 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; 27(35):4860-4; PMID:18408756; https://doi.org/ 10.1038/onc.2008.117 [DOI] [PubMed] [Google Scholar]
- [42].Botrugno OA, Robert T, Vanoli F, Foiani M, Minucci S. Molecular pathways: Old drugs define new pathways: non-histone acetylation at the crossroads of the DNA damage response and autophagy. Clin Cancer Res 2012; 18(9):2436-42; PMID:22512979; https://doi.org/ 10.1158/1078-0432.CCR-11-0767 [DOI] [PubMed] [Google Scholar]
- [43].Zhang J, Hummersone M, Matthews CS, Stevens MF, Bradshaw TD. N3-substituted temozolomide analogs overcome methylguanine-DNA methyltransferase and mismatch repair precipitating apoptotic and autophagic cancer cell death. Oncology 2015; 88(1):28-48; PMID:25277441; https://doi.org/ 10.1159/000366131 [DOI] [PubMed] [Google Scholar]
- [44].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12(1):1-222; PMID:26799652; https://doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3(6):542-5; PMID:17611390; https://doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- [46].Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, Opferman JT, Slack RS. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J 2011; 30(2):395-407; PMID:21139567; https://doi.org/ 10.1038/emboj.2010.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sakakura K, Takahashi H, Kaira K, Toyoda M, Oyama T, Chikamatsu K. Immunological significance of the accumulation of autophagy components in oral squamous cell carcinoma. Cancer Sci 2015; 106(1):1-8; PMID:25338734; https://doi.org/ 10.1111/cas.12559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171(4):603-14; PMID:16286508; https://doi.org/ 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al.. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141(7):1146-58; PMID:20541250; https://doi.org/ 10.1016/j.cell.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cui J, Bai XY, Shi S, Cui S, Hong Q, Cai G, Chen X. Age-related changes in the function of autophagy in rat kidneys. Age (Dordr) 2012; 34(2):329-39; PMID:21455601; https://doi.org/ 10.1007/s11357-011-9237-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Komatsu M, Wang QJ, Holstein GR, Friedrich VL Jr., Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 2007; 104(36):14489-94; PMID:17726112; https://doi.org/ 10.1073/pnas.0701311104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, Rusten TE, Stenmark H, Brech A. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol 2008; 180(6):1065-71; PMID:18347073; https://doi.org/ 10.1083/jcb.200711108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci 2006; 26(31):8057-68; PMID:16885219; https://doi.org/ 10.1523/JNEUROSCI.2261-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert DR, Simonsen A, et al.. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 2011; 7(6):572-83; PMID:21325881; https://doi.org/ 10.4161/auto.7.6.14943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10(6):507-15; PMID:19945408; https://doi.org/ 10.1016/j.cmet.2009.10.008 [DOI] [PubMed] [Google Scholar]
- [56].Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol 2004; 36(12):2445-62; PMID:15325584; https://doi.org/ 10.1016/j.biocel.2004.02.002 [DOI] [PubMed] [Google Scholar]
- [57].Choi EJ, Cho BJ, Lee DJ, Hwang YH, Chun SH, Kim HH, Kim IA. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: Targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer 2014; 14:17; PMID:24418474; https://doi.org/ 10.1186/1471-2407-14-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet 2002; 36:617-56; PMID:12429704; https://doi.org/ 10.1146/annurev.genet.36.060402.113540 [DOI] [PubMed] [Google Scholar]
- [59].Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432(7015):316-23; PMID:15549093; https://doi.org/ 10.1038/nature03097 [DOI] [PubMed] [Google Scholar]
- [60].Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39-85; PMID:15189136; https://doi.org/ 10.1146/annurev.biochem.73.011303.073723 [DOI] [PubMed] [Google Scholar]
- [61].Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004; 11(4):448-57; PMID:14713959; https://doi.org/ 10.1038/sj.cdd.4401359 [DOI] [PubMed] [Google Scholar]
- [62].Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003; 3(5):421-9; PMID:12781359; https://doi.org/ 10.1016/S1535-6108(03)00110-7 [DOI] [PubMed] [Google Scholar]
- [63].Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol 2004; 5(10):792-804; PMID:15459660; https://doi.org/ 10.1038/nrm1493 [DOI] [PubMed] [Google Scholar]
- [64].Signore M, Pelacchi F, di Martino S, Runci D, Biffoni M, Giannetti S, Morgante L, De Majo M, Petricoin EF, Stancato L, et al.. Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell Death Dis 2014; 5:e1223; PMID:24810059; https://doi.org/ 10.1038/cddis.2014.188 [DOI] [PMC free article] [PubMed] [Google Scholar]