


Abstract
Mutator-like transposable elements (MULEs) are widespread across fungal, plant and animal species. Despite their abundance and importance as genetic tools in plants, the transposition mechanism of the MULE superfamily was previously unknown. Discovery of the Muta1 element from Aedes aegypti and its successful transposition in yeast facilitated the characterization of key steps in Muta1 transposition. Here we show that purified transposase binds specifically to the Muta1 ends and catalyzes excision through double strand breaks (DSB) and the joining of newly excised transposon ends with target DNA. In the process, the DSB forms hairpin intermediates on the flanking DNA side. Analysis of transposase proteins containing site-directed mutations revealed the importance of the conserved DDE motif and a W residue. The transposition pathway resembles that of the V(D)J recombination reaction and the mechanism of hAT and Transib transposases including the importance of the conserved W residue in both MULEs and hATs. In addition, yeast transposition and in vitro assays demonstrated that the terminal motif and subterminal repeats of the Muta1 terminal inverted repeat also influence Muta1 transposition. Collectively, our data provides new insights to understand the evolutionary relationships between MULE, hAT and Transib elements and the V(D)J recombinase.
INTRODUCTION
Transposable elements (TEs) are DNA sequences that can move from one locus to another in the genome. TE induced genomic alterations, including insertions, deletions, duplications and translocations have been linked to changes in gene structure and expression (1,2). TEs are found in virtually all eukaryotes where they can comprise the largest proportion of the genome. For example, sequences derived from TEs account for about half of the human genome and more than 70% of the genomes of some grass species (3).
TEs are classified by their transposition intermediate: class 1 TEs use RNA whereas the element itself is the transposition intermediate for class 2 TEs. Class 2 TEs are further classified into superfamilies on the basis of the element encoded transposase that catalyzes their movement (4). A TE family consists of transposase-encoding autonomous elements and non-autonomous elements that do not encode functional transposase. Within a family, elements usually share the terminal inverted repeat (TIR) sequence and the length of the target site duplication (TSD) that is generated upon insertion by the action of transposase (5).
Mutator–like transposable elements (MULEs) are class 2 TEs first identified in maize (6,7) and subsequently found to be widespread with members in plants, animals, protozoans and fungi (8–11). Typical structural features of MULEs include long TIRs (>100 bp) and 8–10 bp flanking TSDs (12). Non-autonomous MULEs often carry various sequences between their TIRs including host gene fragments; such elements are called Pack-MULEs (13). The high transposition frequency of one superfamily member, MuDR, has been widely exploited for gene tagging and mutagenesis in maize (14).
MULE transposases harbor a zinc finger DNA binding domain and a DDE catalytic domain (15–17). Phylogenetic analysis of transposases from all known eukaryotic superfamilies indicates that MULEs and hATs are closely related. For example they share a (C/D) (2) H motif located 15–45 amino acids downstream of the second D of the DDE triad and a W residue upstream of the E residue of the DDE triad (18). MULE and hAT elements also have longer TSDs than most other superfamilies, with the predominant TSD for hAT elements at 8 bp, and 8–10 bp for MULE elements (19). These shared features suggest that their transposition mechanisms may also be similar.
Among eukaryotic class 2 TEs, transposition begins with cleavage of one strand at both ends of the element, the nucleophile is usually H2O and this first cleavage results in exposed 3΄-hydroxyl groups (OH) at either the element end or the flanking (host DNA) end (Figure 1). The free 3΄OH serves as nucleophile for cleavage of the second strand and liberation of the transposon from the donor site (20). Three distinct mechanisms are known to date for second strand cleavage. As an example of the first pathway, Tc1/mariner transposases make double-strand cuts to release the transposon end from the donor site (Figure 1A) (21,22). In the second pathway, first strand cleavage results in a 3΄OH in flanking DNA, which subsequently attacks the opposite strand to generate a hairpin structure (Figure 1B). This pathway is used by hAT and Transib elements in addition to the V(D)J recombinase RAG, which produces the highly diverse repertoire of T-cell receptors, antibodies and immunoglobulins (23–25). In the third pathway, employed by piggyBac elements (26), the 3΄OH generated in first strand cleavage at the transposon end attacks the opposite strand forming a hairpin structure at the transposon end (Figure 1C). After release from the donor site, the 3 ΄ OH at the free transposon ends serve as nucleophiles in a concerted attack on the target DNA in a reaction called strand transfer. Breaks in the target DNA are repaired by host enzymes, thereby generating the TSD flanking the inserted transposon. In contrast, breaks at the donor site are repaired either by end joining, a pathway that often leaves ‘footprints’ at the donor site reflecting element excision (27,28) or homology-dependent gene conversion using a sister chromatid or homolog as template (29).
Figure 1.
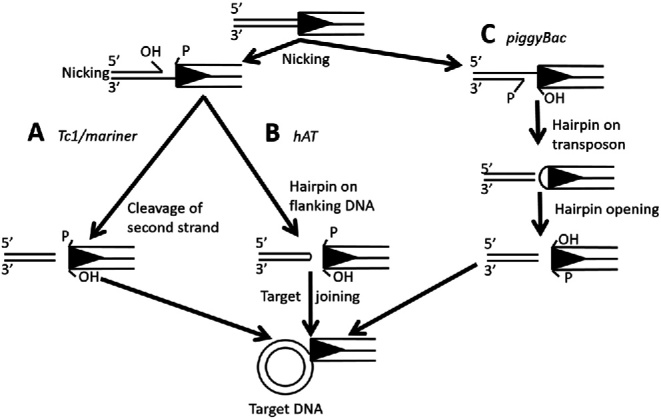
Comparison of transposase-mediated cleavage and target joining mechanisms. Key steps in three DNA cleavage and target joining pathways of TE superfamilies. Black triangle represent the TIR. After transposon ends are released through different DNA cleavage mechanisms, the free 3΄ OH is used for target joining. (A) Tc1/mariner transposases make double-stranded cuts and generate element ends with free 3΄ OH. (B) hAT transposases catalyze the cleavage reaction through formation of a hairpin structure in flanking DNA. (C) piggyBac transposases generate a hairpin structure at the transposon end and release transposon from the donor site.
Because MULEs and hATs have a close evolutionary relationship and share features including the (C/D) (2)H motif, the W residue and long TSDs, it was hypothesized that they utilized similar mechanisms of transposition (18). To test this hypothesis, we studied transposition of the MULE Muta1, isolated from the genome of the mosquito, Aedes aegypti, which can spread dengue fever, yellow fever, chikungunya, zika and many other diseases (30–32). In a previous study we showed that Muta1 transposase catalyzed the transposition of natural and artificial non-autonomous elements in a yeast assay (33). For this study, steps in the Muta1 transposition mechanism were dissected, from the binding of purified transposase to Muta1 ends, to cleavage of Muta1 ends from the donor site, and finally, to the joining of cleaved Muta1 ends to target DNA. Furthermore, mutagenesis analysis revealed that the conserved DDE triad and the W residue, shared by MULE and hAT transposases, plays critical roles in all steps of Muta1 transposition. Finally, the terminal motif and subterminal repeats of the Muta1 TIR also impact transposition reactions.
MATERIALS AND METHODS
Muta1 transposase expression and purification
The 1512-bp Muta1 transposase coding sequence was isolated by polymerase chain reaction (PCR) amplification from pMuta1-PAG415 (Supplementary Figure S3A) (33). In the PCR process, codons of 6 histidines were added to the N-terminal of the coding sequence and cloned between the NcoI and PstI sites of plasmid pBAD-Myc-HisC (Invitrogen) (which has a C-terminal His-tag) to generate pBAD-Muta1-His-NC. This fusion construct has 6 His residues on both the N and C termini of the Muta1 coding sequence. Escherichia coli Top10 (Invitrogen) cells containing pBAD-Muta1-His-NC were grown with shaking at 30°C in LB medium containing 100 mg/ml carbenicillin to OD600 of 0.5. The culture was induced with 0.2% L-arabinose for 18 h at 16°C and cells were harvested by centrifugation at 8000 rpm for 5 min and resuspended in 8 ml of phosphate buffered saline buffer (50 mM NaH2PO4, 500 mM NaCl and 10 mM imidazole, pH 7.5). After addition of lysozyme (0.2 g/l of culture), cells were incubated on ice for 30 min before lysis by sonication for 30 min (Fisher Scientific). A total of 6 ml of lysate was loaded onto a pre-equilibrated column containing 1 ml Ni2+ resin (Invitrogen). The column was washed with 35 ml of Tris-buffered saline (TBS) buffer with 50 mM imidazole followed by 35 ml of TBS buffer with 100 mM imidazole. Muta1–His fusion protein was eluted with 7 ml of TBS buffer with 500 mM imidazole (Supplementary Figure S1), dialyzed twice against 35 ml volume of TBS buffer, concentrated using centrifuge filtration (Amicon system) to a final volume of 100 ul and glycerol added to a final concentration of 15% (v/v) for storage at −80°C.
DNA binding assay
DNAs substrates (Supplementary Table S1) were 5΄ end radiolabeled on both strands with gamma-P32-ATP and T4 polynucleotide kinase (NEB). Muta1 transposase (15 ng) and 2 nM radiolabeled DNA were incubated in 25 mM HEPES, 5% (v/v) glycerol, 0.01% bovine serum albumin (BSA) and 4 mM dithiothreitol (DTT) in the presence of a 100-fold molar excess of sheared herring sperm DNA at room temperature for 30 min. Unlabeled full length Muta1 L-TIR DNA was used in 100 or 500 molar excess as competitor DNA. All products were resolved on 5% native acrylamide gels and gels were dried and exposed to X-ray film.
Double strand break and end joining reactions
The DSB assay was conducted as previously described (23,26). Muta1 transposase (15 ng) was incubated with 1.5 nM radiolabeled Muta1 L-TIR or other end fragments in 25 mM HEPES (pH 8.0), 3 mM Tris (pH 8.0), 75 mM NaCl, 2 mM DTT, 10 mM MgCl2, 0.01% BSA, 5% glycerol and 10 nM pUC19 in a final volume of 20ul at 30°C for different time intervals. Reactions were stopped by incubation in 1% sodium dodecyl sulphate and 20 mM ethylenediaminetetraacetic acid for 30 min at 65°C and displayed on 5% native and denaturing (with 7 M urea) acrylamide gels.
Following published protocols (23,26), 200 nM of Muta1 transposase was incubated with 1.5 nM of radiolabeled Muta1 L-TIR or other end fragments in 25 mM HEPES (pH 8.0), 3 mM Tris (pH 8.0), 75 mM NaCl, 2 mM DTT, 10 mM MgCl2, 0.01% BSA, 5% glycerol and 10 nM pUC19 in a final volume of 20 ul at 30°C for 30 min. Products were displayed on native or denaturing (with 50 mM NaOH) 1% agarose gels.
Mutagenesis of Muta1 transposase
Site-directed mutagenesis was used to generate mutant versions of Muta1 transposase. One pair of primers was used for each mutation site, and PBAD-Muta1-His-NC plasmid (described in first section of ‘Materials and Methods’ section) was used as template. PCR products were digested with Dpn1 to remove template, and the resulting plasmid was sequenced to confirm that mutations occurred as expected.
Yeast transposition assay
The yeast transposition assay using Saccharomyces cerevisiae strain DG2523 and the pWL89A vector was described previously (33). Plasmid pMuta1-PAG415 (Supplementary Figure S3A, (33)) was used to express transposase. Mutant versions of Muta1NA1 were generated by PCR (primers in Supplementary Table S1). All non-autonomous elements were inserted in the XhoI site of pWL89A (Supplementary Figure S3A and B) through homologous recombination in yeast as previously described (33). Transformation was performed using the Frozen-EZ Yeast Transformation kit (Zymo research). Transformants were grown in 5 ml liquid media of CSM-leu-ura with 2% dextrose. After growth to saturation (36 h), cells were washed twice with 5 ml H2O, resuspended in 0.5 ml H2O and plated onto CSM-his-leu-ade with 2% galactose. Colonies were counted after incubation at 30°C for 15 days and viable counts were made by plating 100 μl of a 1 × 105 and 1 × 106 dilution on YPD plates.
Colony PCR was performed on ADE2 revertant colonies using primers (Supplementary Table S1) flanking the insertion sites and PCR products were gel extracted (Zymoclean Gel DNA Recovery Kit) and sequenced for footprint analysis.
RESULTS
Muta1 transposase binds the transposon end
Muta1 transposase harbors a N-terminal FLYWCH type zinc-finger domain and a C-terminal DDE catalytic domain (Figure 2A). The 504 amino acid transposase was cloned, expressed and purified from E. coli as a His-tagged derivative. Initially, a C-terminal His-tag was added, however, because the fusion protein had poor affinity for the nickel column, a N-terminal His-tag was also added, and the double His-tag fusion protein was used in all experiments (see ‘Materials and Methods’ section).
Figure 2.

In vitro analysis of DNA binding of Muta1 transposase. (A) The 3198-bp Muta1 encodes a 504 amino acid transposase with the predicted zinc finger and catalytic (DDE) domains shown. White boxes indicate exons and grey boxes indicate non-coding regions. Black arrowheads represent the TIR whose substructure is expanded below. In this and all subsequent figures open arrows represent the 10-bp terminal palindromic motif, and black arrows are the 12-bp subterminal tandem repeat (Supplementary Figure S2). (B) The 165-bp DNA substrate used in DNA binding assays is 5΄ radiolabeled on both strands (asterisk) and contains a 20-bp flanking DNA segment (white box) and the full length Muta1 L-TIR. (C) Muta1 tpase and DNA binding assay. Lane 1: negative control without tpase protein. Lanes 2–4: fastest migrating band present in all lanes is DNA substrate, slower migrating bands (>300 bp) are nucleoprotein complexes. Competitor DNA is unlabeled DNA in (B) which was added in 100 (+) or 500 (++) molar excess. One hundred molar excess of non-specific DNA is added in all reactions.
Muta1 is 3198 bp, flanked by 8 or 9 bp TSDs and a 145-bp TIR comprised of a 10 bp imperfect palindromic terminal motif and 9 copies of a 12 bp subterminal tandem repeat separated by 3–4 bp linkers (Figure 2A & B and Figure S2). The Muta1 L-TIR and R-TIR share high sequence identity (97%) with all sequence differences located in the linkers DNA between subterminal repeats. For this reason the L- and R-TIR were considered symmetric and only L-TIR was used in this study.
Before determining transposition reaction intermediates (Figure 1), we tested whether Muta1 transposase (purified from E. coli) could bind to L-TIR. A 5΄ P32 radiolabeled 165 bp fragment containing the full length L-TIR and 20 bp of flanking sequence was used as substrate in DNA binding assays (Figure 2B). The result indicates that Muta1 transposase binds to DNA fragments containing Muta1 L-TIR in the presence of 100 molar excess of non-specific DNA. The binding is specific as addition of competitor DNA reduced the binding strength (Figure 2C, lanes 2–4). The many bands (ranging from 350 bp to >700 bp) may represent binding of oligomeric transposase proteins (Figure 2C, lanes 2–4).
Muta1 transposase catalyzes double strand breaks through a hairpin intermediate on the flanking DNA side
As shown in Figure 1, each of the characterized cleavage pathways produces distinct reaction intermediates that can be determined experimentally. For the donor cleavage assay, Muta1 transposase was incubated with a 5΄ radiolabeled 188 bp fragment containing the full length Muta1 L-TIR and 43 bp of flanking sequence (Figure 3A). The reaction products were examined on both native and denaturing polyacrylamide gels to detect formation of a hairpin intermediate. On a native gel, two cleavage products were observed: the 145 bp band derives from the Muta1 L-TIR and the 43 bp band from flanking DNA (Figure 3B, lanes 5–8, diagram on right). Both fragments increase in amount with increased time of incubation (Figure 3B, lanes 5–8).
Figure 3.

In vitro analysis of Muta1 double strand cleavage. (A) The 188-bp DNA substrate is 5΄ radiolabeled and contains full length Muta1 L-TIR (145 bp) and a 43 bp flanking segment. The possible reaction outcomes of the three DNA cleavage pathways in Figure 1 are shown. (B) Reaction products on a native polyacrylamide showing 43 and 145 bp Muta1 L-TIR products (lanes 5–8). Diagram on right indicates the predicted structure and size of each band. (C) Reaction products on a denaturing polyacrylamide gel showing the 145 and 86 nt bands (lanes 4–8).
On a denaturing gel, two reaction products were detected (Figure 3C, lanes 4–8). The size of the two products is consistent with the middle pathway in Figure 3A: the 145 nt band reflects the Muta1 L-TIR, and the 86 nt band reflects a linearized DNA hairpin containing both strands of the flanking DNA (43 + 43 nt, Figure 3C diagram and lanes 4–8). The amount of both fragments increases with increased incubation (from 5 to 120 min, Figure 3C, lanes 4–8). These results indicate that Muta1 donor cleavage involves formation of a hairpin structure in flanking DNA (Figure 3A, model 2).
Muta1 transposase joins the transposon end to target DNA
Donor site cleavage of Muta1 generates a free 3΄ OH of the L-TIR (Figure 3A, middle cleavage model), which could serve as a nucleophile in an end joining reaction. To test if Muta1 transposase could join the Muta1 L-TIR to target DNA, a ‘pre-cleaved’ L-TIR fragment, that is, with its 3΄ OH already exposed, was constructed to serve as substrate with the target as intact pUC19 plasmid (2.6 kb). The 5΄ radiolabeled substrate contains the full length L-TIR with 20 bp of Muta1 internal sequence (Figure 4A).
Figure 4.

In vitro analysis of Muta1 end joining with target DNA. (A) The 165-bp DNA substrate contains full length Muta1 L-TIR with 20 bp of Muta1 internal sequence. Two lines indicate the DNA double strands. On the bottom strand, the 5΄ P is radiolabeled and the 3΄ OH is exposed. (B) Reaction products on a native agarose gel reveal two bands at 4.1 and 2.8 kb, which reflect joining product SEJ (nicked circular plasmid formed by joining of one transposon end to one plasmid strand) and DEJ (linearized plasmid formed by concerted joining of two transposon ends to two plasmid strands), as indicated in the diagram on right. (C) Reaction products on a denaturing agarose gel, where only the 2.8 kb is seen, which reflects both DEJ and SEJ products.
Labeled substrate and pUC19 plasmid were incubated with Muta1 transposase and reaction products displayed on native and denaturing agarose gels (Figure 4A and B). Two bands of 2.8 and 4.1 kb were observed on the native gel (Figure 4B, lanes 3–5). The 2.8 kb band reflects double-end join (DEJ), which is generated by concerted joining of L-TIR substrate to each strand of target DNA, thereby forming a linear, double-stranded DNA molecule (Figure 4B diagram). The 4.1 kb band reflects a slower migrating nicked circular plasmid, which is the product of single-end join (SEJ), generated by joining one L-TIR substrate to one strand of the target DNA. In SEJ product, one strand of pUC19 is cleaved by the joining of one L-TIR fragment and the other plasmid strand is intact (Figure 4B diagram). The amount of SEJ and DEJ products increase over time (from 20 to 120 min, Figure 4B, lanes 3–5). On a denaturing gel, only one product is observed, consistent with DEJ and the linearized SEJ products, as the double-stranded SEJ product is denatured (Figure 4C, lanes 3–5). These results indicate that Muta1 transposase is able to join the Muta1 L-TIR fragment to target DNA in vitro.
Subterminal repeats impact Muta1 transposition reactions in vitro
We next turned our attention to the cis requirements for Muta1 transposition beginning with the distinctive long TIRs. Muta1 can be classified as a type 2 foldback element because its TIR is composed of subterminal tandem repeats capable of forming secondary structures (Figure 2A) (34,35). Although foldback elements are abundant in animal and plant species in the MULE and P superfamilies (11,34–36), the function of the subterminal repeats is largely unknown. For the experiments described above, DNA fragments containing the full length L-TIR (with nine copies of the subterminal repeat) served as substrate (Figures 2–4). The function of subterminal repeats was tested by reducing repeat number in substrates used in DNA binding, donor site cleavage and end joining assays.
DNA binding with full length L-TIR resulted in multiple bands, which suggested binding of oligomeric transposase (Figure 2C). To test if the binding of multiple transposase proteins correlated with the presence of multiple copies of subterminal repeats, we generated a series of DNA fragments consisting of variable copies of the repeat and analyzed their binding activity (Figure 5). All six DNA substrates contained the same 20 bp flanking sequence but different sequences derived from Muta1 L-TIR, corresponding to one copy of the subterminal repeat (Figure 5A, lanes 1 and 2), the terminal motif alone (lanes 3 and 4), the terminal motif plus one, two or three copies of the subterminal repeat (lanes 5–10), and the full length TIR (lanes 11 and 12). Although the substrates vary in size (Figure 5A, see the variation in the rapidly migrating bands in lanes 1–12), binding to transposase is specific as addition of competitor DNA reduced binding of labeled substrate (lanes 1, 2, 5–12). A single band of ∼350 bp was observed for substrates containing the terminal motif and zero or one copy of the subterminal repeat (lanes 1, 2, 5 and 6). In contrast, no binding was detected with substrate containing only the subterminal repeat and 20 bp of flanking DNA (lanes 3 and 4). Additional bands (>350 bp) were detected with substrates containing two or more copies of the subterminal repeat (lanes 7–12). These results suggest that the Muta1 transposase binds specifically to fragments containing the 10 bp terminal motif and 20 bp flanking DNA (lanes 1 and 2) but not to the fragment containing only one copy of the subterminal repeat and flanking DNA (lanes 3 and 4). Increasing the number of the subterminal repeats promotes binding and generates additional bands (lanes 1, 2, 5–12).
Figure 5.

In vitro analysis of the impact of the Muta1 subterminal repeats on transposase binding, double strand break and end joining reactions. (A) DNA binding assay. Diagrams at the top are structures of DNA substrates, all contain 20-bp flanking DNA segment (open box), subterminal repeats and terminal motif. The unlabeled 165-bp DNA containing 20-bp flanking DNA and the full length Muta1 L-TIR is added in 100 (+) molar excess as competitor DNA. Reaction products are resolved on a native polyacrylamide gel. Fast migrating bands are DNA substrates (30–165 bp). (B) Donor cleavage assay. Diagrams at the top are the structures of DNA substrates, diagrams at the bottom are predicted structures of cleavage products. Reaction products are resolved on a native polyacrylamide gel. (C) End joining assay. Diagrams at the top are the structures of DNA substrates, intact pUC19 plasmid is used as target DNA, reaction products are resolved on a native agarose gel.
To investigate the cis requirements for the donor site cleavage reaction, three different DNA fragments containing 100 bp of flanking DNA and either or both the terminal motif and the subterminal repeat were used as substrate (Figure 5B diagram). On a native polyacrylamide gel, the cleavage product (100 nt) was only detected when the substrate contained both motifs (Figure 5B, lane 3 and bottom diagram) after 60 min incubation with transposase.
In the end joining assay, three different DNA fragments containing either one of the two motifs or both and 20 bp of Muta1 internal sequence were used as substrates (Figure 5C diagram). On a native agarose gel, DEJ and SEJ products were only detected when the substrates contained the terminal motif, with or without the subterminal repeat. (Figure 5C, lanes 2 and 6).
Terminal palindromic motif impacts Muta1 transposition reactions in vitro
The sequence of the terminal motif of the Muta1 TIR, GGGTCTACCC, is an imperfect palindrome with the fifth and sixth nucleotides unpaired. To determine if the sequence and the palindromic pattern of this motif impacts transposition, mutations in the terminal motif were tested for DNA binding, donor site cleavage and target joining reactions. For DNA binding, fragments used as substrates all contained 20 bp of flanking sequence and one copy of the subterminal repeat, and mutant versions of the terminal motif (Figure 6A diagram). This substrate was chosen because it was sufficient to bind transposase but did not form multisubunit complexes (Figure 5A, lanes 5 and 6), thus simplifying the interpretation of results.
Figure 6.

In vitro analysis of the impact of the Muta1 terminal palindromic motif on DNA binding, double strand break and end joining reactions. (A) DNA binding assay. Diagram shows the structure of the DNA substrate. Underlined letters indicate mutations in the terminal motif. Reactions products are resolved on a native polyacrylamide gel. The unlabeled 165-bp DNA containing 20-bp flanking DNA and the full length Muta1 L-TIR is added in 100 (+) molar excess as competitor DNA. Fast migrating bands are the DNA substrates (∼50 bp). (B) Quantification of the DNA binding of substrates used in (A). Strength of binding was quantified with ImageJ software and repeated five times to generate the standard deviation. (C) Cleavage assay. Diagram shows the structure of DNA substrates and cleavage products. Sequences of substrates are shown; underlined letters indicate mutations in the terminal motif. Reactions products are resolved on a native polyacrylamide gel. (D) End joining assay. Diagram shows the structure of the DNA substrate. Underlined letters indicate mutations in the terminal motif. Intact pUC19 plasmid is used as target DNA and reaction products are resolved on a native agarose gel.
Mutations introduced in the terminal motif were selected to disrupt or restore the palindromic pattern (Figure 6A and B, underlined letters). For example, the first mutant has a G to C substitution of the first nucleotide (Figure 6A, lane 2) and the palindrome is restored in the second mutant by a C to A substitution of the tenth position (Figure 6A, lane 3). Quantitation of band strengths indicated that mutations in the terminal motif reduce binding activity. However, in each case, second-site mutations that restored the palindromic pattern increase binding (Figure 6A, lanes 3–12, Figure 6B). These data indicate that the palindromic pattern is important for transposase binding and that the weaker binding caused by single mutations has more impact when the mutation is closer to the end of the TIR.
For the donor site cleavage assay, all DNA substrates contain a 100-bp flanking segment, one copy of the subterminal repeat and the terminal motif with the mutations indicated as underlined (Figure 6C). Cleavage products (100 nt) were observed for all mutant substrates but reduced in amount compared with the wild-type terminal motif (Figure 6C, lanes 2–12). These data demonstrate that terminal mutations reduced cleavage activity, but that cleavage did not always correlate with binding because mutations that restored the palindromic pattern did not increase cleavage.
For the end joining assay, substrates contain one copy of the subterminal repeat and the terminal repeat with the mutations indicated as underlined (Figure 6D). Based on the amount of SEJ and DEJ products, mutations in the third and fourth/fifth nucleotide (Figure 6D, lanes 7 and 12) have a more negative impact on end joining activity than mutations in other positions. Further, restoration of the palindromic pattern does not restore end joining activity (Figure 6D, lanes 2–12).
Identification of the catalytic core of the Muta1 transposase
Prior mutagenesis analysis identified several functionally important residues of the Muta1 transposase including the DDE triad (D214, D283, E419) and W401 shared by transposases from the MULE and hAT superfamilies (18, 33). To probe the function, if any, of these amino acids in Muta1 transposition, transposases with D214A, D283A, E419A, W401A or W401F mutations were purified and assayed for DNA binding, donor site cleavage and end joining activities.
Although still capable of binding specifically to the Muta1 L-TIR fragment (Figure 7A, lanes 2–13), the alanine substitution mutant transposases lost activity in all DNA cleavage and joining reactions, while the W401F mutation retained partial activity. First, in the cleavage assay, no cleavage products were detected after 120 min incubation with alanine substitution mutant transposases (Figure 7B, lanes 3–6), and the W401F mutant generated less cleavage products than wild-type transposase (Figure 7B, lanes 2 and 7). The alanine substitution mutants were also defective in the end joining reaction (Figure 7C, lanes 3–6), as no joining products were detected, while the W401F mutation only retained partial activity (Figure 7C, lanes 2 and 7). These results indicate that the DDE triad and the W residue are involved in donor site cleavage and end joining of Muta1-mediated transposition.
Figure 7.

In vitro mutagenesis analysis of the role of conserved residues of Muta1 transposase in transposition. (A) DNA binding assay using wild-type and mutant transposases. Diagram shows the structure of the DNA substrate. The unlabeled 165-bp DNA containing 20-bp flanking DNA and the full length Muta1 L-TIR is added in 100 (+) molar excess as competitor DNA. Binding products are resolved on a native polyacrylamide gel. (B) Cleavage assay using wild-type and mutant transposases. Diagram shows the structure of DNA substrates and predicted cleavage product. Reaction products are resolved on a native polyacrylamide gel. (C) End joining assay using wild-type and mutant transposases. Intact pUC19 plasmid is used as target DNA. Reaction products are resolved on a native agarose gel.
Prior studies showed that the conserved DDE triad of many transposases (hAT Tc1/mariner, piggyback) and the RAG1 recombinase were organized in an RNaseH-like fold to form the active site (20,37–41). In the typical RNase H-like fold of β1-β2-β3-α1-β4-α2/3-β5-α4-α5/6, the DDE triad is usually located on specific secondary structures, with the first D on β1, the second D right after β4 and the E on α4 (Supplementary Figure S4, RAG1, (20)). However, the secondary structure of most A. aegypti MULE transposases differ from the typical RNase H-like fold, because of the lack of β-strand adjacent to the second D of the DDE triad and the addition of four α-helices between the second D and the E (Supplementary Figure S4). These differences may be explained by the limitation of the secondary structure prediction programs, alternatively, the secondary structure predictions suggests the DDE triad may be brought together to form the active center through a non-standard RNase-H like fold, or through formation of oligomer.
Subterminal repeats and terminal motif impact Muta1 transposition in yeast
Yeast transposition assays were performed to determine if the in vitro results presented above correlate with changes in transposition frequency. The assay employs a Muta1 transposase expression vector and a reporter vector containing a non-autonomous element inserted in the 5΄ UTR that blocks expression of the ADE2 gene (Supplementary Figure S3A). Transposase-mediated element excision restores ADE2 expression and permits cells to grow on minimal plates (Supplementary Figure S3B).
To assess the contribution, if any, of the number of subterminal repeats on the frequency of excision in yeast (33), we constructed a series of non-autonomous elements with different termini. Elements were generated by modifying Muta1NA1, a natural non-autonomous derivative of Muta1 isolated from the A. aegypti genome and shown previously to transpose in yeast (Figure 8A, (33)). The ends of Muta1NA1 are not symmetric, containing three and seven copies of the subterminal repeat. Constructs were generated that contain the 315 bp internal sequence of Muta1NA1 flanked by variable numbers of the two motifs, as indicated by the number of ‘Ts’ (Terminal motif) and ‘Rs’ (subterminal Repeat). For example, 1T0R+1T7R represents an element with one copy of the terminal motif but no subterminal repeat at the left end and one copy of the terminal motif and seven copies of the subterminal repeat at the right end. Excision frequencies of the artificial elements indicate that having the terminal motif and one copy of the subterminal repeat at both ends is sufficient for transposition, and increasing the copy number of the subterminal repeat increases element excision (Figure 8B).
Figure 8.

Analysis of the impact of Muta1 subterminal repeats and terminal motif on transposition in yeast. (A) Muta1NA1 contains the 10-bp terminal motif at both ends; three and seven copies of the subterminal repeats on the left and right ends, respectively, and 315 bp of internal DNA sequence. (B) Excision frequencies of Muta1NA1 derivative elements generated by altering the copy number of the two motifs at both ends of Muta1NA1. TIR composition of each element is shown at the bottom (see text for details). Six replicates were used for each. (C) Excision frequencies of mutant versions of Muta1NA1. Mutations were introduced at both ends of the terminal motif of Muta1NA1, as indicated by underlined letters. Six replicates were used for each.
To assess the impact of mutations of the terminal motif on excision frequency in yeast, the terminal motifs at both ends of Muta1NA1 were replaced with the mutations analyzed previously (Figure 6). When compared to the excision frequency of Muta1NA1, all mutant elements showed a reduction in excision frequency with no apparent increase in excision frequency when the palindromic pattern is restored (Figure 8C). Consistent with the in vitro results, mutations closer to the end of the terminal motif had a more severe reduction in excision frequency (Figure 8C).
DISCUSSION
Results of this study provide evidence that the Muta1 transposase catalyzes a double strand break mechanism involving formation of a hairpin structure in the flanking DNA, thereby releasing the transposon from the donor site (Figures 1B and 3C). Similar mechanisms have been reported for hAT and Transib elements, the V(D)J recombinase RAG1 and a subset of retroviral integrases (Figure 1B) (20,38–41). These findings support the close evolutionary relationship between MULEs and hATs, first revealed by phylogenetic analysis (18).
After transposon excision, repair of the donor site requires opening the hairpin structure. Experiments presented in this study suggest that this step is probably not performed by Muta1 transposase because the hairpin product accumulates and cleaved hairpin is not detected as the reaction time is extended (Figure 3C). Similar findings were also reported for Hermes, Transib and RAG1 (23–25). The hairpin may be opened by host enzymes like Artemis, which open similar structures (23,42). Subsequent joining of the opened hairpins for hAT, Transib and RAG1 reactions may be achieved by the non-homologous end joining pathway, which often leaves footprints at the donor site (23–25). Muta1 may utilize a different gap repair mechanism because the majority of excision events (>90%) observed in a yeast transposition assay were precise (the element was removed as well as a single copy of the TSD, (33)). Interestingly, Muta1 footprints were detected more frequently when TSDs do not flank the donor element (33), similar to another MULE (rice Os3378 (43). The different footprint pattern suggests the gap repair mechanism of Muta1 and other MULEs may be distinct from hAT elements. It was hypothesized that Os3378 utilizes a transposase-independent replication slippage pathway for the donor site gap repair, which requires at least 8 bp of homologous DNA flanking the break point (43), this may explain the dependence of precise excision on the presence of donor site TSD.
Mutagenesis analysis confirms the importance of the DDE triad in Muta1 transposition reactions (Figure 7 A–C). Direct involvement of the DDE triad in DSB and end joining reactions has also been shown for hAT, Transib and piggyBac transposases and for the RAG1 recombinase (23–26). Although the spatial organization of Muta1 transposase is not the typical RNaseH-like fold that characterizes the transposases of other superfamilies, the multiple critical residues including the DDE triad and the W may be closely positioned to form the active center through more complex folding or the formation of transposase oligomers.
One explanation for the fact that MULE, hAT, Transib and V(D)J reactions all form hairpin structures in flanking DNA is that besides the DDE triad, they share a few functionally important amino acids that are critical for hairpin formation (18). Crystallographic analysis and in vitro assays revealed that W318 in the Hermes transposase plays an important role in positioning flanking DNA, which ensures that the DSB occurs at the correct position during the excision reaction (38). Furthermore, crystallographic analysis revealed that mutation of the corresponding W893 of RAG1 could destabilize its structure (39). Although the transposase of Transib elements were not reported to contain this W, we identified the W from multiple alignment analysis (Supplementary Figure S5). For Muta1, alanine substitution of the W401A mutation abolished all DNA cleavage and joining reactions, while W401F reduced activity (Figure 7 B and C) and decreased the ratio of precise excision in yeast from 90 to 50% (33), indicating that W401F mutation could lead to inaccurate Muta1 excision. Collectively, our data suggests this W plays a critical role in Muta1 transposition, possibly in hairpin formation, which is similar to its role in hAT elements (38).
Prior to this study the function of the subterminal repeats of foldback transposons was unclear (34–36). Several lines of evidence presented in this study indicate the importance of subterminal repeats for transposition of foldback elements. First, increasing the number of subterminal repeats strengthens Muta1 transposase binding. The generation of additional bands suggests that the presence of multiple copies of subterminal repeats could enhance transposase binding (Figure 5A). Second, in the donor site cleavage assay, cleavage product was only detected when the substrate contained the subterminal repeat (Figure 5B). Finally, increasing the copy number of the subterminal repeat increased excision frequency in yeast (Figure 8B). Collectively, these data provide the first experimental evidence that subterminal repeats are critical for transposition of foldback elements.
Lastly, our date demonstrate that the palindromic pattern of the Muta1 terminal motif (GGGTCTACCC) impacts transposase binding (Figure 6A–D). Several restriction enzymes also bind to palindromic sequences (44). Muta1 termini recognition may utilize a similar mechanism, where the terminal palindromic motif provides structural hints to identify the end of the element. Mutations in the Muta1 terminal motif also reduced donor site cleavage activity and excision frequency in yeast, suggesting its sequence may be optimal for transposition. Similarly, mutagenesis of the end of the TIR of the maize Ds element greatly reduced excision activity (27).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lu Lu, Robert Hice and Peter Atkinson for their technical assistance and advice.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
W.M. Keck Foundation (to S.R.W.). Funding for open access charge: W.M. Keck Foundation (to S.R.W.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Kazazian H.H. Mobile elements: drivers of genome evolution. Science. 2004; 303:1626–1632. [DOI] [PubMed] [Google Scholar]
- 2. Finnegan D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989; 5:103–107. [DOI] [PubMed] [Google Scholar]
- 3. Chénais B., Caruso A., Hiard S., Casse N.. The impact oftransposable elements on eukaryotic genomes: from genome size increaseto genetic adaptation to stressful environments. Gene. 2012; 509:7–15. [DOI] [PubMed] [Google Scholar]
- 4. Feschotte C., Pritham E.J.. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007; 41:331–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wicker T., Sabo F., Hua-Van A., Bennetzen J.L., Capy P., Chalhoub B., Flavell A., Leroy P., Morgante M., Panaid O. et al. . A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007; 8:973–982. [DOI] [PubMed] [Google Scholar]
- 6. Robertson D.S. Characterization of a mutator system in maize. Mutat Res. 1978; 51:21–28. [Google Scholar]
- 7. Lisch D. Regulation of the Mutator System of Transposons in Maize. Methods Mol. Biol. 2013; 1057:123–142. [DOI] [PubMed] [Google Scholar]
- 8. Neuveglise C., Chalvet F., Wincker P., Gailardin C., Casaregola S.. Mutator-like element in the yeast Yarrowia lipolytica displays multiple alternative splicings. Eukaryot. Cell. 2005; 4:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pritham E.J., Feschotte C., Wessler S.R.. Un-expected diversity and differential success of DNA transposons in four species of entamoeba protozoans. Mol. Biol. Evoanl. 2005; 22:1751–1763. [DOI] [PubMed] [Google Scholar]
- 10. Lopes F.R., Silva J.C., Benchimol M., Costa G.G., Pereira G.A., Carareto C.M.. The protist Trichomonas vaginalis harbors multiple lineages of transcriptionally active Mutator-like elements. BMC Genomics. 2009; 10:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marquez C.P., Pritham E.J.. Phantom, a new subclass of Mutator DNA transposons found in insect viruses and widely distributed in animals. Genetics. 2010; 185:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lisch D. Mutator transposons. Trends Plant Sci. 2002; 7:498–504. [DOI] [PubMed] [Google Scholar]
- 13. Jiang N., Bao Z., Zhang X., Eddy S.R., Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants. Nature. 2004; 431:569–573. [DOI] [PubMed] [Google Scholar]
- 14. Lisch D., Jiang N.. Bennetzen JL, Hake SC. Mutator and MULE transposons. Handbook of Maize: Genetics and Genomics. 2009; NY: Springer; 277–306. [Google Scholar]
- 15. Babu M.M., Iyer L.M., Balaji S., Aravind L.. The natural history of the WRKY-GCM1 zinc fingers and the relationship between transcription factors and transposons. Nucleic Acids Res. 2006; 34:6505–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nesmelova I.V., Hackett P.B.. DDE transposases: Structural similarity and diversity. Adv. Drug Deliv. Rev. 2010; 62:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eisen J.A., Benito M.I., Walbot V.. Sequence similarity of putative transposases links the maize Mutator autonomous element and a group of bacterial insertion sequences. Nucleic Acids Res. 1994; 22:2634–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yuan Y., Wessler S.R.. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:7884–7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rubin E., Lithwick G., Levy A.A.. Structure and evolution of the hAT transposon superfamily. Genetics. 2001; 158:949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hickman A.B., Chandler M., Dyda F.. Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit. Rev. Biochem. Mol. Biol. 2010; 45:50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plasterk R.H., Izsvák Z., Ivics Z.. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999; 15:326–332. [DOI] [PubMed] [Google Scholar]
- 22. Feng X., Colloms S.D.. In vitro transposition of ISY100, a bacterial insertion sequence belonging to the Tc1/mariner family. Mol. Microbiol. 2007; 65:1432–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou L., Mitra R., Hickman A.B., Dyda F., Craig N.L.. Transposition of hAT elements links transposable elements and V(D)J recombination. Nature. 2004; 432:995–1001. [DOI] [PubMed] [Google Scholar]
- 24. Hencken C., Li X., Craig N.L.. Functional characterization of an active Rag-like transposase. Nat. Struct. Mol. Biol. 2012; 19:834–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hiom K., Melek M., Gellert M.. DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell. 1998; 94:463–470. [DOI] [PubMed] [Google Scholar]
- 26. Mitra R., Fain-Thornton J., Craig N.L.. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J. 2008; 27:1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weil C.F., Kunze R.. Transposition of maize Ac/Ds transposable elements in the yeast Saccharomyces cerevisiae. Nat. Genet. 2000; 26:187–190. [DOI] [PubMed] [Google Scholar]
- 28. Plasterk R.H. The origin of footprints of the Tc1 transposon of Caenorhabditis elegans. EMBO J. 1991; 10:1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Engels W.R., Johnson-Schlitz D.M., Eggleston W.B., Sved J.. High-frequency P element loss in Drosophila is homolog dependent. Cell. 1990; 62:515–525. [DOI] [PubMed] [Google Scholar]
- 30. Womack M. The yellow fever mosquito, Aedes aegypti. Wing Beats. 1993; 5:112–125. [Google Scholar]
- 31. Marchette N.J., Garcia R., Rudnick A.. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am. J. Trop. Med. Hyg. 1969; 18:411–415. [DOI] [PubMed] [Google Scholar]
- 32. Nene V., Wortman J.R., Lawson D., Haas B., Kodira C., Tu Z.J., Loftus B., Xi Z., Megy K., Grabherr M. et al. . Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007; 316:1718–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu K., Wessler S.R.. Functional characterization of the active Mutator-like transposable element, Muta1 from the mosquito Aedes aegypti. Mobile DNA. 2017; 8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rebatchouk D., Narita J.O.. Foldback transposable elements in plants. Plant Mol. Biol. 1997; 34:831–835. [DOI] [PubMed] [Google Scholar]
- 35. Potter S., Truett M., Phillips M., Maher A.. Eucaryotic transposable elements with inverted terminal repeats. Cell. 1980; 17:429–439. [DOI] [PubMed] [Google Scholar]
- 36. Windsor A.J., Waddell C.S.. FARE, a new family of foldback transposons in Arabidopsis. Genetics. 2000; 156:1983–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Craig N., Craigie R., Gellert M., Lambowitz A.. Mobile DNA II. 2002; Washington DC: ASM. [Google Scholar]
- 38. Hickman A.B., Ewis H.E., Li X., Knapp J.A., Laver T., Doss A.L., Tolun G., Steven A.C., Grishaev A., Bax A. et al. . Structural basis of hAT transposon end recognition by Hermes, an octameric DNA transposase from Musca domestica. Cell. 2014; 158:353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim M.S., Lapkouski M., Yang W., Gellert M.. Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature. 2015; 518:507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parker J.S., Roe S.M., Barford D.. Crystal structure of a PIWI protein suggests mechanisms for siRNA recognition and slicer activity. EMBO J. 2004; 23:4727–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Song J.J., Smith S.K., Hannon G.J., Joshua-Tor L.. Crystal structure of Argonaute and its implications for RISC slicer activity. Science. 2004; 305:1434–1437. [DOI] [PubMed] [Google Scholar]
- 42. Colot V., Haedens V., Rossignol J.L.. Extensive, nonrandom diversity of excision footprints generated by Ds-like transposon ascot-1 suggests new parallels with V(D)J recombination. Mol. Cell. Biol. 1998; 18:4337–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao D., Ferguson A., Jiang N.. Transposition of a rice Mutator-like element in the yeast Saccharomyces cerevisiae. Plant Cell. 2015; 27:132–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pingoud A., Jeltsch A.. Recognition and cleavage of DNA by type-II restriction endonucleases. Eur. J. Biochem. 1997; 246:1–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.