


Abstract
Pre-mRNA splicing is catalyzed by the spliceosome, a multi-megadalton ribonucleoprotein machine. Previous work from our laboratory revealed the splicing factor SRSF1 as a regulator of the SUMO pathway, leading us to explore a connection between this pathway and the splicing machinery. We show here that addition of a recombinant SUMO-protease decreases the efficiency of pre-mRNA splicing in vitro. By mass spectrometry analysis of anti-SUMO immunoprecipitated proteins obtained from purified splicing complexes formed along the splicing reaction, we identified spliceosome-associated SUMO substrates. After corroborating SUMOylation of Prp3 in cultured cells, we defined Lys 289 and Lys 559 as bona fide SUMO attachment sites within this spliceosomal protein. We further demonstrated that a Prp3 SUMOylation-deficient mutant while still capable of interacting with U4/U6 snRNP components, is unable to co-precipitate U2 and U5 snRNA and the spliceosomal proteins U2-SF3a120 and U5-Snu114. This SUMOylation-deficient mutant fails to restore the splicing of different pre-mRNAs to the levels achieved by the wild type protein, when transfected into Prp3-depleted cultured cells. This mutant also shows a diminished recruitment to active spliceosomes, compared to the wild type protein. These findings indicate that SUMO conjugation plays a role during the splicing process and suggest the involvement of Prp3 SUMOylation in U4/U6•U5 tri-snRNP formation and/or recruitment.
INTRODUCTION
Most eukaryotic genes transcribed by RNA polymerase II give rise to precursor messenger RNAs (pre-mRNA) that contain exons and introns. Removal of introns and joining of exons to form mature mRNA, i.e. pre-mRNA splicing, is catalyzed by the spliceosome. This dynamic macromolecular machine is composed of five small nuclear ribonucleoprotein particles (snRNPs) termed U1, U2, U5 and U4/U6, and many non-snRNP splicing factors. Each snRNP consists of one small nuclear RNA (snRNA) or two in the case of U4/U6, a common set of seven Sm proteins (B/B΄, D3, D2, D1, E, F and G) and a variable number of particle-specific proteins (1).
Spliceosomes are assembled stepwise by the recruitment of snRNPs and other proteins to the pre-mRNA. Initially, U1 snRNP is recruited to the 5΄ splice site (ss) and U2 snRNP to the branch site of the pre-mRNA, forming the A complex (also known as the pre-spliceosome). Subsequently, the U4/U6•U5 tri-snRNP binds, generating the pre-catalytic B complex. After numerous RNA and protein rearrangements, including the dissociation of the U1 and U4 snRNPs, the spliceosome is converted first into an activated (Bact) complex and then into a catalytically-active complex (B* complex). The latter catalyzes the first step of the splicing reaction (i.e. cleavage at the 5'ss and intron lariat formation). Further rearrangements yield the C complex, which in turn catalyzes the second step, during which the intron is excised and the flanking 5΄ and 3΄ exons are ligated. Following this two-step catalytic process, the spliceosome disassembles.
Splicing catalysis is largely an RNA-based process (2,3). However, different proteins, such as Prp8 (4), are essential for the formation of the spliceosome's active site. During all transitions of the splicing process, the spliceosome's underlying RNA-protein interaction network is compositionally and conformationally remodeled. This remodeling extends all the way to the snRNPs, and consequently, several must be re-assembled after each splicing reaction in order to engage in further rounds of splicing. For example, U4/U6 is completely disrupted during catalytic activation (5), and the U4/U6•U5 tri-snRNP is reassembled after dimerization of the U4 and U6 snRNPs, and subsequent association with U5 snRNP (6,7). The association of the U4 and U6 snRNPs is mediated in part by base pairing between their respective snRNAs. Reannealing of U4 and U6 snRNAs after splicing requires Prp24 (8), an assembly chaperone in yeast, or its ortholog SART3 (7) in human. In addition, the U4/U6-specific Prp3 protein is essential for splicing, and is required for U4/U6 di-snRNP and U4/U6•U5 tri-snRNP formation (9). However the molecular mechanisms underlying its functions are unclear. Human (h) Prp3 forms a complex with the Prp4 protein (10,11) and also interacts with U5-specific proteins (12). Moreover, hPrp3 interacts directly with the U4/U6 snRNAs (13), which are extensively base paired within the U4/U6 di-snRNP complex (5).
In addition to the snRNPs, numerous non-snRNP proteins play essential roles during pre-mRNA splicing. Such is the case with SR proteins, which are well-described regulators of both constitutive and alternative splicing. Members of this protein family, and in particular SRSF1 (previously known as SF2/ASF), perform both nuclear and cytoplasmic regulatory tasks at different steps of mRNA metabolism (14). Moreover, our laboratory has shown that SRSF1 functions as a regulator of the SUMO conjugation pathway (15).
The process known as SUMO conjugation or SUMOylation is a rapid, reversible post-translational modification (PTM) consisting of the covalent attachment of a small ubiquitin-related modifier (SUMO) peptide to a lysine residue in the target protein. There are three well-characterized functional SUMO isoforms encoded by the human genome (SUMO1, 2 and 3), which modify distinct but overlapping sets of substrates. While still unclear whether SUMO4 is indeed conjugated to cellular proteins, SUMO5 has been recently identified as a novel, primate- and tissue-specific SUMO variant (16–20). Like ubiquitin, SUMO is conjugated to its targets by an isopeptide bond between its C-terminal glycine and the ε-NH2 group of the target lysine residue. In general, SUMOylation substrates contain a consensus sequence defined as ΨKxD/E, where Ψ is a large, hydrophobic amino acid, K is the target lysine, x is any amino acid, E is glutamic acid and D aspartic acid. However, many SUMOylated proteins deviate from this consensus sequence or even lack one (21).
The steps involved in the SUMO pathway resemble those of the ubiquitin pathway. Before being conjugated, SUMO is cleaved by specific proteases (SENPs), exposing its C-terminal Gly-Gly motif (22). After this step, mature SUMO is activated by the SUMO E1 activating enzyme, the heterodimer ‘AOS1-UBA2’, in an ATP-dependent manner and then transferred to the catalytic Cys residue of ‘Ubc9’, the SUMO-specific E2 conjugating enzyme. Finally, an isopeptidic bond is formed between the C-terminal Gly residue of SUMO and a Lys residue in the target protein. This step is generally aided by SUMO E3 ligases and among those characterized so far, some of them display substrate specificity while others display SUMO isoform preferences (17). SUMOylated proteins are substrates for SENPs, which deconjugate SUMO, ensuring the reversibility and dynamic nature of the process. Most frequently, SUMO conjugation regulates intra- or intermolecular interactions, altering either the conformation of the modified protein or the recruitment of its partners (17). In several cases, SUMOylation fosters new associations by non-covalent interaction of SUMO with proteins harboring SUMO-interaction motifs (SIMs). The establishment of SUMO–SIM interactions exerts a variety of effects, ranging from intramolecular structural rearrangements, as reported for thymine DNA glycosylase, to the assembly of multi-protein complexes, as described for nuclear PML bodies (23). In addition, SUMOylation can also interfere with protein stability by triggering ubiquitylation of poly-SUMO-modified proteins through the recruitment of SUMO-targeted ubiquitin ligases (STUbL) (24).
The biological relevance of protein SUMOylation was clearly documented by reports showing that inactivation of SUMO in Saccharomyces cerevisiae or of the unique E2 SUMO-conjugating enzyme Ubc9 in mice is lethal (25,26). Consistent with this, multiple studies have shown that SUMOylation regulates a wide range of cellular functions, including intracellular transport, maintenance of genome integrity, formation of nuclear subdomains (18), and also some aspects of rRNA or snoRNA metabolism (27–29). Furthermore, SUMO conjugation affects not only the stability, localization and activity of transcriptional regulators, but also the activity of DNA and histone modifiers, leading to changes in chromatin structure and therefore in gene expression (30).
Ubc9 has been found to localize in nuclear speckles (31), which are thought to coordinate splicing and gene expression, as they contain not only splicing factors but also other proteins involved in mRNA metabolism, such as transcription factors, RNA polymerase II subunits, cleavage and polyadenylation factors, and RNA export proteins (32). Ubc9's sub-nuclear localization suggests that nuclear speckles might also serve as ‘SUMOylation factories’ where proteins are modified by SUMO. In addition, the SUMO protease USLP1 has been found to localize in Cajal bodies, which contain high concentrations of snRNPs and other RNA processing factors, suggesting that they are sites for assembly and/or posttranscriptional modification of the splicing machinery within the nucleus (31,33).
Proteomic approaches revealed that RNA-related proteins are the predominant group among SUMO substrates. These studies reported that several splicing factors comprising members of the hnRNP and SR protein families, as well as different spliceosomal proteins are SUMOylated in human cells (34–36). Furthermore, SUMO conjugation has been found to regulate different aspects of mRNA metabolism, such as pre-mRNA 3΄ end processing and RNA editing, by modifying the function of poly(A) polymerase, symplekin and CPSF-73 in the former case, and ADAR1 in the latter (37,38). Previous studies from our laboratory demonstrated that the splicing factor SRSF1 interacts with Ubc9, promoting the SUMOylation of specific substrates, and that SRSF1 regulates the SUMO E3 ligase activity of PIAS1 (15), which co-purifies with the spliceosome (39). Strikingly, ubiquitylation/de-ubiquitylation has been implicated in the regulation of the splicing process. In particular, non-proteolytic ubiquitylation of the U4/U6 protein Prp3, promoted by the Prp19 complex, is required for stabilization of the U4/U6•U5 tri-snRNP, while de-ubiquitylation of Prp3 by Usp4/Sart3 is required for U4 dissociation and recycling (40). However, the role, if any, of SUMOylation/de-SUMOylation in pre-mRNA splicing remains unclear.
In this study, we have investigated the effects of SUMOylation on the splicing machinery. We found that addition of SENP1 to an in vitro splicing reaction, which led to a decrease in protein SUMOylation, also decreased the efficiency of pre-mRNA splicing. We identified several spliceosomal proteins that are SUMOylated in the context of both in vitro splicing and cultured cells. We further demonstrated that the level of SUMO conjugation of some of these proteins is regulated by the splicing factor SRSF1. Focusing on the spliceosomal protein Prp3, we have mapped its SUMO attachment sites and generated a SUMOylation-deficient mutant, Prp3 2KR. This mutant was unable to restore splicing of several pre-mRNAs to the levels observed with the wild type protein when they were transfected into cultured cells depleted of the endogenous Prp3 protein. Consistent with this, Prp3 2KR exhibited reduced interaction with RNP complexes containing U2 and U5, as well as a diminished recruitment to active co-transcriptional spliceosomes.
MATERIALS AND METHODS
In vitro splicing
[32P]-labeled MINX pre-mRNA (41) was transcribed in vitro using T7 RNA polymerase (Ambion). HeLa nuclear extract (NE) was prepared according to (42). Splicing reactions contained 40% (v/v) HeLa nuclear extract in buffer D [20 mM HEPES–KOH at pH 7.9, 100 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 20% (v/v) glycerol, 0.5 mM DTT, 0.5 mM PMSF], 20 mM creatine phosphate and 2 mM ATP, and the end concentration of KCl and MgCl2 was adjusted to 65 mM and 3 mM, respectively. Reactions were incubated at 30°C for the times indicated in each figure. RNA was extracted using Trizol (Invitrogen) and analyzed on a 14% polyacrylamide gel, followed by autoradiography. SENP1 (500 ng, ∼300 nM), Ubc9 (500 ng, ∼500 nM) and SAE1/SAE2 (500 ng, ∼10 nM), recombinant proteins purified from Escherichia coli and purchased from ENZO Life Sciences, were added to NE, and pre-incubated under splicing conditions for 10 min at 30°C before the addition of MINX pre-mRNA. ‘Mock’ conditions were achieved by addition of either heat-inactivated SENP1 (500 ng) or SENP1 storage buffer, according to the information provided by the manufacturer (50 mM Tris, 150 mM NaCl, 0.5 mM DTT, pH 7.5). Radioactivity in RNA bands was quantified with a PhosphorImager (Molecular Dynamics). Splicing efficiency was calculated by dividing the amount of products (mRNA + excised lariat-intron) by the sum of the precursor, intermediates and products. To analyze spliceosomal complex formation, 10 μl of the splicing reaction were combined with 2.5 μl of loading buffer [1× TBE, 30% (v/v) glycerol, 1.25 mg/ml heparin] at the time points indicated, and then placed on ice. Spliceosomal complexes were separated on 2% (w/v) agarose gels (43). In vitro splicing reactions were alternatively monitored by RT-qPCR, using primers that specifically amplify the substrate and the mature product (listed in Supplementary Table S3). In this case, splicing efficiency was calculated as the ratio of amplified mRNA over amplified (mRNA + pre-mRNA) after 60 min of splicing.
MS2 affinity selection of spliceosomal complexes
Purified MS2-maltose binding protein (MS2-MBP) fusion protein was incubated with 32P-labeled MS2-tagged MINX pre-mRNA and then added to a 1 ml splicing reaction, as described above, except containing 10 nM MS2-tagged MINX pre-mRNA. Spliceosomal complexes were allowed to assemble at 30°C for the indicated time points, and an aliquot was analyzed for splicing complex formation on a 2% agarose gel. The reaction was complemented with 10 mM N-ethyl maleimide (NEM), 10 mM iodoacetamide (IAA) and 125 mM NaCl, and then loaded onto an amylose resin column (New England Biolabs), equilibrated with buffer G (20 mM HEPES–KOH at pH 7.9, 150 mM NaCl, 1.5 mM MgCl2). After extensive washing with buffer G, the spliceosomal complexes were eluted dropwise with elution buffer (buffer G containing 12 mM maltose). RNA was isolated from the eluates by proteinase K treatment, phenol extraction and ethanol precipitation, separated by denaturing polyacrylamide gel electrophoresis on an 8.3 M urea/10% (w/v) polyacrylamide gel, and visualized by silver staining (snRNAs) or autoradiography (substrate, intermediates and products of the splicing reaction).
Anti-SUMO immunoprecipitation with immobilized antibodies
Monoclonal anti-SUMO2 8A2 or mouse IgG (Invitrogen) was added to 1 ml of Protein G–agarose (Roche) equilibrated in 20 mM NaPO4, pH 7.0. To cross-link the antibodies to Protein G, 50 mM borate buffer, pH 9.0, containing 20 mM DMP (dimethyl pimelimidate, Thermo Scientific) was freshly prepared and directly added to the agarose. After 1 h incubation, the cross-linker was quenched with 50 mM Tris–HCl, pH 8.0. Before use, beads were washed once with 200 mM acetic acid, pH 2.7 and 500 mM NaCl, and twice with 20 mM NaPO4, pH 7.0. For immunoprecipitation of SUMOylated proteins in MS2 affinity-purified splicing complexes, the latter were incubated with buffer containing 20 mM NaPO4, pH 7.4, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate, 5 mM EDTA, 5 mM EGTA, 10 mM NEM and protease inhibitor (Complete, Roche) in order to disrupt protein-protein and protein–RNA interactions as well as to inactivate SUMO proteases. 50 μl of antibody-bound beads were then added and the mixture incubated overnight at 4°C. Beads were washed three times with IP buffer (20 mM NaPO4, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate, 5 mM EDTA, 5 mM EGTA, 10 mM NEM and Complete protease inhibitor, Roche). A final wash with three bead volumes of high-salt IP buffer (500 mM NaCl instead of 150 mM) was carried out for 30 min at 37°C on a rotating wheel. Protein elution was performed at 37°C on a rotating wheel for 30 min, using three bead volumes of SDS sample buffer.
Mass spectrometry (MS)
Proteins precipitated by the control IgG or anti-SUMO2 antibodies were resolved by SDS-PAGE. Each lane was cut into equal pieces before undergoing in-gel trypsin digestion. The extracted peptides were analyzed by liquid chromatography (LC)–coupled mass spectrometry (MS) under standard conditions on a LTQ orbitrap Xl instrument (Thermo Fisher Scientific). Proteins were identified by searching fragment spectra against the Uniprot database, using Mascot as a search engine.
Transfection of plasmids and siRNAs
Plasmid DNA and siRNAs were transfected into HEK 293T and HeLa cells with Lipofectamine 2000 according to manufacturer's instructions (Thermo Fisher). The siRNA against SRSF1 used was previously described (44). The sequences corresponding to the three different siRNAs (Invitrogen) targeting the human PRP3 3΄UTR are shown in Supplementary Table S3. An siRNA targeting luciferase was used as a control, siRNA LUC: 5΄-CUUACGCUGAGUACUUCGA(dT)(dT)-3΄.
Western blots and antibodies
Protein samples were resolved by SDS-PAGE and transferred to PVDF or nitrocellulose membranes (BioRad). Membranes were blocked and then incubated with the primary antibody. After washing, membranes were incubated with HRP-conjugated (BioRad) or IRDye® 800CW (LI-COR Biosciences) secondary antibodies. Bound antibody was detected using ECL plus reagent (GE Healthcare) or an Odyssey imaging system (LI-COR Biosciences). Western blots were performed at least three times, and representative images are shown in each case. The antibodies used were mouse monoclonal anti-βactin C4 (Santa Cruz Biotechnology), mouse monoclonal anti-SUMO2 8A2, mouse monoclonal anti-T7 tag (Novagen), rabbit monoclonal anti Prp3 (St John's Laboratory), rabbit polyclonal anti-60K/Prp4 (45), rabbit polyclonal anti-SF3a120 (46) and rabbit polyclonal anti-Snu114 (47).
Site-directed mutagenesis
Site-directed mutagenesis was performed by the DpnI method, based on Stratagene's QuickChange specifications. The primers used to mutate putative SUMO sites from Lys to Arg are listed in Supplementary Table S3. Mutations were always verified by sequencing.
Purification of His-SUMO- or His–ubiquitin-conjugated proteins
HEK 293T cells were transfected in 35-mm culture wells with the indicated plasmids. After 48 h, His-SUMO2 or His–ubiquitin conjugates were purified under denaturing conditions using Ni-NTA-agarose beads according to the manufacturer's instructions (Qiagen). Briefly, transfected cells were harvested in ice-cold PBS plus 100 mM IAA. An aliquot was taken as input and the remaining cells were lysed in 6M guanidinium-HCl containing 100 mM Na2HPO4/NaH2PO4, 10 mM Tris–HCl pH 8.0, 5 mM imidazole and 10 mM iodoacetamide. Samples were sonicated to reduce the viscosity and after centrifugation for 20 min at 12 000 ×g, proteins in the supernatants were purified using Ni-NTA beads (Qiagen) according to (48). Samples were subsequently washed with wash buffer I (8M urea, 10 mM Tris–HCl, 100 mM Na2HPO4/NaH2PO4, 5 mM imidazole, 10 mM iodoacetamide, pH 8.0), wash buffer II (8 M urea, 10 mM Tris–HCl, 100 mM Na2HPO4/NaH2PO4, 0.2% Triton X-100, 5 mM imidazole, 10 mM iodoacetamide, pH 6.3), and wash buffer III (8 M urea, 10 mM Tris–HCl, 100 mM Na2HPO4/NaH2PO4, 0.1% Triton X-100, 5 mM imidazole, 10 mM iodoacetamide, pH 6.3). Samples were eluted in 2× Laemmli sample buffer containing 300 mM imidazole for 5 min at 95°C.
Quantitative PCR for cellular RNAs (RT-qPCR)
Total cellular RNA was isolated by using 500 μl of Tri-Reagent (MRC) and measured with a NanoDrop 1000 spectrophotometer (Thermo Scientific). From this RNA preparation, 2 μg were reverse transcribed to cDNA with a random deca-oligonucleotide primer mix using MMLV Reverse Transcriptase (Invitrogen). Quantitative PCRs (qPCRs) were performed using SYBR Green dye, 1/20 dilution of cDNA sample and Taq DNA polymerase (Invitrogen) in a Mastercycler® ep realplex PCR device (Eppendorf). The annealing temperature was 60°C and the elongation time at 72°C was 30 s. Relative RNA abundances from cDNAs and no-reverse transcription controls were estimated employing internal standard curves with a PCR efficiency of 100 ± 10% for each set of primers, in each experiment. Realplex qPCR software was used to analyze the data. The specific primers are listed in Supplementary Table S3. Splicing efficiency was assessed according to the following ratio: mRNA/(pre-mRNA+mRNA).
Immunoprecipitation of RNP complexes (RIP)
Cells were harvested and lyzed in RIPA buffer (50 mM Tris–HCl (pH 7.5), 1% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 0.05% (w/v) SDS, 1 mM EDTA, 150 mM NaCl) containing complete protease inhibitor and RNasin (Promega). Extracts were sonicated with a Bioruptor at high amplitude with three 30-s bursts, and insoluble material was pelleted. The supernatant was pre-cleared with GammaBind G sepharose beads for 30 min at 4°C before addition of anti-T7 antibody and incubation overnight. Complexes were incubated with GammaBind G sepharose beads for 1 h and washed three times in RIPA buffer. One half of the sample was resuspended in SDS sample buffer for western blot analysis followed by quantification with Image Studio Software (LI-COR Biosciences). The other half was subject to total RNA extraction followed by RT-qPCR. cDNA was prepared from one-half of the RNA using 10-mer random primers. The cDNA and no-reverse transcription control were analyzed by quantitative PCR with primer pairs spanning snRNAs as detailed in the primer list (Supplementary Table S3). For each RNA analyzed, the average ratio of immunoprecipitated RNA divided by the input, as well as the standard deviation were calculated from three independent RIP experiments.
Chromatin immunoprecipitation
Cells were cross-linked with 1% (v/v) formaldehyde (final concentration), washed twice with cold PBS, scraped and collected. Cell pellets were resuspended in 2 ml of SDS lysis buffer (1% w/v SDS, 10 mM EDTA, 50 mM Tris–HCl, pH 8.1) containing Complete Protease Inhibitor Cocktail (Roche) and incubated for 10 min on ice. Cell extracts were sonicated with a Branson sonicator W-450 D at 30% amplitude with fifteen 10-s bursts, resulting in ∼500-nt chromatin fragments, and then centrifuged for 10 min at 12 000 g. A 50 μl sample of the supernatant was saved as input DNA and the remainder was diluted 1:10 in ChIP dilution buffer (0.01% w/v SDS, 1.1% v/v Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl pH 8.1, 167 mM NaCl) containing protease inhibitors. The chromatin solution was precleared at 4°C with Protein G Dynabeads® for 1 h before incubating overnight at 4°C with anti-T7 antibody. Complexes were incubated with Dynabeads® Protein G beads for 1 h at 4°C. The beads were washed by rocking for 4 min, once in each of the following buffers: low salt immune complex wash buffer (0.1% w/v SDS, 1% v/v Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 150 mM NaCl), high-salt immune complex wash buffer (same as low salt buffer, except with 500 mM NaCl) and LiCl immune complex wash buffer (0.25 M LiCl, 1% v/v NP-40, 1% w/v deoxycholic acid, 1 mM EDTA, 10 mM Tris–HCl, pH 8.1), and then twice in TE (10 mM Tris–HCl, 1 mM EDTA). The bound complexes were eluted in 1% (w/v) SDS and 50 mM NaHCO3 and cross-links reversed by incubating for 6 h at 65°C. Samples were digested with proteinase K for 1 h at 45°C and the DNA extracted using a Qiagen PCR purification kit. DNA retrieved by ChIP was analyzed by quantitative real-time PCR with primers spanning introns as detailed in Supplementary Table S3. Data sets were normalized to ChIP input values.
RESULTS
SUMOylation affects the efficiency of pre-mRNA splicing in vitro
Previous studies pointed to a connection between the splicing and SUMO machineries. To study the effect of spliceosomal protein SUMOylation on pre-mRNA splicing, we performed in vitro splicing with HeLa nuclear extract (NE) and a 32P-labeled pre-mRNA substrate (‘MINX’ pre-mRNA) (41), which consists of two exons separated by an intron with canonical splice sites. The kinetics of the splicing reaction and assembly of the spliceosomal complexes A, B and C, is shown in Figure 1A and B. Western blot analysis of protein SUMOylation during the splicing reaction showed that SUMO conjugation increases with time, suggesting that HeLa NE possesses SUMOylation activity (Figure 1C). We further confirmed that this was indeed the case by performing in vitro SUMOylation reactions in which HeLa NE was incubated with purified, recombinant His-SUMO2, under splicing conditions and in the absence of any other exogenously added SUMO pathway component (Supplementary Figure S1). These results indicate that spliceosome-associated proteins may be actively modified during the in vitro splicing reaction. We therefore altered the level of SUMO conjugation by adding purified, recombinant SENP1 or Ubc9 to the reaction, and assayed the effect on pre-mRNA splicing (Figure 1D). Remarkably, when SUMO conjugation was diminished by addition of exogenous SENP1 (Figure 1F), there was a clear reduction in splicing efficiency, as evidenced by the lower amount of splicing product formation (mRNA and excised lariat-intron) at each time point relative to the control reaction (Figure 1D and E). Furthermore, in the presence of SENP1, the splicing intermediate peaks at a later time point compared to the mock reaction (black arrow in panel D). In contrast, when SUMO conjugation levels were enhanced by addition of the E2 conjugating enzyme Ubc9 (Figure 1F), no significant changes were observed (Figure 1D and E). Similar effects of SENP1 on splicing efficiency were obtained when substrate (MINX pre-mRNA) disappearance and product (MINX mRNA) accumulation over time was monitored by reverse transcription-real time PCR with specific primers as shown in Supplementary Figure S2. Furthermore, when heat-inactivated SENP1 was added to the splicing reaction, not only were SUMO conjugation levels no longer diminished, but also the reduction in splicing efficiency after 60 min was no longer observed (Supplementary Figure S2C and D). The decrease in splicing efficiency observed upon addition of an active SENP1 was abolished when SUMO conjugation levels were recovered by adding both E1 activating (SAE1/SAE2) and E2 conjugating (Ubc9) enzymes to the reaction (Figure 1G and H). As seen in Figure 1H, the combined addition of SENP1, SAE1/SAE2 and Ubc9 enzymes even seems to enhance splicing efficiency above the levels achieved by SAE1/SAE2 plus Ubc9, possibly due to a requirement of SUMOylation/de-SUMOylation cycles. This observation clearly warrants further investigation.
Figure 1.

Addition of the SUMO deconjugating enzyme SENP1 inhibits in vitro splicing. Pre-mRNA splicing was performed with HeLa nuclear extract (NE) and radiolabeled MINX pre-mRNA for 0–60 min. Splicing intermediates and products were analyzed on a denaturing polyacrylamide gel (A), and spliceosomal complex formation was analyzed by native agarose gel electrophoresis (B). The pre-mRNA, lariat-intron-3΄ exon, lariat-intron and mRNA, as well as spliceosomal complexes H, A, B and C, are indicated on the right of each panel. (C) SUMOylation of nuclear proteins at different times of the splicing reaction (0-60 min) was analyzed by western blot with an antibody against SUMO2. (D) As in (A), except splicing reactions were pre-incubated with SENP1, Ubc9 or SENP1 storage buffer (mock) for 10 min before adding MINX pre-mRNA. The black arrow points to the lariat-intron-3΄ exon intermediate that peaks at later times upon SENP1 treatment. (E) Splicing efficiency defined as products divided by (substrate+intermediates+products) derived from quantification of the corresponding radiolabeled bands at the indicated time points. Error bars were calculated from two independent experiments. (F) Western blot analysis of SUMO conjugation levels in NE after pre-incubation for 10 min with recombinant SENP1, Ubc9 or mock-treated (SENP1 storage buffer), before addition of MINX pre-mRNA. (G) As in (F) but additionally with Ubc9 and SAE1/SAE2 or SENP1, Ubc9 and SAE1/SAE2. (H) MINX splicing efficiency as determined by RT-qPCR after 60 min of splicing under mock conditions (SENP1 storage buffer), or in the presence of the indicated SUMO pathway enzymes. Splicing efficiency was calculated as pre-mRNA/(pre-mRNA+mRNA). Western blots in panels C and F were visualized with ECL reagent, and panel G with an Odyssey imaging system.
The fact that SUMOylation occurs during the splicing reaction, that addition of a catalytically active SENP1 reduces splicing efficiency and that this effect can be rescued by addition of SUMO activating and conjugating enzymes, indicates that SUMO conjugation plays a role during the in vitro splicing process.
Protein components of the spliceosome are SUMO substrates
To address whether SUMO-conjugated proteins are present in spliceosomes assembled in vitro, we performed an MS2-based pull down of splicing complexes formed at different times of the splicing reaction (Supplementary Figure S3). For this purpose, we took advantage of the three binding sites for the MS2 viral coat protein present at the 3΄ end of the MINX pre-mRNA used as the splicing substrate. After combining MS2–MBP (maltose binding protein) fusion protein with the MINX pre-mRNA, splicing complexes were allowed to form in HeLa NE for 2–20 min and were then purified via incubation with amylose beads and subsequent elution with maltose. Western blot analysis of the eluates with an anti-SUMO2 antibody showed that the amount of SUMOylated spliceosome-associated proteins increases with time, with a marked increase between 2 and 7 min (Figure 2A). Analysis of splicing complex formation (Figure 2B) and the RNA content of the purified splicing complexes (Figure 2C) indicates that after 7 min, large amounts of B complex are assembled as a consequence of the U4/U6•U5 tri-snRNP interaction with the pre-spliceosomal A complex. Based on the intensity of the MINX pre-mRNA and U2 snRNA, similar amounts of spliceosomes were purified at the 2 and 7 min time points. The increase in SUMO conjugates observed in the eluate of the 7 min reaction could be due to either enhanced SUMOylation of pre-mRNA associated proteins and/or the recruitment of additional SUMO-conjugated proteins to the spliceosome during tri-snRNP integration. These results again suggest that spliceosomal proteins are actively modified during the splicing process.
Figure 2.

Identification of SUMOylated proteins in spliceosomes formed in vitro. (A) Spliceosomal complexes were allowed to form on radiolabelled MINX pre-mRNA containing MS2 binding sites bound by MBP–MS2 fusion protein under in vitro splicing conditions for 0–20 min. The RNA–protein complexes were purified by MBP pull-down with amylose beads and, after elution with maltose, proteins in the purified spliceosomal complexes (eluates), as well as in the input and flow-through, were analyzed by WB with anti-SUMO2 antibody visualized by ECL reagent. (B) The kinetics of spliceosomal complex formation was monitored by native agarose gel electrophoresis. (C) RNA composition of affinity-purified splicing complexes was analyzed by denaturing PAGE. MINX-MS2 pre-mRNA and the U1-U6 snRNAs were visualized by silver staining. The marker (M) in lane 1: RNA from anti-m3G (2,2,7-trimethylguanosine-containing cap structure of snRNAs), affinity-purified human snRNPs. (D) SUMOylated proteins within the affinity-purified spliceosomal complexes were immunoprecipitated with an anti-SUMO2 antibody (S2), after disruption of the RNP complexes, and then analyzed by denaturing SDS-PAGE. As a control (C), immunoprecipitation was also performed with IgG.
To gain further insight into actual SUMO conjugation targets, we performed anti-SUMO immunoprecipitation of spliceosomal complexes affinity-purified at different time points of the splicing reaction. To isolate individual SUMOylated proteins (and not their binding partners), purified spliceosomal complexes were first sonicated and denatured to completely disrupt RNP and protein-protein interactions, while NEM (N-ethyl maleimide) was added to inactivate SUMO isopeptidases (16,49). Afterwards, the samples were subjected to immunoprecipitation with either anti-SUMO2 antibody or control IgG (Supplementary Figures S3 and S4). A number of proteins were enriched in the SUMO2 immunoprecipitates relative to the control, as revealed by coomasie staining (Figure 2D and Supplementary Figure S4). Immunoprecipitated proteins were then identified by mass spectrometry (MS). A variety of splicing-related proteins were enriched by this strategy as shown in Table 1 and Supplementary Table S1. Many of the identified proteins were reported as putative SUMOylation targets in other proteomic studies (Supplementary Table S2). To the best of our knowledge, this is the first report revealing several spliceosomal proteins as potential SUMO conjugation substrates, such as SF3A3 (SF3a60), SF3B3 (SF3b130) and Prp19 among others. In addition, our results further indicate that the identified proteins are present in a SUMOylated state within the spliceosome, in in vitro splicing reactions.
Table 1. Summary of spliceosome-associated proteins enriched by anti-SUMO2 immunoprecipitation.
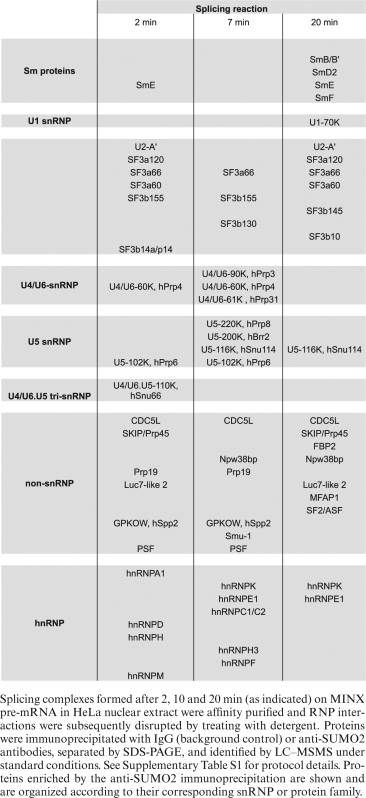
|
Spliceosomal proteins are SUMOylated in cultured cells
After identifying SUMOylated spliceosomal proteins in in vitro assembled spliceosomes, we investigated whether a subset of these proteins are also modified by SUMO2 in cultured cells. To address this, cell extracts from HEK 293T transfected with His-SUMO2 were subjected to nickel affinity purification to enrich for SUMOylated proteins. Pulled-down proteins were analyzed by western blot with specific antibodies against the different spliceosomal proteins, or with anti-tag antibodies in the case of transfected spliceosomal proteins when specific antibodies were not available. We were able to detect SUMO conjugation to Snu114 (Figure 3A and B) and Prp3 (Figure 3C and D). In agreement with our previous findings (15), SRSF1 overexpression strongly enhanced SUMO conjugation to Snu114 (Figure 3A) and to Prp3 to a lesser extent (Figure 3C), while depletion of endogenous SRSF1 by RNAi clearly diminished both Snu114 and Prp3 SUMOylation (Figure 3B and D). In addition, and consistent with previous reports (15,36,50,51), the amount of these SUMO conjugates is modulated by co-transfection of His-SUMO2 with Ubc9 or SENP1, corroborating that they correspond to SUMOylated forms of the spliceosomal proteins Snu114 and Prp3 (Figure 3B and C).
Figure 3.

The spliceosomal proteins Snu114 and Prp3 are SUMOylated in cultured cells. (A–D) HEK 293T cells were transfected with the siRNAs and/or DNA expression vectors as indicated at the top of each panel. After 48 h, cells were lysed and cell lysates were subjected to Nickel affinity chromatography (Ni-NTA). Aliquots of the cell lysates and eluates (Ni-NTA) were analyzed by western blot with the antibodies indicated below each panel. Western blots in panels A and B were visualized with ECL reagent, while panels C and D with an Odyssey imaging system.
The spliceosomal protein Prp3 is SUMOylated at lysines 289 and 559
With the goal of determining the involvement of these modifications during the spliceosome assembly/disassembly cycle, we first used bioinformatic predictions to define potential canonical SUMO attachment sites within the identified spliceosomal proteins (Sumo sp 2.0 <www.sumosp.biocuckoo.org>; SUMOplotTMPrediction <www.abgent.com/tools/toSumoplot>).
Considering the reported involvement of non-proteolytic ubiquitylation of Prp3 in the splicing cycle (40), we focused on the SUMOylation of this particular protein. After searching for potential SUMO attachment sites within human Prp3, four lysine residues within the consensus ΨKxD/E were found (Figure 4B). We then performed site-directed mutagenesis replacing each of these lysine residues by arginine followed by nickel affinity purification of SUMOylated proteins from cells co-expressing His-SUMO2 and the different T7-Prp3 mutants. Mutation of lysine 289 or 559 showed only a slight decrease in SUMO conjugation levels (Figure 4A). We thus generated several Prp3 double mutants. Only the combined replacement of Lys 289 and 559 by Arg residues (here after referred as the Prp3 2KR mutant) led to a drastic reduction in Prp3 SUMOylation levels (Figure 4A). Mutation of both proximal acidic residues to Ala (Glu291Ala, Glu561Ala) within the corresponding SUMO consensus motifs led to a similar decrease in SUMOylation (Figure 4A), further confirming that Lys 289 and 559 are bona fide SUMO attachment sites.
Figure 4.

Prp3 is SUMOylated at lysines 289 and 559. (A) HEK 293T cells were transfected with expression vectors encoding wt or mutated T7-tagged Prp3, and His-SUMO2, as indicated above each lane. After 48 h, cell lysates were subjected to Nickel affinity chromatography (Ni-NTA). Aliquots of the cell lysates and eluates (Ni-NTA) were analyzed by western blot with an anti-T7 antibody to detect the T7-Prp3 variants. (B) Putative SUMO attachment sites in Prp3, predicted in silico, and the surrounding SUMO consensus sequences. Ψ, bulky, hydrophobic amino acid; X, any amino acid; E, glutamic acid; D, aspartic acid. (C) As in (A) but co-transfection of T7-Prp3 variants with His-Ubiquitin (Ub). (D) Domain architecture of Prp3 and localization of detected SUMO conjugation sites. The western blot in panel A was visualized with an Odyssey imaging system and panel C with ECL reagent.
Consistent with previous reports (40), Prp3 is also modified by ubiquitin (Figure 4C). Interestingly, the Prp3 SUMOylation-deficient mutant 2KR is still conjugated to ubiquitin to the same extent as wt Prp3 (Figure 4C), which indicates that these mutated lysines are not likely to be ubiquitin targets. Remarkably, Lys 559 belongs to the DUF1115 domain within Prp3 (PFAM ID PF06544; Figure 4D), which is highly conserved from yeast to human. Within this domain, not only the indicated Lys residue, but also the surrounding consensus sequence is preserved (13). Lys 289, while less evolutionary conserved than Lys 559, is also present at an equivalent position in Prp3 from X. laevis, D. rerio and D. melanogaster (Supplementary Figure S5).
The Prp3 SUMOylation-deficient mutant displays diminished interaction with spliceosomal components
To determine whether loss of Prp3 SUMOylation affects its function, we first analyzed the interaction of wild type (wt) Prp3 and Prp3 2KR with endogenous spliceosomal snRNPs in HEK 293T cells. For this purpose, we performed co-immunoprecipitation (co-IP) and RNA immunoprecipitation (RIP) assays with an anti-T7 antibody and whole cell extracts derived from T7-tagged Prp3 transfected cells. The co-precipitation of selected snRNP proteins was assayed via western blotting. The U2 snRNP protein SF3a120/SF3A1, the U5 snRNP protein Snu114, and the U4/U6-60K (Prp4) protein, as well as U2, U5, U4 and U6 snRNAs were co-precipitated with wt T7-Prp3 (Figure 5). With the SUMOylation-deficient mutant, T7-Prp3 2KR, the levels of co-precipitated U2-SF3a120/SF3A1 and U5-Snu114, as well as U2 and U5 snRNAs, were drastically reduced. In contrast, the amount of co-precipitated U6 and U4 snRNAs was not reduced (Figure 5C) while that of U4/U6-60K/Prp4 protein, a direct interaction partner of Prp3, was only marginally reduced (Figure 5A and B), suggesting that Prp3 2KR, like the wt protein, is still assembled into the U4/U6 di-snRNP. Thus, much less of the Prp3 2KR mutant appears to associate with RNP complexes containing U5, such as the U4/U6•U5 tri-snRNP, as well as complexes containing additionally U2 (potentially endogenous spliceosomal B complexes). Taken together these results suggest that Prp3 SUMOylation might be important for the interaction of the U4/U6 di-snRNP with U5 during tri-snRNP formation.
Figure 5.

Decreased association of a Prp3 SUMOylation-deficient mutant with U2 and U5-containing RNP complexes. Overexpressed wt or 2KR T7-Prp3 was immunoprecipitated from HEK 293T cell lysates. (A) Co-IP of endogenous U2-SF3a120, U5-Snu114 and U4/U6-60K was assessed by western blot with antibodies specific for each protein and visualized with an Odyssey imaging system. (B) Quantification corresponding to three independent co-IP experiments as the one shown in (A) was performed with Image Studio Software (LI-COR Biosciences), according to the following calculation: ‘fold change’ = [IP/input]wt / [IP/input]2KR. (C) Co-IP of U4, U6, U2 or U5 snRNA was quantified by RT-qPCR with primers specific for each snRNA. Data are represented as mean ± S.E. (n = 3, ***P ≤ 0.001; Student's t test).
A lack of Prp3 SUMOylation affects splicing efficiency in cultured cells
To explore whether Prp3 SUMOylation has indeed any relevance for the splicing process within living cells, we analyzed the splicing efficiency of a subset of different housekeeping or highly expressed genes. To this end, the levels of their corresponding pre-mRNAs and mature mRNAs were measured by RT-qPCR with specific primers that either anneal to intron–exon boundaries or to introns in the former case, or to exon–exon junctions in the latter. Initially, we analyzed splicing efficiency upon transfecting cultured cells with expression vectors for either wt or different Prp3 mutants. We found that the splicing efficiency of two different genes, HPRT1 and Akt1 (Supplementary Figure S6), calculated as the mRNA/(pre-mRNA+mRNA) ratio, was comparable when cells overexpressed either T7-Prp3 wt or T7-Prp3 K244/376R, a double mutant that showed no reduction in SUMO conjugation levels (Figure 4A). However, splicing efficiency was significantly reduced upon introduction of a SUMOylation-deficient mutant, either T7-Prp3 2KR or the SUMO-consensus double mutant E291/561A (Supplementary Figure S6). These results clearly indicate that Prp3 SUMO conjugation levels have an impact on the splicing process and even suggest that the SUMOylation-deficient Prp3 may function in a dominant negative manner upon overexpression.
To strengthen these findings, we not only expanded the set of genes analysed, but also performed knock-down/rescue experiments. For this purpose, endogenous Prp3 was knocked-down by a pool of three different siRNAs targeted to its 3΄UTR, and cells were subsequently transfected with siRNA-resistant, wild type or 2KR T7-Prp3. The levels of endogenous, as well as transfected Prp3 proteins were assessed by western blot with an anti-Prp3 antibody (Figure 6A), while the levels of pre-mRNAs and mRNAs for the selected set of genes was quantified by RT-qPCR as indicated above. Although the endogenous Prp3 was efficiently knocked down as shown in Figure 6A, splicing was not fully abrogated, but clearly reduced for all of the genes analysed (Figure 6B). Furthermore, transfected wt T7-Prp3 was able to restore splicing efficiency or even enhance it over basal levels, consistent with the observation that the levels of the transfected Prp3 proteins were higher than the endogenous one (Figure 6A). In contrast, transfected 2KR T7-Prp3 was either unable to restore splicing to basal levels (four genes on the right side of panel 6B) or failed to enhance it to the levels achieved by the wt protein (four genes on the left side of panel 6B). These results suggest that different splicing events display differential sensitivity not only to Prp3 levels, but also to the extent of Prp3 SUMOylation. Remarkably, in every case the splicing efficiency was lower when the rescue was performed with the SUMOylation-deficient mutant than with the wt Prp3 protein (Figure 6B). These results are consistent with the idea that SUMOylation of Prp3 plays an important role in the splicing process.
Figure 6.

Prp3 2KR mutation affects pre-mRNA splicing efficiency and Prp3 recruitment to chromatin in cultured cells. (A and B) HeLa cells were transfected with a control siRNA (siC) or three siRNAs targeting Prp3 (siPrp3). After 48 h, cells were transfected with either a control plasmid (pcDNA), or expression vectors encoding wildtype (wt) Prp3 or the SUMOylation-deficient mutant Prp3 2KR. After 72 h, protein lysates were subjected to western blot with an anti-Prp3 antibody and visualized with an Odyssey Imaging System (A). Total RNA was extracted and the splicing efficiency (the ratio of mRNA over pre-mRNA + mRNA) of ACTB, GAPDH, RSP9, RPS26, Akt, TBP, HPRT1 and HSPCB was determined by RT-qPCR (B). (C) HeLa cells were transfected with either a control plasmid (pcDNA) or expression vectors encoding wildtype Prp3 or the SUMOylation-deficient mutant Prp3 2KR. After 48 h, ChIP analysis was performed against the T7-Prp3 variants with anti-T7 antibodies. Quantification of immunoprecipitated intronic DNA regions was assessed by qPCR with specific primers for introns corresponding to the indicated, actively transcribed genes. Data are represented as mean ± S.E. (n = 3, *P < 0.05; **P ≤ 0.01; ***P ≤ 0.001; Student's t test). (D) Western blot for input and IP of the T7-Prp3 variants subjected to ChIP analysis, visualized by ECL reagent.
To provide additional evidence for an effect of the 2KR mutation on Prp3 function during splicing, we assessed the recruitment of either T7-Prp3 wt or T7-Prp3 2KR to chromatin where co-transcriptional splicing occurs (52). For this purpose, we performed a chromatin immunoprecipation (ChIP) experiment with an anti-T7 antibody, and measured by qPCR the amount of precipitated intronic sequences of several actively transcribed genes. This reflects the extent of Prp3 recruitment to spliceosomes that cotranscriptionally catalyze the splicing of the corresponding pre-mRNAs. In agreement with the results presented above, significantly less of the SUMOylation-deficient T7-Prp3 was associated with chromatin in all of the assayed genes, as compared to the T7-Prp3 wt protein (Figure 6C), even though similar expression levels of wt and 2KR T7-Prp3 were observed (Figure 6D). As Prp3 is recruited to the spliceosome solely as part of the U4/U6•U5 tri-snRNP, these results are consistent with the idea that the lack of SUMOylation of the Prp3 2KR protein leads to impaired tri-snRNP formation.
DISCUSSION
In this study, we have investigated the conjugation of SUMO to spliceosomal proteins both in an in vitro splicing system, as well as in cultured human cells. By performing in vitro splicing, we found that splicing efficiency was compromised when SUMOylation was severely reduced by adding recombinant SENP1. Addition of Ubc9 had no detectable effect, in agreement with the fact that under splicing conditions there was extensive SUMOylation in nuclear extracts, even in the absence of added Ubc9 (Figure 1). These results clearly show that SUMOylation of one or more spliceosomal proteins is required for efficient splicing in vitro.
Affinity purification of spliceosomal complexes formed at different stages of an in vitro splicing reaction, followed by immunoprecipitation with an anti-SUMO2 antibody and subsequent MS analysis, indicated that a variety of proteins present in spliceosomes are SUMOylated (Table 1 and Supplementary Table S1). These included splicing essential proteins of the U2 snRNP (SF3B1, SF3B3, SF3A1, SF3A2, SF3A3), which aid in the proper and stable recruitment of the U2 snRNP to the pre-mRNA branch site (53). In addition, the U4/U6 proteins Prp3 and Prp31, which are both required for tri-snRNP formation were also identified as likely SUMO conjugation substrates. Finally, the functionally important U5 proteins Brr2, an RNA helicase required for spliceosome activation (54), the GTPase Snu114 (55), and the catalytic core scaffold protein Prp8, were identified as likely SUMO substrates. We subsequently confirmed in cultured cells that several of them are indeed SUMOylated (Figure 3). It is possible that SUMO conjugation regulates the activity and/or the interaction of these proteins with other spliceosomal components. Indeed, as discussed below, inhibition of Prp3 SUMOylation via mutagenesis of its SUMO conjugation sites appears to prevent the interaction of the U4/U6 di-snRNP with U5 to form the tri-snRNP, leading to reduced splicing efficiency. Many of the SUMOylated proteins identified in in vitro assembled spliceosomes were detected in previous proteomic studies of SUMOylated proteins (Supplementary Table S2). However, it was not clear from those studies whether they are present in a SUMOylated state within the spliceosome, as our studies now reveal.
Upon close inspection of the different SUMO substrates identified by MS, we were able to find lysine residues within established SUMO consensus sequences, pointing to putative SUMO attachment sites. By site-specific mutagenesis, we obtained a mutant version of the spliceosomal protein Prp3 that is unable to conjugate to SUMO without affecting its conjugation to Ubiquitin. Prp3 is a component of the U4/U6 di-snRNP and also of the U4/U6•U5 tri-snRNP, and is recruited to the spliceosome at the B complex stage (56). Upon spliceosome activation (i.e. Bact formation), Prp3 is displaced together with the U4 snRNA (56). The fact that interactions of this mutant with U4 and U6 snRNAs, as well as with the U4/U6 snRNP protein 60K/Prp4, do not seem to be significantly affected (Figure 5) suggests that the U4/U6 di-snRNP is still properly assembled in the presence of the Prp3 SUMOylation-deficient mutant. However, this mutant displayed diminished interaction with U2 and U5 snRNP proteins, as well as with U2 and U5 snRNAs (Figure 5). This indicates that SUMOylation of Prp3 may be required for tri-snRNP formation and/or for the proper recruitment of U4/U6 di-snRNP as part of the tri-snRNP to active spliceosomes. SUMOylation/de-SUMOylation cycles might modulate interactions of Prp3 with U5 snRNP proteins, first stabilizing the U4/U6 interaction with U5, and subsequently allowing for U4/Prp3 release during spliceosome activation. Given the fact that only a small fraction of Prp3 appears to be SUMOylated at a given time, as indicated by the fact that in general no SUMO-conjugates are observed in input fractions of our anti-T7 Prp3 western blot assays (Figure 3C), and as already reported for most targets of this particular post-translational modification (17,19,57), it is not clear whether the attachment of SUMO is required only transiently to establish and/or stabilize Prp3 interactions (e.g. those needed for tri-snRNP formation), or throughout the splicing cycle.
Pre-mRNA splicing occurs simultaneously with transcription (52) and thus we assessed by ChIP analysis whether the SUMOylation-deficient Prp3 mutant is recruited as efficiently as the wild type, transfected Prp3 protein to chromatin. Consistent with SUMOylation facilitating Prp3 recruitment to active spliceosomes, ChIP analyses of various genes revealed reduced levels of Prp3 2KR associated with chromatin. Our data showing that upon depletion of endogenous Prp3, overexpression of the Prp3 2KR mutant does not restore splicing of a variety of pre-mRNAs to the levels achieved by overexpression of the wild type protein, are also consistent with this conclusion (Figure 6). We observed differential effects of Prp3 knockdown and overexpression of wild type and SUMOylation-deficient Prp3 on the splicing of several pre-mRNAs in cells. This is consistent with recent reports showing that depletion, mutation or even drug-mediated inactivation of core spliceosomal components do not necessarily lead to a general inhibition of the splicing process, but instead have differential effects on different splicing events (58,59). This seems to arise due to the differential sensitivity of individual pre-mRNAs for splicing factors.
The experimental approach for determining the relevance of SUMO conjugation to specific substrates has always been the mutation of Lys residues, putative targets of SUMOylation, to Arg. Even though the fact that SUMOylation is decreased to the same extent after mutating acidic residues within the canonical SUMO consensus sequence suggests that the mutated Lys residues are bona fide SUMOylation sites, there is still a possibility that these mutations affect protein folding, which could decrease the stability and/or functionality of the mutants. In the case of Prp3, it has been demonstrated that its C-terminal region, including the DUF1115 domain, binds regions of the U4/U6 di-snRNA, including the 3΄ end of U6 (13). According to our RIP experiment (Figure 5), the K289/559R mutations do not seem to affect Prp3 binding to U4 or U6 snRNAs. This is consistent with previous studies showing that single mutations within the C-terminal domain do not have serious consequences for Prp3 binding to the U4/U6 snRNA duplex (13). The 2KR mutation also did not appear to affect the binding of Prp3's direct interaction partner in the U4/U6 snRNP, Prp4. There is also no indication of reduced Prp3 2KR levels in the transfected cells, as evidenced by western blotting (Figure 4). Taken together, these results suggest that the 2KR mutation does not significantly affect the folding nor the stability of Prp3.
Prp3 was previously reported to be ubiquitylated (40). Studies by Song et al. showed that ubiquitylated Prp3 has a higher affinity for the U5 snRNP component Prp8, which stabilizes the interaction of U4/U6 with U5, and thus the U4/U6•U5 tri-snRNP. Prp3 is ubiquitylated by the Prp19 complex and de-ubiquitylated by Usp4Sart3, which might facilitate the release of Prp3 together with the U4 snRNA from the spliceosome during maturation of its active site (40). Thus, as with lack of SUMOylation, it is likely that lack of ubiquitylation of Prp3 leads to a reduction in tri-snRNP formation, which in turn inhibits the formation of precatalytic spliceosomes. Even though ubiquitylation sites in Prp3 have not been yet identified, our current results indicate that Lys289 and Lys559 are not target residues for this modification. However, a possible cross-talk between both post-translational modifications cannot be ruled out and warrants further investigation.
SUMOylation is an attractive mechanism to aid in the structural rearrangements of the spliceosome. As this PTM often alters protein interactions, it is possible that its attachment or removal from splicing factors could trigger the changes in the composition and structure of the spliceosome observed at nearly all stages of the splicing reaction (5). Moreover, the recycling of spliceosomal components after a completed round of splicing would require reversibility of this protein modification, which could be easily achieved by SENPs. An emerging concept in the SUMO field is ‘group SUMOylation’, which refers to the requirement for simultaneous modification of multiple targets involved in the same biological process (50), which could be the case for spliceosomal proteins. Thus, it is tempting to suggest that altering SUMO conjugation to several spliceosomal proteins, in addition to the one analyzed in detail in this study, may have even more drastic consequences for the splicing process. Furthermore, it has been shown that the spliceosome is modulated by ubiquitylation (40,60), phosphorylation and acetylation (61,62). However, hardly anything is known about the regulation of proteins involved in the splicing process by SUMO conjugation. This is indeed curious considering that RNA-related proteins are the most abundant group among SUMOylation substrates (34–36). On the other hand, even in those cases where the targets for PTMs are known, the enzymes responsible for these modifications have rarely been characterized. Our identification of the splicing factor SRSF1 as a new component of the SUMOylation pathway (15) also provides an attractive link between SUMO and the splicing machinery. SRSF1 not only displays certain characteristics of SUMO E3 ligases but also affects the activity of a known member of this latter group, PIAS1, which has interestingly been found to co-purify with the spliceosome (39). In this study, we have shown that SRSF1 affects SUMOylation levels of certain spliceosomal proteins (Figure 3), indicating that, besides its other mechanisms of action, SRSF1 may also regulate the splicing process by affecting SUMO conjugation.
Deciphering splicing at the molecular level is not only important for understanding the regulation of gene expression, but it is also of medical relevance, as aberrant pre-mRNA splicing is the basis of many human diseases or contributes to their severity (63,64). Furthermore, increasing experimental evidence points to a link between alterations and/or mutations in core spliceosomal components and specific pathological conditions (65–68). It is interesting to note that Prp3 and Prp8 are mutated in a familial form of retinitis pigmentosa (69,70). It is tempting to speculate that misregulation of the spliceosome, caused by mutations that could interfere with SUMOylation of spliceosomal protein components, might also contribute to human disease.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Federico Pelisch for inspiring ideas and encouraging discussion, Kuan-Ting Pan for help with the MS analysis, Valeria Buggiano, Gabi Heyne and Monika Raabe for technical help, Anzhalika Sidarovich for help in setting up in vitro splicing assays, and Alberto Kornblihtt and members of his laboratory for helpful discussion.
Author contributions: B.P. was a Bunge Born/Max Planck travelling fellowship beneficiary; B.P., P.M. and L.B. are recipients of doctoral fellowships from the Consejo Nacional de Investigaciones Científicas y Técnicas de Argentina (CONICET) and G.R. was a postdoctoral fellow from the same institution. Cindy L. Will, Henning Urlaub and Reinhard Lührmann are Max Planck investigators and Anabella Srebrow is a career investigator from CONICET.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agencia Nacional de Investigaciones Científicas y Tecnológicas of Argentina (ANPCyT) [2012-0136, 2014-2888 to A.S.]; University of Buenos Aires, Argentina (UBACyT) [20020130100157BA to A.S.]; European Alternative Splicing Network (EURASNET) to (R.L. and A.S.); Deutsche Forschungsgemeinschaft (DFG) [LU294/15-1 to R.L.]. The open access publication charge for this paper has been waived by Oxford University Press - NAR.
Conflict of interest statement. None declared.
REFERENCES
- 1. Will C.L., Lührmann R.. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011; 3:a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fica S.M., Tuttle N., Novak T., Li N.S., Lu J., Koodathingal P., Dai Q., Staley J.P., Piccirilli J.A.. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013; 503:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anokhina M., Bessonov S., Miao Z., Westhof E., Hartmuth K., Lührmann R.. RNA structure analysis of human spliceosomes reveals a compact 3D arrangement of snRNAs at the catalytic core. EMBO J. 2013; 32:2804–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galej W.P., Oubridge C., Newman A.J., Nagai K.. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013; 493:638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wahl M.C., Will C.L., Lührmann R.. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009; 136:701–718. [DOI] [PubMed] [Google Scholar]
- 6. Raghunathan P.L., Guthrie C.. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 1998; 8:847–855. [DOI] [PubMed] [Google Scholar]
- 7. Bell M., Schreiner S., Damianov A., Reddy R., Bindereif A.. p110, a novel human U6 snRNP protein and U4/U6 snRNP recycling factor. EMBO J. 2002; 21:2724–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raghunathan P.L., Guthrie C.. A spliceosomal recycling factor that reanneals U4 and U6 small nuclear ribonucleoprotein particles. Science. 1998; 279:857–860. [DOI] [PubMed] [Google Scholar]
- 9. Anthony J.G., Weidenhammer E.M., Woolford J.L. Jr. The yeast Prp3 protein is a U4/U6 snRNP protein necessary for integrity of the U4/U6 snRNP and the U4/U6.U5 tri-snRNP. RNA. 1997; 3:1143–1152. [PMC free article] [PubMed] [Google Scholar]
- 10. Ayadi L., Callebaut I., Saguez C., Villa T., Mornon J.P., Banroques J.. Functional and structural characterization of the prp3 binding domain of the yeast prp4 splicing factor. J. Mol. Biol. 1998; 284:673–687. [DOI] [PubMed] [Google Scholar]
- 11. Gonzalez-Santos J.M., Wang A., Jones J., Ushida C., Liu J., Hu J.. Central region of the human splicing factor Hprp3p interacts with Hprp4p. J. Biol. Chem. 2002; 277:23764–23772. [DOI] [PubMed] [Google Scholar]
- 12. Liu S., Rauhut R., Vornlocher H.P., Lührmann R.. The network of protein-protein interactions within the human U4/U6.U5 tri-snRNP. RNA. 2006; 12:1418–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu S., Mozaffari-Jovin S., Wollenhaupt J., Santos K.F., Theuser M., Dunin-Horkawicz S., Fabrizio P., Bujnicki J.M., Lührmann R., Wahl M.C.. A composite double-/single-stranded RNA-binding region in protein Prp3 supports tri-snRNP stability and splicing. Elife. 2015; 4:e07320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Long J.C., Caceres J.F.. The SR protein family of splicing factors: master regulators of gene expression. Biochem. J. 2009; 417:15–27. [DOI] [PubMed] [Google Scholar]
- 15. Pelisch F., Gerez J., Druker J., Schor I.E., Munoz M.J., Risso G., Petrillo E., Westman B.J., Lamond A.I., Arzt E. et al. . The serine/arginine-rich protein SF2/ASF regulates protein sumoylation. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:16119–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Becker J., Barysch S.V., Karaca S., Dittner C., Hsiao H.H., Berriel Diaz M., Herzig S., Urlaub H., Melchior F.. Detecting endogenous SUMO targets in mammalian cells and tissues. Nat. Struct. Mol. Biol. 2013; 20:525–531. [DOI] [PubMed] [Google Scholar]
- 17. Gareau J.R., Lima C.D.. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010; 11:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Geiss-Friedlander R., Melchior F.. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell. Biol. 2007; 8:947–956. [DOI] [PubMed] [Google Scholar]
- 19. Impens F., Radoshevich L., Cossart P., Ribet D.. Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liang Y.C., Lee C.C., Yao Y.L., Lai C.C., Schmitz M.L., Yang W.M.. SUMO5, a novel poly-SUMO isoform, regulates PML nuclear bodies. Sci. Rep. 2016; 6:26509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matic I., Schimmel J., Hendriks I.A., van Santen M.A., van de Rijke F., van Dam H., Gnad F., Mann M., Vertegaal A.C.. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol. Cell. 2010; 39:641–652. [DOI] [PubMed] [Google Scholar]
- 22. Hickey C.M., Wilson N.R., Hochstrasser M.. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell. Biol. 2012; 13:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jentsch S., Psakhye I.. Control of nuclear activities by substrate-selective and protein-group SUMOylation. Annu. Rev. Genet. 2013; 47:167–186. [DOI] [PubMed] [Google Scholar]
- 24. Geoffroy M.C., Hay R.T.. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat. Rev. Mol. Cell. Biol. 2009; 10:564–568. [DOI] [PubMed] [Google Scholar]
- 25. Johnson P.R., Hochstrasser M.. SUMO-1: Ubiquitin gains weight. Trends Cell Biol. 1997; 7:408–413. [DOI] [PubMed] [Google Scholar]
- 26. Nacerddine K., Lehembre F., Bhaumik M., Artus J., Cohen-Tannoudji M., Babinet C., Pandolfi P.P., Dejean A.. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell. 2005; 9:769–779. [DOI] [PubMed] [Google Scholar]
- 27. Panse V.G., Kressler D., Pauli A., Petfalski E., Gnadig M., Tollervey D., Hurt E.. Formation and nuclear export of preribosomes are functionally linked to the small-ubiquitin-related modifier pathway. Traffic. 2006; 7:1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Westman B.J., Verheggen C., Hutten S., Lam Y.W., Bertrand E., Lamond A.I.. A proteomic screen for nucleolar SUMO targets shows SUMOylation modulates the function of Nop5/Nop58. Mol. Cell. 2010; 39:618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Finkbeiner E., Haindl M., Raman N., Muller S.. SUMO routes ribosome maturation. Nucleus. 2011; 2:527–532. [DOI] [PubMed] [Google Scholar]
- 30. Cubenas-Potts C., Matunis M.J.. SUMO: a multifaceted modifier of chromatin structure and function. Dev. Cell. 2013; 24:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ihara M., Stein P., Schultz R.M.. UBE2I (UBC9), a SUMO-conjugating enzyme, localizes to nuclear speckles and stimulates transcription in mouse oocytes. Biol. Reprod. 2008; 79:906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spector D.L., Lamond A.I.. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 2011; 3:a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schulz S., Chachami G., Kozaczkiewicz L., Winter U., Stankovic-Valentin N., Haas P., Hofmann K., Urlaub H., Ovaa H., Wittbrodt J. et al. . Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep. 2012; 13:930–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blomster H.A., Hietakangas V., Wu J., Kouvonen P., Hautaniemi S., Sistonen L.. Novel proteomics strategy brings insight into the prevalence of SUMO-2 target sites. Mol. Cell. Proteomics. 2009; 8:1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vertegaal A.C., Ogg S.C., Jaffray E., Rodriguez M.S., Hay R.T., Andersen J.S., Mann M., Lamond A.I.. A proteomic study of SUMO-2 target proteins. J. Biol. Chem. 2004; 279:33791–33798. [DOI] [PubMed] [Google Scholar]
- 36. Golebiowski F., Matic I., Tatham M.H., Cole C., Yin Y., Nakamura A., Cox J., Barton G.J., Mann M., Hay R.T.. System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2009; 2:ra24. [DOI] [PubMed] [Google Scholar]
- 37. Vethantham V., Rao N., Manley J.L.. Sumoylation modulates the assembly and activity of the pre-mRNA 3΄ processing complex. Mol. Cell. Biol. 2007; 27:8848–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Desterro J.M., Keegan L.P., Jaffray E., Hay R.T., O’Connell M.A., Carmo-Fonseca M.. SUMO-1 modification alters ADAR1 editing activity. Mol. Biol. Cell. 2005; 16:5115–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rappsilber J., Ryder U., Lamond A.I., Mann M.. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002; 12:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Song E.J., Werner S.L., Neubauer J., Stegmeier F., Aspden J., Rio D., Harper J.W., Elledge S.J., Kirschner M.W., Rape M.. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010; 24:1434–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zillmann M., Zapp M.L., Berget S.M.. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Mol. Cell Biol. 1988; 8:814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dignam J.D., Lebovitz R.M., Roeder R.G.. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983; 11:1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Das R., Reed R.. Resolution of the mammalian E complex and the ATP-dependent spliceosomal complexes on native agarose mini-gels. RNA. 1999; 5:1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blaustein M., Pelisch F., Tanos T., Munoz M.J., Wengier D., Quadrana L., Sanford J.R., Muschietti J.P., Kornblihtt A.R., Caceres J.F. et al. . Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat. Struct. Mol. Biol. 2005; 12:1037–1044. [DOI] [PubMed] [Google Scholar]
- 45. Makarova O.V., Makarov E.M., Urlaub H., Will C.L., Gentzel M., Wilm M., Lührmann R.. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J. 2004; 23:2381–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Will C.L., Urlaub H., Achsel T., Gentzel M., Wilm M., Lührmann R.. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J. 2002; 21:4978–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fabrizio P., Laggerbauer B., Lauber J., Lane W.S., Lührmann R.. An evolutionarily conserved U5 snRNP-specific protein is a GTP-binding factor closely related to the ribosomal translocase EF-2. EMBO J. 1997; 16:4092–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tatham M.H., Rodriguez M.S., Xirodimas D.P., Hay R.T.. Detection of protein SUMOylation in vivo. Nat. Protoc. 2009; 4:1363–1371. [DOI] [PubMed] [Google Scholar]
- 49. Pillai R.S., Will C.L., Lührmann R., Schumperli D., Muller B.. Purified U7 snRNPs lack the Sm proteins D1 and D2 but contain Lsm10, a new 14 kDa Sm D1-like protein. EMBO J. 2001; 20:5470–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Flotho A., Melchior F.. Sumoylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013; 82:357–385. [DOI] [PubMed] [Google Scholar]
- 51. Pelisch F., Pozzi B., Risso G., Munoz M.J., Srebrow A.. DNA damage-induced heterogeneous nuclear ribonucleoprotein K sumoylation regulates p53 transcriptional activation. J. Biol. Chem. 2012; 287:30789–30799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Listerman I., Sapra A.K., Neugebauer K.M.. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat. Struct. Mol. Biol. 2006; 13:815–822. [DOI] [PubMed] [Google Scholar]
- 53. Dybkov O., Will C.L., Deckert J., Behzadnia N., Hartmuth K., Lührmann R.. U2 snRNA-protein contacts in purified human 17S U2 snRNPs and in spliceosomal A and B complexes. Mol. Cell Biol. 2006; 26:2803–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Absmeier E., Wollenhaupt J., Mozaffari-Jovin S., Becke C., Lee C.T., Preussner M., Heyd F., Urlaub H., Lührmann R., Santos K.F. et al. . The large N-terminal region of the Brr2 RNA helicase guides productive spliceosome activation. Genes Dev. 2015; 29:2576–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bartels C., Klatt C., Lührmann R., Fabrizio P.. The ribosomal translocase homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the spliceosome. EMBO Rep. 2002; 3:875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wahl M.C., Lührmann R.. SnapShot: spliceosome dynamics I. Cell. 2015; 161:1474. [DOI] [PubMed] [Google Scholar]
- 57. Hay R.T. SUMO: a history of modification. Mol. Cell. 2005; 18:1–12. [DOI] [PubMed] [Google Scholar]
- 58. Corrionero A., Minana B., Valcarcel J.. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011; 25:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Papasaikas P., Tejedor J.R., Vigevani L., Valcarcel J.. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell. 2015; 57:7–22. [DOI] [PubMed] [Google Scholar]
- 60. Bellare P., Small E.C., Huang X., Wohlschlegel J.A., Staley J.P., Sontheimer E.J.. A role for ubiquitin in the spliceosome assembly pathway. Nat. Struct. Mol. Biol. 2008; 15:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gunderson F.Q., Merkhofer E.C., Johnson T.L.. Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:2004–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M.. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325:834–840. [DOI] [PubMed] [Google Scholar]
- 63. Ward A.J., Cooper T.A.. The pathobiology of splicing. J. Pathol. 2010; 220:152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wahl M.C., Lührmann R.. SnapShot: spliceosome dynamics III. Cell. 2015; 162:690. [DOI] [PubMed] [Google Scholar]
- 65. Bonnal S., Vigevani L., Valcarcel J.. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012; 11:847–859. [DOI] [PubMed] [Google Scholar]
- 66. Mozaffari-Jovin S., Wandersleben T., Santos K.F., Will C.L., Lührmann R., Wahl M.C.. Novel regulatory principles of the spliceosomal Brr2 RNA helicase and links to retinal disease in humans. RNA Biol. 2014; 11:298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ramsay A.J., Rodriguez D., Villamor N., Kwarciak A., Tejedor J.R., Valcarcel J., Lopez-Guillermo A., Martinez-Trillos A., Puente X.S., Campo E. et al. . Frequent somatic mutations in components of the RNA processing machinery in chronic lymphocytic leukemia. Leukemia. 2013; 27:1600–1603. [DOI] [PubMed] [Google Scholar]
- 68. Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R., Sato Y., Sato-Otsubo A., Kon A., Nagasaki M. et al. . Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478:64–69. [DOI] [PubMed] [Google Scholar]
- 69. Chakarova C.F., Hims M.M., Bolz H., Abu-Safieh L., Patel R.J., Papaioannou M.G., Inglehearn C.F., Keen T.J., Willis C., Moore A.T. et al. . Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2002; 11:87–92. [DOI] [PubMed] [Google Scholar]
- 70. Boon K.L., Grainger R.J., Ehsani P., Barrass J.D., Auchynnikava T., Inglehearn C.F., Beggs J.D.. prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast. Nat. Struct. Mol. Biol. 2007; 14:1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.