

ABSTRACT
Large granular lymphocyte leukemia (LGLL) is a rare incurable chronic disease typically characterized by clonal expansion of CD3+ cytotoxic T-cells. Two signal transducer and activator of transcription factors, STAT1 and STAT3, are constitutively active in T-LGLL. Disruption of this activation induces apoptosis in T-LGLL cells. Therefore, considerable efforts are focused on developing treatments that inhibit STAT activation. Calcitriol, the active form of vitamin D, has been shown to decrease STAT1 and STAT3 phosphorylation in cancer cell lines and autoimmune disease mouse models. Thus, we investigated whether calcitriol could be a valid therapeutic for T-LGLL. Calcitriol treatment of the TL-1 cell line (model of T-LGLL) led to decreased phospho-Y701 STAT1 and phospho-Y705 STAT3 and increased vitamin D receptor (VDR) levels. Doses of 10 and 100 nM calcitriol also significantly decreased the inflammatory cytokine IFN-γ in the TL-1 cell line. The overall cell viability did not change when the TL-1 cell line was treated with 0.1 to 1000 nM calcitriol. Studies with primary T-LGLL patient peripheral blood mononuclear cells showed that the majority of T-LGLL patients have detectable VDR and activated STATs in contrast to normal donor controls. Treatment of primary T-LGLL patient cells with calcitriol recapitulated findings from the TL-1 cell line. Overall, our results suggest that calcitriol may reprogram T-cells to decrease essential STAT activation and pro-inflammatory cytokine output. These data support further investigation into calcitriol as an experimental therapeutic for T-LGLL.
KEYWORDS: Calcitriol, inflammatory cytokine, LGL leukemia, peripheral blood mononuclear cells, phosphorylation, STAT1, STAT3, vitamin D, vitamin D receptor
Abbreviations
- AML
acute myeloid leukemia; cyclosporine A (CsA)
- EAE
experimental allergic encephalomyelitis
- IFN-γ
interferon gamma
- IL
interleukin
- JAK
Janus kinase
- LGL
large granular lymphocyte
- LGLL
large granular lymphocyte leukemia
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- PBMC
peripheral blood mononuclear cell
- PHA
phytohemagglutinin
- RXR
retinoid X receptor
- STAT
signal transducer and activator of transcription
- SH2
Src homology 2
- VDR
vitamin D receptor
- VDRE
vitamin D response element
Introduction
Large granular lymphocyte leukemia (LGLL) is a rare chronic disease, with the major subtype characterized by clonal expansion of CD3+ cytotoxic T-LGL cells displaying an activated phenotype. Typically, LGLs will undergo expansion upon antigen stimulation, then go through activation induced cell death. In LGLL, the LGLs have multiple survival pathways activated and are able to evade apoptosis.1 LGLs typically account for 10-15% of peripheral blood mononuclear cells (PBMCs) in healthy persons, therefore a greater persisting LGL percentage can be indicative of LGLL. The disease often presents with neutropenia, anemia, and autoimmune diseases including rheumatoid arthritis.2 The usual treatments include methotrexate, cyclophosphamide (Cytoxan), or cyclosporine A (CsA); these are not curative and can have harsh side effects such as tissue injury, including renal and hepatic, or even induction of other cancers.3
One dysregulated pathway in LGLL is the Janus kinase (JAK) – signal transducer and activator of transcription (STAT) pathway. JAKs phosphorylate STAT monomers at a conserved tyrosine residue, which allows STAT dimerization and transcription of target genes.4 Research from our laboratory and with collaborators has shown constitutive activation of STAT1 and STAT3 in T-LGLL.5 Furthermore, roughly 40% of LGLL patients harbor an activating STAT3 mutation.6-8 Inhibition of STAT activation via an inhibitor selective for JAK family tyrosine kinases was shown previously to cause apoptosis in LGLL patient cells5 prompting ongoing efforts to develop new therapies to inhibit STAT activation. Moreover, this dysregulation can cause continual transcription of target genes of the pathway, including inflammatory cytokines.4 Therefore, finding therapeutics to target this pathway to kill the leukemic LGLs has been one major focus in LGLL research.
The vitamin D receptor (VDR) is a nuclear transcription factor that binds its ligand 1,25-(OH)2-D3, the active form of vitamin D, also called calcitriol. This allows a conformational change in VDR which typically leads to heterodimerization with the retinoid X receptor (RXR), allowing transcription of genes with the vitamin D response element (VDRE).9 Traditionally, vitamin D is associated with bone metabolism, however in the past 30 y it has become apparent that it plays an important role in normal immune system function.9,10 During this time, studies in leukemia cell lines and in lymphocytes from patients with rheumatoid arthritis have shown that VDR is upregulated in these lymphocytes.11,12 However in T-cells, VDR is only detectable if the cells have undergone activation.11 In the early 1980s, it was shown that calcitriol inhibited proliferation in phytohemagglutinin (PHA)-activated PBMCs, partially due to suppression of interleukin-2 (IL-2) production.13 Cantorna and Waddell recently published a review of the evidence that vitamin D and VDR are needed to turn off chronically activated T-cells,14 which is particularly applicable to T-LGLL, given that the T-LGLs are constitutively activated T-cells. Therefore, the literature clearly shows that calcitriol can act on T-cells and regulate processes other than calcium homeostasis.
Vitamin D has been shown to decrease JAK/STAT activation in cancer cell lines and autoimmune disease mouse models. Calcitriol treatment of esophageal squamous cell carcinoma cell lines suppressed IL-6 induced STAT3 phosphorylation.15 When mice with experimental allergic encephalomyelitis (EAE), a model of multiple sclerosis, were treated with calcitriol, they showed decreased STAT1 phosphorylation in microglia and decreased phosphorylation of JAK2, TYK2, STAT3 and STAT4 in T-cells.16 In addition, the clinical severity and duration of EAE was significantly decreased in mice treated with calcitriol, and importantly was associated with decreased IFN-γ production and T-cell proliferation.16 Treatment of mice with diabetic periodontitis with calcidiol, the precursor vitamin D metabolite of calcitriol, increased VDR expression while inhibiting JAK1/STAT3 signaling in gingival epithelium.17
Recent research on vitamin D has been focused on understanding how vitamin D signaling changes in disease as well as whether vitamin D could be a valid therapeutic. Deficient circulating vitamin D metabolites have been reported for autoimmune diseases and cancers.9,10,18 Gocek et al reviewed the potential therapeutic effects of calcitriol in acute myeloid leukemia (AML), which are achieved by inducing differentiation and inhibiting proliferation of the AML blasts.19 While this potential therapeutic response is promising, the high amount of vitamin D supplement needed to see these effects in patients can cause calciotropic effects, therefore vitamin D analogs may be a better alternative.10 Vitamin D analogs are currently being developed and utilized in clinical trials to circumvent the adverse effects of large doses of vitamin D.20
Our present study was prompted by anecdotal evidence from T-LGLL patients stating that voluntary vitamin D supplementation improved their complete blood count parameters, namely absolute neutrophil count. Given the research showing calcitriol's therapeutic effects in AML and autoimmune diseases we wanted to explore the mechanistic basis for calcitriol effects in LGLL and determine whether calcitriol could be a valid therapeutic for LGLL. We performed a western blot survey of cryo-stored T-LGLL patient PBMCs which showed detectable VDR, STAT1 and/or STAT3 protein in the majority of patients. Treatment of the TL-1 cell line with calcitriol significantly decreased STAT1 and STAT3 tyrosine phosphorylation, significantly increased VDR expression, and significantly decreased IFN-γ output. Experiments with T-LGLL PBMCs showed similar data compared to the TL-1 cell line. Overall, our work demonstrates that calcitriol, a natural substance, can decrease activation of STAT1 and STAT3 and decrease inflammatory cytokine output. This indicates vitamin D supplementation could be a viable therapeutic option for T-LGLL.
Results
VDR and STATs are present in LGLL samples
We began the study by retrieving cryo-stored PBMCs from T-LGLL patients who participate in our LGLL Registry. This screening of samples encompassed T-LGLL patients and 2 normal donor PBMC samples. The cells were not stimulated by any cell culture media, serum, or cytokines. They were washed, lysed, then subjected to gel electrophoresis and protein gel blotting. Relevant clinical data for the patient samples used in this study can be found in Table 1.
Table 1.
Clinical data summary.
| Patient | other disease/information | age | sex | TCR | LGL phenotype and purity | ANC | STAT3 | Treatment |
|---|---|---|---|---|---|---|---|---|
| T-LGLL#1 | asymptomatic | 71 | F | αβ | CD3+CD4+ 90% | 2690 | K658F | no |
| T-LGLL#2 | 20 | M | γδ | CD3+CD8+ 24% | 810 | Y640F | no | |
| T-LGLL#3 | celiac | 69 | F | αβ | CD3+CD8+ 81% | 1090 | WT | no |
| T-LGLL#4 | 61 | F | αβ | CD3+CD8+ 90% | 620 | D661Y | no | |
| T-LGLL#5 | lupus | 39 | F | αβ | CD3+CD8+ 35%, CD3+76% | 1000 | WT | no |
| T-LGLL#6 | HA, transf/dep | 58 | M | αβ | CD3+CD8+ 74% | 2130 | Y640F | CsA |
| T-LGLL#7 | 61, 59# | F | β | CD8+ 43% | 1800, 2100# | WT | no | |
| T-LGLL#8 | 60 | F | + marrow | CD3+CD8+CD57+ 15%, CD45+ bright 38% | 490 | WT | no | |
| T-LGLL#9 | 53, 55# | F | + | CD3+CD8+ 24% | 690, 250# | WT | no | |
| T-LGLL#10 | PRCA | 45 | M | γ | CD3+CD8+ 89% | 700 | WT | MTX |
| T-LGLL#11 | 43 | M | αβ | CD3+CD8+ 94% | 1600 | Y640F, prev I659L | MTX | |
| T-LGLL#12 | agranulocytosis | 38 | M | αβ, γδ | CD3+CD8+ 65% | 0 | WT | CsA, pred. |
| T-LGLL#13 | 70 | F | αβ | CD3+ 86% | 200 | D661V | no | |
| T-LGLL#14 | RA | 56 | F | γδ | CD3+CD8+ 84% | 280 | WT | no |
| T-LGLL#15 | transf/dep until CTX | 64 | M | αβ | CD2+CD3+CD8+ 67% | 900 | WT | no; prev. on CTX |
| T-LGLL#16 | 69 | F | αβ | CD3+CD8+ 55%, CD3+CD56+ 45% | 980 | WT | no | |
| T-LGLL#17 | asymptomatic | 61 | M | γ | CD3+CD8+ 55% | 7000 | WT | no |
| T-LGLL#18 | 51 | F | γ | CD3+CD8+ 96%,CD3+CD57+ 67% | 420 | WT | CsA | |
| T-LGLL#19 | asymptomatic | 38 | F | αβ | 20-30%, bone marrow | 1200 | Y640F | no |
| T-LGLL#20 | 46 | M | αβ | CD3+CD8+ 56%, CD3+CD16+ 34%, CD3+CD57+ 51% | 1000 | Y640F | no | |
| T-LGLL#21 | RA | 34 | F | * | CD3+ 93%, CD8+ 32% | 200 | Y640F | Plaquenil, pred. |
| T-LGLL#22 | stable disease | 64 | F | no data | CD3+CD8+ 30% | 3538 | Y640F | no |
| T-LGLL#23 | 23 | F | αβ | CD3+CD8+ 63%, CD3+CD57+ 29% | 1100 | Y640F | no | |
| T-LGLL#24 | 65 | F | + | CD3+CD8+ 93%, CD3+CD57+ 61% | 730 | D661Y | no | |
| T-LGLL#25 | asymptomatic | 79 | M | αβ | CD3+CD8+CD57+ 44% | 1623 | Y640F | no |
Abbreviations: methotrexate (MTX), Cytoxan (CTX), cyclosporine A (CsA), prednisone (pred.), rheumatoid arthritis (RA), pure red cell aplasia (PRCA), hemolytic anemia (HA), transfusion dependent (transf/dep).
Age = patient age at the time of the sample collection.
Treatments listed include only immunosuppressants for LGLL and/or autoimmune disease. Other medications the patients may be taking are not listed.
Clinical data correspond to laboratory diagnostic tests performed on the date of blood collection or the closest date for which this information is available.
TCR and flow data are from peripheral blood unless otherwise specified.
TCR positivity is sometimes there, sometimes not # data correlate to A, B samples respectively in Fig. 1.
Fig. 1A shows this initial panel of samples in which we probed for VDR, the tyrosine 701 phosphorylated form of STAT1 (pY701-STAT1), and total STAT1. We chose to look at STAT1 due to reports of direct interaction of VDR and STAT1.21,22 The majority of T-LGLL patients showed detectable VDR, and total STAT1, with fewer showing pY701-STAT1. In the normal PBMC samples, we found no detectable VDR or STAT1 regardless of phosphorylation. From this screen, we determined that detectable levels of VDR, pY701-STAT1, and total STAT1 in T-LGLL is abnormal. An interesting finding from these screens was T-LGLL#1 and T-LGLL#6 showed molecular signatures that more closely matched that of the normal donor samples. T-LGLL#1 is a patient described as asymptomatic, while T-LGLL#6 is a patient with concurrent hemolytic anemia on CsA. In general, it appears that any patient expressing VDR will also express at least total STAT1 and possibly have pY701-STAT1.
Figure 1.
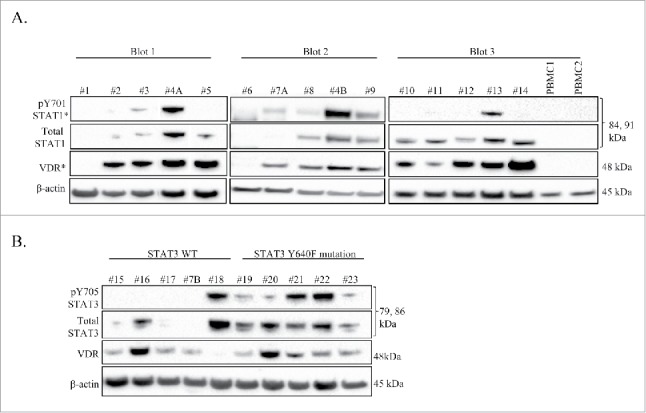
VDR and STAT are detectable in LGLL PBMC patient samples. Western blot analysis was performed on cryo-stored T-LGLL patient primary PBMCs or normal healthy control PBMCs. Total protein loaded per sample was 25 μg (Blots 1, 3) or 35 μg (Blot 2), with β-actin used as a loading control. Relevant clinical data corresponding to these samples can be found in Table 1. (A) An initial panel of T-LGLL and 2 normal healthy donor PBMCs were probed for VDR, and STAT1 (pY701 and total) on 3 separate blots. Some samples had degraded protein and were cropped out of the blots. (B) Patients with WT STAT3 (n = 5) or the Y640F STAT3 mutation (n = 5) were probed for VDR and STAT3 (pY705 and total). pY701-STAT1 and VDR blots were overexposed to show cases of faint protein detection (denoted by *). #4A,B and #7A,B were PBMC samples run from 2 different blood draw dates.
Our lab and collaborators have previously shown that STAT3 has activating mutations in 30-40% of LGLL. Of these activating mutations, Y640F is the most common. This mutation occurs in the Src homology 2 (SH2) domain, and was found to confer increased transcriptional activity6 and correlated with a better therapeutic response to methotrexate therapy.23 Therefore, we sought to correlate STAT3 Y640F mutation with the presence or absence of VDR. To accomplish this, we surveyed 5 WT STAT3 T-LGLL patients and 5 Y640F STAT3 T-LGLL patients (Fig. 1B). This was a new set of patients, except for T-LGLL #7B who was previously surveyed in Fig. 1A; the blood draw dates for these samples were different. Again, most patients expressed detectable VDR, the tyrosine 705 phosphorylated form of STAT3 (pY705-STAT3), and total STAT3. The patients who showed less or no expression of these proteins (T-LGLL#15, T-LGLL#17, and T-LGLL#7B) were previously on immunosuppressive treatment, asymptomatic, and never on treatment, respectively. Patient T-LGLL#19 has been described as asymptomatic, yet had moderate levels of total STAT3, and low levels of pY705-STAT3 and VDR. All the patients with the Y640F STAT3 mutation had a least some detectable level of all 3 proteins (pY705-STAT3, total STAT3, and VDR). Patients with WT STAT3 exhibited detectable levels of only one or 2 of these proteins, however 4 out of the 5 showed detectable VDR. Overall, these results show that presence of VDR does not correlate with STAT3 mutational status, however patients with the Y640F mutation consistently exhibited both total STAT3 and pY705-STAT3. In addition, a general conclusion that may be drawn from Fig. 1 is that most LGLL patients have detectable VDR, STAT1, and/or STAT3, contrary to normal controls.
VDR and phosphorylated STATs are only present in activated CD8+ T-cells
The literature has clearly demonstrated that VDR is not detectable in resting T-cells, but becomes upregulated upon antigen stimulation.11,24 Fig. 2A shows protein levels of VDR, total STAT1, and pY701-STAT1 for 3 normal donors' isolated CD8+ T-cells, where one half of the sample was naïve and the other half was treated with PHA to obtain an activated state. VDR was absent in the naïve T-cells, but present in the activated CD8+ T-cells. Total STAT1 was faintly present in the naïve cells, but increased with activation. pY701-STAT1 was absent in the naïve cells but detectable in the activated cells. We probed lysates of donors A and B for pY705-STAT3 and total STAT3. We found similar results, with total STAT3 being present in both naïve and activated T-cells, while pY705-STAT3 was absent in the naïve cells and present in the activated cells. Therefore, these experiments further confirm that the presence of VDR in LGLL samples is indicative of an activated state.
Figure 2.
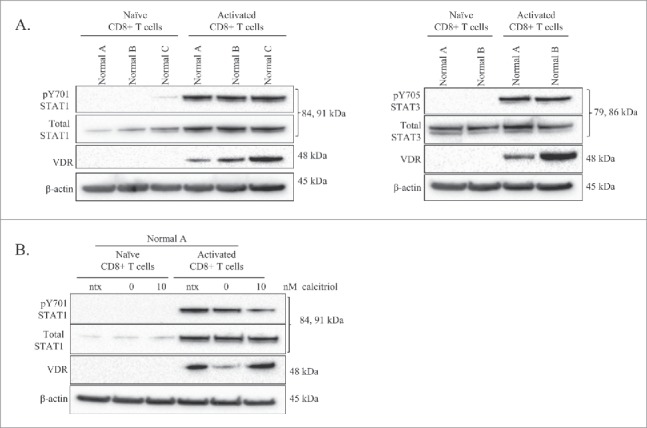
Comparison of VDR and STAT levels in naïve vs. activated normal CD8+ T-cells, and the effects of calcitriol treatment on each cell type. Naïve CD8+ T-cells were isolated from 3 healthy normal donors. One half of each sample was cultured as naïve cells while the other half was activated with PHA for 24 hours. Total loaded protein per sample was 25μg, with β-actin used as a loading control. (A) Western blotting of VDR, STAT1 (pY701, total) and STAT3 (pY705, total) was performed. (B) The naïve and activated cells from Normal Donor #0 were cultured for 24 hours, with or without calcitriol treatment, lysates were probed on a protein gel blot for VDR and STAT1 (pY701, total).
The naïve and activated cells from normal donor A were treated with calcitriol or vehicle. Literature review indicated a range of calcitriol doses in cell culture which are considered physiological.13,25 Due to a limited amount of cells, we chose the 10 nM concentration for this experiment. The controls included media only (ntx) and vehicle only (0 nM). We chose a 24 timepoint based on previous work indicating that activation and addition of calcitriol causes upregulation of VDR at 24 hours.14 The naïve T-cells had no detectable VDR, total STAT1, or pY701-STAT1 with or without calcitriol treatment; however, when we treated activated T-cells with calcitriol (10 nM), pY701-STAT1 decreased while VDR increased (Fig. 2B). Taken together, the results in Fig. 2 confirm that VDR expression is limited to activated cells, which illustrates that most patient samples in Fig. 1 display an activated phenotype.
Treatment of the TL-1 cell line with calcitriol decreased STAT phosphorylation and increased VDR
We next performed experiments with the TL-1 cells, a model of T-LGLL, to determine if calcitriol treatment decreased STAT phosphorylation as we saw in the normal donor sample. For our cell line experiments, we utilized calcitriol doses of 0.1, 1, 10, and 100 nM, along with the media only (ntx) and ethanol (0 nM) negative controls. Cells were harvested at 24 and 48 hours. The 48 hour data are not shown; the cell lines' IL-2 dependence every 48 hours resulted in a natural diminishment of STAT phosphorylation, regardless of calcitriol treatment. The results from these experiments showed a significant increase in the level of VDR with 10 nM or 100 nM treatment (Fig. 3A,C). The ratio of pY701-STAT1/total STAT1 declined in a dose-dependent manner and became significant with the 100 nM dose (p=0.0652 for 10 nM treatment) (Fig. 3A, C). The ratio of pY705-STAT3/total STAT3 decreased significantly at 10 nM calcitriol and was trending toward significant decrease (p = 0.0553) at 100 nM calcitriol (Fig. 3B ,C). From these cell culture studies, we conclude that calcitriol significantly decreases STAT1 and STAT3 tyrosine phosphorylation while significantly increasing VDR levels in TL-1 cells.
Figure 3.

The effects of calcitriol treatment on the TL-1 cell line, a model of T-LGLL. The TL-1 cell line was treated with the indicated doses of calcitriol, or appropriate negative controls for 24 hours. Total protein loaded per sample was 25 μg, with β-actin used as a loading control. (A) Representative western blots for VDR, pY701-STAT1, and total STAT1 are shown. (B) Additional representative protein gel blots for pY705 STAT3 and total STAT3 are shown. (C) Data from 3 independent experiments were quantified to determine relative expression levels. Student's t-test was used to determine significance of calcitriol treatment compared to 0 nM vehicle control (ns = not significant, *p < 0 .05, **p < 0 .01).
Calcitriol treatment causes decreased STAT phosphorylation in the nucleus and cytoplasm while increasing VDR exclusively in the nucleus
Because both STAT and VDR proteins are transcription factors that can shuttle between the nucleus and cytoplasm, we investigated subcellular localization of VDR and STATs in the TL-1 cell line upon treatment with calcitriol. We treated the cells with 10 nM calcitriol or negative controls, then prepared nuclear and cytoplasmic fractions at 24 hours. In order to assess fraction purity, we used histone H3 and HSP90 as markers of nucleus and cytoplasm, respectively. We observed a decrease in pY701-STAT in both the nucleus and cytoplasm when comparing 10 nM calcitriol to 0 nM control (Fig. 4A). We found the same result for pY705-STAT3, a decrease in both the nucleus and cytoplasm (Fig. 4B). VDR increased with 10 nM calcitriol and was exclusively in the nucleus (Fig. 4A). Altogether, these data demonstrate that dephosphorylated STAT proteins can be localized to either the cytoplasm or nucleus, and that VDR stays localized in the nucleus. The nuclear VDR and dephosphorylated nuclear STAT proteins show readouts of increased and decreased transcriptionally active proteins, respectively.
Figure 4.

Nuclear and cytoplasmic fractionation of TL-1 cells with and without calcitriol treatment. The TL-1 cell line was treated with 10 nM calcitriol, or appropriate negative controls for 24 hours. Subcellular fractionation was performed to isolate cytoplasmic and nuclear proteins. Total protein loaded per sample was 25 μg, with HSP90 and histone H3 used as markers of cytoplasm and nucleus, respectively. Two identical separate blots were used to probe for all targets. The subcellular fractions were probed for (A) VDR and STAT1 (pY701 and total) and (B) STAT3 (pY705, total). These are representative western blots of three independent experiments.
RXR-β is the only RXR isoform present in the TL-1 cell line
RXR is the heterodimerizing binding partner of VDR which mediates nuclear transcription of genes with the VDRE. We sought to verify that RXR was indeed present in the TL-1 cell line. There are 3 isoforms of RXR (α, β, γ) which are differentially expressed depending on tissue type.26 We utilized whole cell lysates from the experiment of Fig. 3, and probed for the RXR isoforms. Fig. 5A shows that RXR-β is the isoform expressed in the TL-1 cell line. When comparing the treated cells with the 0 nM control, it is apparent that RXR-β increased with calcitriol treatment (Fig. 5A). Since two of the isoforms were not detectable, we probed other cell lines to determine their isoform distribution. We used a panel of AML cell lines to compare AML with LGLL, as well as the breast adenocarcinoma cell line MCF-7, and the multidrug resistant epidermal carcinoma cell line KB-3-1. We also probed for VDR to establish whether upregulation of this protein is seen in these other cancers. Fig. 5B shows that MCF-7, KB-3-1, HL-60/ABTR, MOLM-14, OCI-AML 2, THP1, KG-1, KG-1-A, Kasumi, and TL-1 all expressed the RXR-β isoform. Interestingly, MOLM-14 and THP-1 showed robust levels of RXR-α, while MCF-7, KB-3-1, HL-60/VCR, HL-60/ABTR, OCI-AML 2, and Kasumi cells showed moderate levels of RXR-α. None of the cell lines we tested showed detectable RXR-γ. There are 2 known isoforms of VDR, with the 48 kDa isoform, seen in our LGLL cell lines, being the best characterized. This isoform was expressed in all the cell lines, with the lowest amounts seen in KG-1 and KG-1-A. Most interestingly only KB-3-1, MOLM-14, OCI-AML 2, and THP-1 showed detectable levels of the 54 kDa VDR isoform. These results demonstrate that VDR is detectable in the cancer cell lines we tested. Furthermore, some cell lines have 2 RXR isoforms and both VDR isoforms, suggesting that VDR signaling in these cancers may occur differently than in LGLL.
Figure 5.
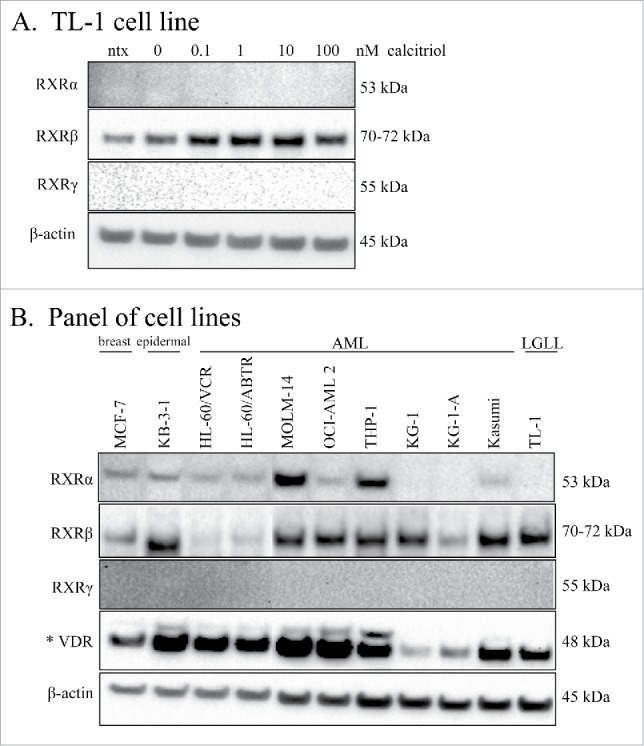
RXR isoform distribution in LGLL and other cell lines. Lysates from the experiment in Fig. 3 were run on independent blots and probed for the 3 RXR isoforms. Total protein loaded per sample was 25 μg, with β-actin used as a loading control. The TL-1 cell line (A) was probed for the RXR isoforms: α, β, and γ. (B) Western blotting of lysates from MCF-7, KB-3-1, 9 AML cell lines, and the TL-1 cell line was performed for the RXR isoforms (α, β, and γ), and VDR. The VDR blot is shown overexposed (denoted by *) to visualize the 54 kDa isoform. One AML cell line lysate was degraded and therefore was cropped out of this blot.
Calcitriol treatment of the TL-1 cell line causes a decrease in IFN-γ production
Since STATs are known to transcribe cytokine genes,4 we aimed to determine if the decrease in phospho-STAT in the TL-1 cell line would correspond with a decrease in cytokine production, as this has been shown to occur in the EAE mouse model upon calcitriol treatment.16 We used the Luminex platform to screen for changes in IFN-γ, IL-2, IL-4, IL-5, IL-10, and IL-17, using the culture media from the ntx, 0 nM, and 10 nM calcitriol at 24 hours (corresponds to the experiment from Fig. 3). Since the cells require IL-2 supplementation to survive, we considered this a confounding factor and did not take the IL-2 results into consideration. When comparing 0 nM to 10 nM, the most notable observation was the nearly 50% decrease in IFN-γ (Fig. 6A). Since this cytokine had the most drastic change, we repeated this analysis on 3 independent experiments to quantify the reduction more completely. In this experiment, we assessed the 0, 10, and 100 nM conditions. Indeed, IFN-γ decreased significantly (p < 0 .01) at both 10 and 100 nM calcitriol treatments compared to the 0 nM control (Fig. 6B). Overall these findings establish that calcitriol causes a significant decrease in IFN-γ in TL-1 cells.
Figure 6.

Analysis of cytokine output by the LGLL cell lines with and without calcitriol treatment. Cell culture media samples from TL-1 cells were analyzed for cytokine production. (A) Six cytokines (IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-17) were measured from cells treated with 10 nM calcitriol compared to the negative controls (ntx, 0 nM) at 24 hours. Cytokine levels were measured in pg/mL; the y-axis shows the appropriate range for each cytokine and thus varies highly between cytokines. (B) Further analysis of IFN-γ was performed (n = 3). The 10 and 100 nM calcitriol treatments were compared to the 0 nM control. IFN-γ was significantly decreased, with p < 0 .01 for 10 and 100 nM calcitriol. (Student's t-test).
The TL-1 cell viability does not change when treated with calcitriol
We next performed a cell viability assay at 24 and 48 hours with the TL-1 cells to see if calcitriol changed the viability in a dose-dependent manner. Data were adjusted to the 0 nM (ethanol) control so that it was equal to 100% viability. We tested the same range of calcitriol doses, but added a 10-fold higher dose of 1000 nM as some studies reported seeing effects in this range.16 Calcitriol treatment did not change cell viability at 24 hours (Fig. 7). Similar results were seen at 48 hours (data not shown). From these data we conclude that calcitriol doses from 0.1 to 1000 nM do not change viability in the TL-1 cell line.
Figure 7.
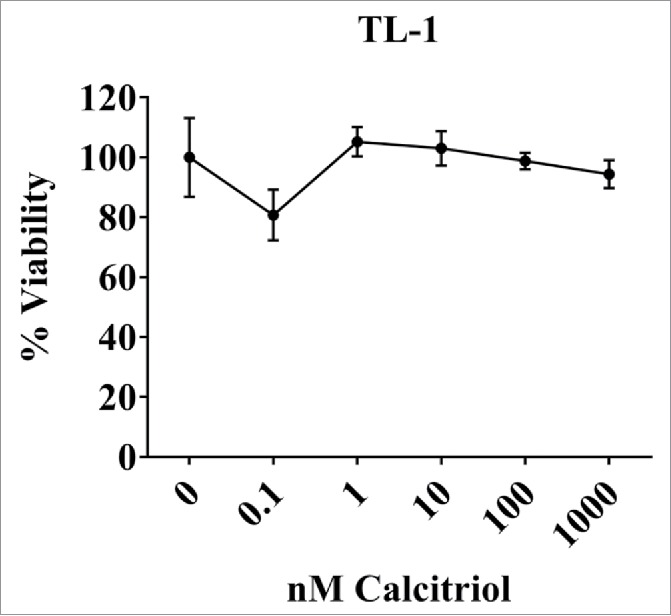
Cell viability of the TL-1cell line with and without calcitriol treatment. TL-1 were treated with the indicated doses of calcitriol, or appropriate negative controls. Each dose was done in quadruplicate. MTS reagent was added at 24 to assess cell viability. Data were normalized to the 0 nM control and are shown as mean with standard deviation. Treatments were not significantly changed compared to the 0 nM control (Student's t-test).
Treatment of LGLL patient PBMCs with calcitriol
We treated 4 T-LGLL patient PBMC samples with calcitriol to see if effects matched those observed in the TL-1 cell line. Each patient exhibited different STAT3 mutational status (Table 1, Fig. 8). The cells were treated under ntx, 0 nM, and 10 nM calcitriol conditions for 24 hours, then harvested. Since two studies have shown VDR and STAT1 can directly bind,21,22 we chose to assess STAT1 status over STAT3, if the protein amount was limited. The baseline samples described for this set of experiments were uncultured PBMCs and prepared as the samples described in Fig. 1.
Figure 8.

The effects of calcitriol treatment on PBMCs from LGLL patients. Fresh primary T-LGLL patient PBMCs were treated with the indicated doses of calcitriol, or appropriate negative controls for 24 hours. Patient samples from (A) (T-LGLL#13) and (B) (T-LGLL#17) were previously surveyed in Fig. 1, while patient samples from (C) (T-LGLL #24) and (D) (T-LGLL #25) were unique patients. Each blot (A-C) was probed for VDR, pY701-STAT1, and total STAT1. Patient T-LGLL #25 (D) was probed for the additional targets of pY705-STAT3 and total STAT3 on a separate blot. β-actin was used as a loading control. In some cases, pico ECL reagent was used for detection (denoted by *). Relevant clinical data for each patient can be found in Table 1.
PBMCs from T-LGLL#13, a patient whose sample was previously used in the cryo-panel (Fig. 1), were treated with calcitriol (Fig. 8A). At the time of this experiment, the patient was not on any treatment and had the D661V STAT3 mutation. VDR increased with calcitriol treatment while STAT1 (pY701 and total) remained unchanged. Of note, the patients' cells showed detectable levels of VDR and STAT1 (pY701 and total) in the baseline sample (data not shown). PBMCs from T-LGLL #17, another patient whose sample was previously used in the cryo sample survey, were treated with calcitriol (Fig. 8B). At the time of this experiment, the patient was not on any treatment and was WT for STAT3. VDR increased with calcitriol treatment while STAT1 (pY701 and total) remained unchanged. This patient's cell sample did not exhibit detectable VDR or STAT1 prior to being cultured (data not shown). PBMCs from T-LGLL#24, a patient sample not previously tested in the cryo-panel, were treated with calcitriol (Fig. 8C). At the time of this experiment, the patient was not on any treatment and had the D661Y STAT3 mutation. VDR dramatically increased with treatment, while both pY701- and total STAT1 decreased. Of note, this patient had no detectable VDR and low STAT1 (pY701 and total) prior to the cells being cultured (data not shown). PBMCs from T-LGLL#25, a patient sample not previously tested in the cryo-panel, were treated with calcitriol (Fig. 8D). At the time of this experiment, the patient was not on any treatment and had the Y640F STAT3 mutation. VDR did not change with treatment, while pY701- and total STAT1 decreased. A surplus of protein allowed probing for pY705 and total STAT3, which decreased slightly and stayed the same, respectively. The patient's cells did not show detectable VDR, but phosphorylated and total STATs were faintly present prior to the cells being cultured (data not shown). Overall, treatment of primary patient PBMCs with calcitriol showed clear decrease of STAT1 phosphorylation in 2 of the 4 patients, substantial reduction in STAT3 phosphorylation in the only patient tested, and an increase in VDR in 3 of the 4 patients. With the inherent variation of human samples, with LGL percent purity and viability upon recovery from cryo storage as major confounding factors, it is reasonable to see diverse results but also highly encouraging that most of the results recapitulate data seen from TL-1 experiments.
Discussion
This study shows that calcitriol treatment of T-LGLL cells decreases activation of STAT1 and STAT3, 2 potential drivers of T-LGLL. A western blot survey of T-LGLL patients indicated that the majority have detectable VDR and phosphorylated/activated STAT1 and STAT3. This molecular signature most closely resembles that of an activated immune cell, which is expected as T-LGLL cells are chronically activated immune cells. The effects of calcitriol treatment in TL-1 cells showed significantly decreased IFN-γ output. In addition VDR levels increased while STAT1 and STAT3 phosphorylation decreased in the nucleus, showing the effects are happening where transcription occurs.
The timepoints in this study were chosen because previous work showed that decreases in inflammatory cytokines occurred at 48 hours.14 However, one caveat is that the TL-1 cell line is dependent on exogenous IL-2 for survival, and needs media change with fresh IL-2 every 48 hours. IL-2 then signals through its receptor and ultimately causes STAT phosphorylation. We observed decreased STAT phosphorylation at 48 hours across all treatments and controls (data not shown), demonstrating that this was an effect of IL-2 depletion rather than calcitriol. Although this is a confounding factor, we think the 48 hour timepoint was still informative in our experiments as it showed that although phosphorylated STATs decreased, total STATs remained rather constant.
Phosphorylation of a conserved tyrosine residue in the SH2 domain of STAT proteins is needed for dimerization and subsequent transcription of target genes.4 Phosphorylation of this tyrosine is a direct measure of STAT activation. Data from whole lysate experiments in the TL-1 cell line (Fig. 3) showed activation of both STATs decreased upon calcitriol treatment, with cell fractionation experiments (Fig. 4) confirming this occurs in the nucleus. Three mechanisms could explain the decrease in phospho-STAT levels. The first could be decreased phosphorylation of STATs by JAKs; this could be due to activity of suppressor of cytokine signaling proteins.27 The second is removal of the phosphate by a tyrosine phosphatase enzyme, which can occur in either the nucleus or cytoplasm.28 The third mechanism could be protein degradation; with the exception of total STAT3 decreasing with calcitriol treatment in the TL-1 cell line, total STAT remained stable in whole cell lysate experiments, suggesting that degradation is not the likely mechanism. To address which mechanism is most responsible, future experiments will include elucidating the activation states of JAK-STAT pathway regulators as a result of calcitriol treatment. Additionally, we plan to perform experiments with cycloheximide to confirm that the half-life of STAT proteins is unaffected by calcitriol.
Two isoforms of VDR (48 and 54 kDa) 29,30 and 3 isoforms of RXR (α, β, γ)26 have been identified. We found the TL-1 cell line expressed the 48 kDa VDR and RXR-β isoforms. A survey of other cell lines (Fig. 5B) revealed that other combinations of isoform expression can occur in cancer. Most interestingly, our survey of AML cell lines, which model another type of blood cancer, showed differential expression of VDR and RXR isoforms. Previous work found that Jurkat cells, which models acute T-cell leukemia, express RXR-α.31 In addition, RXR-α and β are both present at the mRNA level in human PBMCs.32 Therefore, it is possible that RXR isoform expression becomes unbalanced in cancer and that diverse VDR-RXR combinations may result in different cancer phenotypes.
Results from two experiments in our study support the hypothesis that the TL-1 cells are becoming reprogrammed from an inflammatory to non-inflammatory phenotype upon calcitriol treatment: the significant decrease in IFN-γ production and the unchanged viability. Since T-cells and LGLs are the only known cells that can make IFN-γ,33 and activated CD8+ lymphocytes (the T-LGLL phenotype) have the highest levels of VDR compared to other immune cells,24 this shows calcitriol can directly target the leukemic LGLs. The phenomenon of calcitriol decreasing IFN-γ output in PHA-activated human PBMCs was published almost 30 y ago.34 T-helper 1 cells make IFN-γ and other inflammatory cytokines which prevents naïve T-cells from being able to differentiate into T-helper 2 cells or other such cells that produce non-inflammatory cytokines, therefore the inflammatory response is perpetuated as more T-helper 1 cells are made.35 In infectious diseases, the antigen is cleared and the T-cells are able to turn off. However in autoimmune disease, the T-cells continue to recognize an antigen and therefore continue to produce inflammatory cytokines.14 The ability of calcitriol to turn off inflammation in autoimmune disease was shown with the EAE mouse model study, where isolated neural antigen-sensitized spleen cells produced significantly less IFN-γ.16 One mechanism for the decrease in IFN-γ production is the simple fact that STAT1 becomes dephosphorylated, thus inactivated, and can no longer bind the response element for the IFN-γ gene to cause transcription. Another mechanism is that VDR-RXR can bind the promoter of IL-236 or IFN-γ,37 showing direct repression of these inflammatory cytokines is possible. These mechanisms will be addressed by future studies in our laboratory.
Crosstalk between cytokine and vitamin D signaling pathways has been reported previously by two groups.21,22 Both studies demonstrated direct interaction by VDR and STAT1. Since cytokines can activate the JAK-STAT pathway, we considered that this could be an important link in LGLL. Fig. 9 illustrates a working model of what could be happening in a LGL cell without (left panel) or with (right panel) calcitriol. Without calcitriol, an inflammatory cytokine such as IFN-γ binds its receptor and causes subsequent JAK phosphorylation, which phosphorylates STAT1, allowing it to dimerize and translocate to the nucleus, then cause transcription of target genes such as IFN-γ. The cell secretes IFN-γ, thus positively feeding this cycle in an autocrine or paracrine fashion. On the other hand, addition of calcitriol allows VDR to bind its ligand and halt STAT1 transcription of target genes. The potential mechanisms include VDR being released from constitutive STAT1 binding, allowing transcription of VDRE gene targets21,22, or direct inhibition of IFN-y transcription by binding its promoter.37 The ultimate result is a dramatic decrease in IFN-γ production, which was observed in the TL-1 cell line (Fig. 9). Cell viability may remain unchanged throughout this process (as seen in Fig. 7) while the cells gain a new and less inflammatory phenotype.
Figure 9.
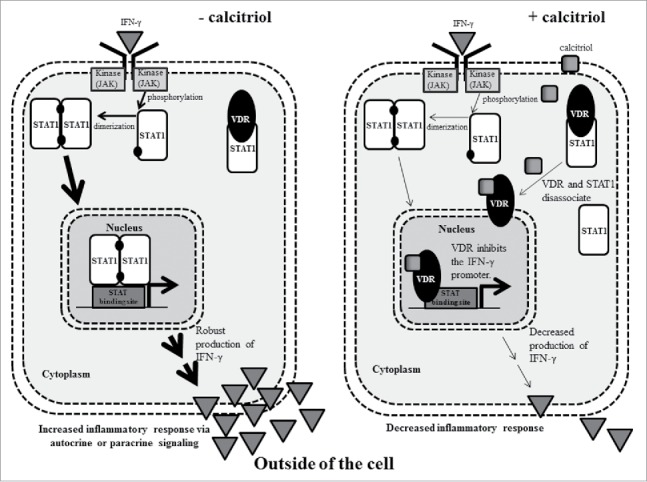
A working model that demonstrates how calcitriol decreases inflammatory cytokine signaling in LGLL cells. In the left panel, IFN-γ binds its receptor which leads to phosphorylation of STAT1. The phosphorylated STAT1 monomer is able to dimerize then enter the nucleus to cause transcription of IFN-γ, increasing IFN-γ output, which could act in an autocrine or paracrine fashion to propagate inflammation. In the right panel, calcitriol enters the cell and binds to VDR. VDR is released from its constitutive STAT1 interaction and translocates to the nucleus. In this example, it binds and inhibits the IFN-γ promoter. Ultimately, this leads to decreased IFN-γ output. The Discussion describes these mechanisms in greater detail.
There are currently no known activating STAT1 mutations in LGLL. We tested a separate cohort of T-LGLL patients (n = 76) for STAT1 mutations in exons 20 and 21, which correspond to the SH2 and transactivation domains, the areas of known STAT3 activating mutations (Fig. S1A, B). We found that all patients in this group were WT for STAT1, while almost 30% exhibited a STAT3 mutation (Fig. S1C). Given that 30-40% of LGLL patients have been previously found to harbor a STAT3 mutation and we found a 29% frequency, this shows a good sampling of the LGLL patients. Furthermore, this indicates that if STAT1 mutations exist, they do not occur at the frequency of STAT3 mutations. Regardless of the patients not harboring an activating mutation in STAT1, studying it is relevant for two reasons. First, we saw high levels of total and phosphorylated STAT1 in most LGLL patients. Elevated STAT1 could become activated via the JAK-STAT pathway, and cause transcription of inflammatory cytokines. Chronic inflammation is a characteristic of both autoimmune diseases and cancer and can promote cancer development.38 Previous work by our laboratory and collaborators showed IFN-γ is significantly upregulated in T-LGLL patient sera compared to normal donor sera, making this a plausible theory.23 Second, constitutive activation of STAT1 in LGLL has previously been reported by our laboratory, as well as the possibility of STAT1-STAT3 heterodimer species.5 It is plausible that VDR and STAT1 could interact in LGLL cells, given the high levels of each in most LGLL patients. Otherwise, it would be reasonable to expect that the high levels of VDR would already be working to turn off inflammation via promotion or inhibition of transcription.
Synergistic effects of vitamin D with other treatments have been reported and highlight another advantage for LGLL patients currently on immunosuppressant treatment. Treatment of T lymphocytes from active ulcerative colitis patients with calcitriol or nonhypercalcemic analogs in combination with CsA reduced cell proliferation in comparison to healthy controls.39 Another study showed a decrease in IL-2 receptor and production in CD4+ cells obtained from rheumatoid arthritis patients when calcitriol was combined with CsA, with the CsA at 100 times below the optimal dosage.40 CsA is an immunosuppressant that is commonly prescribed to treat LGLL. This treatment can be quite toxic, thus vitamin D supplementation may allow patients to take a lower CsA dose, potentially minimizing side effects.
In summary, we have shown that calcitriol, the active form of vitamin D, could be a potential therapeutic for T-LGLL. Further work needs to be done to elucidate the mechanism for the decreased activation of STATs and to understand how the cells are being reprogrammed to a less inflammatory phenotype. If supra-physiological doses of calcitriol are needed to elicit therapeutic effects in humans, the use of calcitriol analogs is another option to reduce calciotropic effects. Our data suggest that clinical investigation of calcitriol supplementation as a therapeutic for T-LGLL may warrant further study.
Materials and methods
Human subjects
All human subjects whose LGLL samples were used in this study consented to join Dr. Loughran's LGLL Registry at the University of Virginia (IRB-HSR#17000). For normal healthy donor samples, blood was purchased from Virginia Blood Services.
Research STAT mutation testing
DNA from PBMCs was extracted on the Anaprep system (BioChain) and amplified with premade primers (Invitrogen, cat# hs00454880_ce for exon 20,19 and hs00405919_ce exon21 in STAT3) in MyTaq master mix (Bioline, cat#BIO-25041). Primers were M13 tailed and a no input M13 PCR was run with every sample to detect previous product carryover. PCR product was cleaned with ExoSAP-IT(USB) (Affymetrix, cat#78250) and submitted to Eurofins for Sanger Sequencing. Untailed forward primers were used to sequence each STAT3. For the 76 patients surveyed for STAT1 mutations in exons 20 and 21, high resolution melt assays were carried out on the CFX384 cycler (BioRad) using a 60°C annealing temperature for 45 cycles and analyzed with Precision Melt Analysis Software with unstated parameters according to the Precision Melt Supermix recommendations (BioRad, cat#1725110). The following primers were used to probe the region of interest for mutations: HRMSTATF1 ctgttaccacctgcttgcc, HRMSTAT1R1 ctgagaatcccctgaagtatct chr2:191,841,539-191,841,646; HRMSTAT1F2 tggtctttgtcaatatttggatac, HRMSTAT1R2 ggttgaaccctacacgaaga chr2:191,841,601-191,841,735; HRMSTAT1F3 tgcgaatgatgtcagggaaa, HRMSTAT1R3 tctgtcctctttcattttggga chr2:191,841,677-191,841,810 (contains common SNP rs206680804, no additional variants seen) HRMSTAT1F3ALT tcagcagccatgactttgta, HRMSTAT1R3ALT ttttagaacctgacttccatgc chr2:191,841,655-191,841,757 (omits common SNP); HRMSTAT1F4 tgtgctcaaactcccactca, HRMSTAT1R4 cctgttgaaggaccagcag chr2:191,843,561-191,843,688.
Reagents
Calcitriol (1,25-(OH)2 vitamin D3) was purchased from Cayman Chemical (cat#71820). RIPA lysis buffer (cat#R0278), protease and phosphatase cocktails (cat#P8340, cat#P5726), and lectin (phytohemaglutinin; PHA, from Phaseolus vulgaris, cat#A6419) were all purchased from Sigma Aldrich. All antibodies were purchased from Cell Signaling Technology. FBS was purchased from Seradigm (cat#97068-085). IL-2 was purchased from Miltenyi Biotec (cat#130-097-743). Clarity enhanced chemiluminescence (ECL) reagent (cat#170-5061) and PVDF membrane and paper stacks (cat#170-4274), were purchased from BioRad. RPMI 1640 (cat#10-040-CV), IMDM (cat#31980030), NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (cat#78833), Pierce bicinchoninic acid (BCA) protein assay kit (cat#PI23225), SuperSignal West Pico Chemiluminescent Substrate (cat#34087), and SuperSignal West Femto Maximum Sensitivity Substrate (cat#34096) were purchased from ThermoFisher Scientific. Polyacrylamide gels (4-12%) were purchased from Life Technologies (cat#NW04125BOX). The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) Cell Proliferation Colorimetric Assay Kit was purchased from BioVision (cat#K300-2500).
Primary cells
PBMCs from either T-LGLL patients or healthy donors were isolated by Ficoll-hypaque gradient separation as described previously.41 Cell viability was determined by Trypan blue exclusion assay. Naïve CD8+ T-cells were isolated from buffy coats of 3 normal donors using MagniSort Human CD8 Naïve T-cell Enrichment Kit (eBioscience, cat#8804-6812-74). After obtaining a cell count, each donor's cells were plated at a density of 2 million cells/mL of RPMI 1640 supplemented with10% FBS. One half of the isolated naïve CD8+ T-cells were treated with 5 mg/mL of PHA. The other half remained untreated to preserve the CD8+ T cells in their naïve state. As a large number of CD8+ T-cells were obtained from one normal donor, a portion of their CD8+ T-cells, both the PHA-treated and non-treated cells, received either media-only, ethanol (vehicle control), or 10 nM calcitriol immediately upon plating with or without PHA. All CD8+ T-cells regardless of their treatment condition were maintained at 37°C, 5% CO2 for 24 hours before their protein was harvested using RIPA lysis buffer with protease and phosphatase inhibitor cocktails as described in the protein gel blot section of the methods. The same procedure was used for primary T-LGLL PBMCs used for experiments except these were plated at a density of 1 million cells/mL.
Cell lines
The TL-1 cell line, a model of T-LGLL, was used for the bulk of the data reported. This cell line was previously derived from a T-LGLL patient by our laboratory and immortalized with the retroviral HTLV-2 tax gene.8 Other cell lines were used as controls for several antibodies tested in this study. The KG-1, KG-1A, and Kasumi-1 cell lines were purchased from American Type Culture Collection (ATCC). The MOLM-14 cell line was generously provided by Dr. Mark A. Levis (Johns Hopkins School of Medicine). The OCI-AML 2 cell line was a gift from Dr. Xiaorong Gu (Cleveland Clinic). The THP-1 cell line was generously provided by Dr. H.G. Wang (Penn State University). The MCF-7 and KB-3-1 cell lines were provided by Dr. Myles Cabot (East Carolina University). The ABT-737-resistant HL-60 (HL-60/ABTR) cell line was generated as previously described.42 Vincristine-resistant HL-60/VCR cells were selected as previously described.43 All cell lines used in this study were authenticated using short tandem repeat DNA profiling (Genetica DNA laboratories).
Cell culture
Cells were plated at a density of 1 million cells/mL for all experiments with TL-1, except for the cell proliferation assay, with those cells seeded at 25,000 cells/100 μl (250,000 cells/mL). Calcitriol was dissolved in 100% ethanol and used in all experiments. Media for all experiments with TL-1 and primary patient cells was RPMI 1640 supplemented with 10% FBS. TL-1 cells were supplemented with IL-2 at 200 U/mL. This cell line and the others listed below were maintained at 37°C, 5% CO2. For experiments with calcitriol, cells were treated for 24 or 48 hours at the doses of calcitriol listed. Appropriate negative controls included media-only as a no treatment condition and ethanol treated as a vehicle control, with the final percentage of ethanol in the media always less than 0.1%. After treatment, cells were harvested, lysed, and protein was extracted. The other cell lines used in this study were grown in varying conditions to maintain optimal growth characteristics. Kasumi-1 cells were grown in RPMI 1640 supplemented with 20% FBS. KG-1 and KG-1A cells were grown in IMDM supplemented with 20% FBS. MCF-7 and KB-3-1 cells were cultured in DMEM with 10% FBS and 110 mg/L sodium pyruvate. MOLM-14, OCI-AML 2, THP-1, HL-60/VCR, and HL-60/ABTR were grown in RPMI 1640 supplemented with 10% FBS. In addition, HL-60/VCR cells were maintained in medium containing 2 µg/ml vincristine (Cayman Chemical, cat#11764).
Western blot
Cells, either treated with calcitriol/vehicle or retrieved from cryo-storage, were washed with 1X PBS then lysed in a RIPA lysis buffer that contained protease and phosphatase inhibitors. Protein content was quantified using the BCA assay. Proteins were electrophoresed on a 4-12% polyacrylamide gel and then transferred to a PVDF membrane, which was probed with primary antibody overnight at 4°C according to manufacturer standards. Primary antibodies used in these studies were: pY701-STAT1 (cat#7649), total STAT1 (cat#9175), pY705-STAT3 (cat#9131), total STAT3 (cat#9139), VDR (cat#12550), RXR-α (cat#3085), RXR-β (cat#8715), and RXR-γ (cat#5629), with β-actin as a loading control (cat#3700). After washes, the membrane was probed with secondary antibody (anti-rabbit IgG-HRP linked #7074 or anti-mouse IgG-HRP linked #7076) for an hour then treated with ECL substrate. For proteins at lower levels either SuperSignal West Pico or Femto Maximum Sensitivity Substrate was used. Images were captured with a ChemiDoc instrument and analyzed using Image Lab software (BioRad).
MTS assay
TL-1 cells were treated with varying doses of calcitriol and appropriate negative controls. Cells were seeded at 25,000 cells/well. The MTS Cell Proliferation Colorimetric Assay Kit was used to assess cell viability. Twenty μl of MTS reagent was added to each well after either 24 or 48 hours of calcitriol treatment. The reaction was incubated at 37°C, 5% CO2 for 1 hour, and formazan product was read on a plate reader at 492 nm, with the absorbance exceeding control wells. Data were normalized to the ethanol (0 nM) vehicle control. All conditions were done in quadruplicate.
Cytokine analysis
After treatment with calcitriol, the cells were pelleted, and aliquots of the media were collected and stored at -80°C prior to analysis by the UVA Flow Cytometry Core by a Luminex MAGPIX bead-based multiplex analyzer.
Cell fractionation assay
Cells were fractionated according to the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents protocol. Briefly, 3 million cells were pelleted after treatment. The cells were washed with PBS then cytoplasm was obtained after lysis with CERI (protease and phosphatase inhibitors added) and CERII buffers. NER buffer was used to obtain the nuclear fraction. After quantification of the fractions, 25 μg of protein was loaded onto a gel for western blot analysis.
Statistical analysis
Data were analyzed using GraphPad Prism software version 7. An unpaired 2-tailed Student's t-test was used to determine significance. P-values of <0 .05 were considered significant.
Sequence alignments
Homo sapiens STAT1 and STAT3 amino acid sequences were retrieved from the National Cancer Institute Protein Database (Accession NP_009330 for STAT1 and Accession NP_644805 for STAT3). The sequences were put into FASTA format and aligned using Clustal Omega software, which is a free online alignment tool curated by the European Bioinformatics Institute. Annotations to the sequences were performed to show mutated and analogous WT amino acids in STAT3 and STAT1, respectively, for the 76 patients. These data can be found in the Supplement.
Contributions
KCO participated in research design, conducted experiments, performed data analysis, and wrote the manuscript. PMK participated in research design, conducted experiments, performed data analysis, and contributed to the writing of the manuscript. TLO participated in research design, conducted experiments, performed data analysis, and contributed to the writing of the manuscript. SFT participated in research design and contributed to the writing of the manuscript. RJR conducted experiments and performed data analysis. DJF participated in research design, performed data analysis, and contributed to the writing of the manuscript. TPL participated in research design, performed data analysis, and contributed to the writing of the manuscript.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Alexander Wendling and Shubha Dighe for support with blood processing to obtain PBMCs and Holly Davis for LGLL Registry support. We thank Cait Hamele and Lindsay Chatfield for support with research STAT mutation testing and Kabir Ahluwalia for technical assistance. We also thank Mike Solga in the UVA Flow Cytometry Core for performing the Luminex assays. The authors wish to extend a special thank you to the LGLL patient who inspired this project.
Funding
This work was funded by the National Cancer Institute of the National Institutes of Health under award numbers R01CA098472, R01CA178393, and P01CA171983 (to TPL), the Bess Family Charitable Fund (to TPL), the LGL Leukemia Foundation (to TPL), the Immunology Training Grant T32AI007496-21 (to PMK), and the UVA Wagner Fellowship (to PMK).
References
- 1.Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program 2012; 2012:652-9; PMID:23233648; 21190991https://doi.org/10.1182/asheducation-2012.1.652 [DOI] [PubMed] [Google Scholar]
- 2.Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood 2011; 117:2764-74; PMID:21190991; https://doi.org/ 10.1182/blood-2010-07-296962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinway SN, LeBlanc F, Loughran TP Jr. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev 2014; 28:87-94; PMID:24679833; https://doi.org/ 10.1016/j.blre.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abroun S, Saki N, Ahmadvand M, Asghari F, Salari F, Rahim F. STATs: An Old Story, Yet Mesmerizing. Cell J 2015; 17:395-411; PMID:26464811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, Li Y, Wang JM, Yang-Yen HF, Karras J, et al.. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest 2001; 107:351-62; PMID:11160159; https://doi.org/ 10.1172/JCI9940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, Lagstrom S, Clemente MJ, Olson T, Jalkanen SE, et al.. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 2012; 366:1905-13; PMID:22591296; https://doi.org/ 10.1056/NEJMoa1114885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, Olson T, Przychodzen B, Afable M, Gomez-Segui I, et al.. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 2012; 120:3048-57; PMID:22859607; https://doi.org/ 10.1182/blood-2012-06-435297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajala HL, Porkka K, Maciejewski JP, Loughran TP Jr., Mustjoki S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann Med 2014; 46:114-22; PMID:24512550; https://doi.org/ 10.3109/07853890.2014.882105 [DOI] [PubMed] [Google Scholar]
- 9.Yang CY, Leung PS, Adamopoulos IE, Gershwin ME. The implication of vitamin D and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol 2013; 45:217-26; PMID:23359064; https://doi.org/ 10.1007/s12016-013-8361-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams JS, Hewison M. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab 2008; 4:80-90; PMID:18212810; https://doi.org/ 10.1038/ncpendmet0716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC One,25-dihydroxyvitamin D3 receptors in human leukocytes. Science 1983; 221:1181-3; PMID:6310748; https://doi.org/ 10.1126/science.6310748 [DOI] [PubMed] [Google Scholar]
- 12.Manolagas SC, Werntz DA, Tsoukas CD, Provvedini DM, Vaughan JH One,25-Dihydroxyvitamin D3 receptors in lymphocytes from patients with rheumatoid arthritis. J Lab Clin Med 1986; 108:596-600; PMID:3023512 [PubMed] [Google Scholar]
- 13.Rigby WF, Stacy T, Fanger MW. Inhibition of T lymphocyte mitogenesis by 1,25-dihydroxyvitamin D3 (calcitriol). J Clin Invest 1984; 74:1451-5; PMID:6332829; https://doi.org/ 10.1172/JCI111557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cantorna MT, Waddell A. The vitamin D receptor turns off chronically activated T cells. Ann N Y Acad Sci 2014; 1317:70-5; PMID:24673331; https://doi.org/ 10.1111/nyas.12408 [DOI] [PubMed] [Google Scholar]
- 15.Chen PT, Hsieh CC, Wu CT, Yen TC, Lin PY, Chen WC, Chen MF. 1alpha,25-Dihydroxyvitamin D3 Inhibits Esophageal Squamous Cell Carcinoma Progression by Reducing IL6 Signaling. Mol Cancer Ther 2015; 14:1365-75; PMID:25824337; https://doi.org/ 10.1158/1535-7163.MCT-14-0952 [DOI] [PubMed] [Google Scholar]
- 16.Muthian G, Raikwar HP, Rajasingh J, Bright JJ One,25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res 2006; 83:1299-309; PMID:16547967; https://doi.org/ 10.1002/jnr.20826 [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Li H, Xie H, Fu M, Guo B, Ding Y, Li W, Yu H. Twenty-five-Hydroxyvitamin D3 attenuates experimental periodontitis through downregulation of TLR4 and JAK1/STAT3 signaling in diabetic mice. J Steroid Biochem Mol Biol 2013; 135:43-50; PMID:23333931; https://doi.org/ 10.1016/j.jsbmb.2013.01.008 [DOI] [PubMed] [Google Scholar]
- 18.Teleni L, Baker J, Koczwara B, Kimlin MG, Walpole E, Tsai K, Isenring EA. Clinical outcomes of vitamin D deficiency and supplementation in cancer patients. Nutr Rev 2013; 71:611-21; PMID:24032365; https://doi.org/ 10.1111/nure.12047 [DOI] [PubMed] [Google Scholar]
- 19.Gocek E, Studzinski GP. The Potential of Vitamin D-Regulated Intracellular Signaling Pathways as Targets for Myeloid Leukemia Therapy. J Clin Med 2015; 4:504-34; PMID:26239344; https://doi.org/ 10.3390/jcm4040504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol Rev 2016; 96:365-408; PMID:26681795; https://doi.org/ 10.1152/physrev.00014.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vidal M, Ramana CV, Dusso AS. Stat1-vitamin D receptor interactions antagonize 1,25-dihydroxyvitamin D transcriptional activity and enhance stat1-mediated transcription. Mol Cell Biol 2002; 22:2777-87; PMID:11909970; https://doi.org/ 10.1128/MCB.22.8.2777-2787.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lange CM, Gouttenoire J, Duong FH, Morikawa K, Heim MH, Moradpour D. Vitamin D receptor and Jak-STAT signaling crosstalk results in calcitriol-mediated increase of hepatocellular response to IFN-α. J Immunol 2014; 192:6037-44; PMID:24821973; https://doi.org/ 10.4049/jimmunol.1302296 [DOI] [PubMed] [Google Scholar]
- 23.Loughran TP Jr., Zickl L, Olson TL, Wang V, Zhang D, Rajala HL, Hasanali Z, Bennett JM, Lazarus HM, Litzow MR, et al.. Immunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the Eastern Cooperative Oncology Group (E5998). Leukemia 2015; 29:886-94; PMID:25306898; 10666315https://doi.org/10.1038/leu.2014.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veldman CM, Cantorna MT, DeLuca HF. Expression of 1,25-dihydroxyvitamin D(3) receptor in the immune system. Arch Biochem Biophys 2000; 374:334-8; PMID:10666315; https://doi.org/ 10.1006/abbi.1999.1605 [DOI] [PubMed] [Google Scholar]
- 25.Weeres MA, Robien K, Ahn YO, Neulen ML, Bergerson R, Miller JS, Verneris MR. The effects of 1,25-dihydroxyvitamin D3 on in vitro human NK cell development from hematopoietic stem cells. J Immunol 2014; 193:3456-62; PMID:25149465; https://doi.org/ 10.4049/jimmunol.1400698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dawson MI, Xia Z. The retinoid X receptors and their ligands. Biochim Biophys Acta 2012; 1821:21-56; PMID:22020178; https://doi.org/ 10.1016/j.bbalip.2011.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsen L, Ropke C. Suppressors of cytokine signalling: SOCS. APMIS 2002; 110:833-44; PMID:12645661; https://doi.org/ 10.1034/j.1600-0463.2002.1101201.x [DOI] [PubMed] [Google Scholar]
- 28.Bohmer FD, Friedrich K. Protein tyrosine phosphatases as wardens of STAT signaling. JAKSTAT 2014; 3:e28087; PMID:24778927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sunn KL, Cock TA, Crofts LA, Eisman JA, Gardiner EM. Novel N-terminal variant of human VDR. Mol Endocrinol 2001; 15:1599-609; PMID:11518809; https://doi.org/ 10.1210/mend.15.9.0693 [DOI] [PubMed] [Google Scholar]
- 30.Gardiner EM, Esteban LM, Fong C, Allison SJ, Flanagan JL, Kouzmenko AP, Eisman JA. Vitamin D receptor B1 and exon 1d: functional and evolutionary analysis. J Steroid Biochem Mol Biol 2004; 89-90: 233-8; PMID:15225777; 12097375https://doi.org/10.1016/j.jsbmb.2004.03.078 [DOI] [PubMed] [Google Scholar]
- 31.Ishaq M, Fan M, Wigmore K, Gaddam A, Natarajan V. Regulation of retinoid X receptor responsive element-dependent transcription in T lymphocytes by Ser/Thr phosphatases: functional divergence of protein kinase C (PKC)theta; and PKC α in mediating calcineurin-induced transactivation. J Immunol 2002; 169:732-8; PMID:12097375; https://doi.org/ 10.4049/jimmunol.169.2.732 [DOI] [PubMed] [Google Scholar]
- 32.Szabova L, Macejova D, Dvorcakova M, Mostbock S, Blazickova S, Zorad S, Walrand S, Cardinault N, Vasson MP, Rock E, et al.. Expression of nuclear retinoic acid receptor in peripheral blood mononuclear cells (PBMC) of healthy subjects. Life Sci 2003; 72:831-6; PMID:12479981; https://doi.org/ 10.1016/S0024-3205(02)02307-X [DOI] [PubMed] [Google Scholar]
- 33.Young HA, Hardy KJ. Interferon-gamma: producer cells, activation stimuli, and molecular genetic regulation. Pharmacol Ther 1990; 45:137-51; PMID:2105509; https://doi.org/ 10.1016/0163-7258(90)90012-Q [DOI] [PubMed] [Google Scholar]
- 34.Rigby WF, Denome S, Fanger MW. Regulation of lymphokine production and human T lymphocyte activation by 1,25-dihydroxyvitamin D3. Specific inhibition at the level of messenger RNA. J Clin Invest 1987; 79:1659-64; PMID:2884234; https://doi.org/ 10.1172/JCI113004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boissier MC, Assier E, Falgarone G, Bessis N. Shifting the imbalance from Th1/Th2 to Th17/treg: the changing rheumatoid arthritis paradigm. Joint Bone Spine 2008; 75:373-5; PMID:18571969; https://doi.org/ 10.1016/j.jbspin.2008.04.005 [DOI] [PubMed] [Google Scholar]
- 36.Alroy I, Towers TL, Freedman LP. Transcriptional repression of the interleukin-2 gene by vitamin D3: direct inhibition of NFATp/AP-1 complex formation by a nuclear hormone receptor. Mol Cell Biol 1995; 15:5789-99; PMID:7565732; https://doi.org/ 10.1128/MCB.15.10.5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cippitelli M, Santoni A. Vitamin D3: a transcriptional modulator of the interferon-gamma gene. Eur J Immunol 1998; 28:3017-30; PMID:9808170; https://doi.org/ 10.1002/(SICI)1521-4141(199810)28:10%3c3017::AID-IMMU3017%3e3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- 38.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140:883-99; PMID:20303878; https://doi.org/ 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stio M, Treves C, Celli A, Tarantino O, d'Albasio G, Bonanomi AG. Synergistic inhibitory effect of cyclosporin A and vitamin D derivatives on T-lymphocyte proliferation in active ulcerative colitis. Am J Gastroenterol 2002; 97:679-89; PMID:11922564; https://doi.org/ 10.1111/j.1572-0241.2002.05549.x [DOI] [PubMed] [Google Scholar]
- 40.Gepner P, Amor B, Fournier C One,25-dihydroxyvitamin D3 potentiates the in vitro inhibitory effects of cyclosporin A on T cells from rheumatoid arthritis patients. Arthritis Rheum 1989; 32:31-6; PMID:2521453; https://doi.org/ 10.1002/anr.1780320106 [DOI] [PubMed] [Google Scholar]
- 41.Lamy T, Liu JH, Landowski TH, Dalton WS, Loughran TP Jr. Dysregulation of CD95/CD95 ligand-apoptotic pathway in CD3(+) large granular lymphocyte leukemia. Blood 1998; 92:4771-7; PMID:9845544 [PubMed] [Google Scholar]
- 42.Doi K, Liu Q, Gowda K, Barth BM, Claxton D, Amin S, Loughran TP Jr., Wang HG. Maritoclax induces apoptosis in acute myeloid leukemia cells with elevated Mcl-1 expression. Cancer Biol Ther 2014; 15:1077-86; PMID:24842334; https://doi.org/ 10.4161/cbt.29186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGrath T, Latoud C, Arnold ST, Safa AR, Felsted RL, Center MS. Mechanisms of multidrug resistance in HL60 cells. Analysis of resistance associated membrane proteins and levels of mdr gene expression. Biochem Pharmacol 1989; 38:3611-9; PMID:2573357; https://doi.org/ 10.1016/0006-2952(89)90134-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.