

Abstract
Eukaryotic genomes show tremendous size variation across taxa. Proximate explanations for genome size variation include differences in ploidy and amounts of noncoding DNA, especially repetitive DNA. Ultimate explanations include selection on physiological correlates of genome size such as cell size, which in turn influence body size, resulting in the often-observed correlation between body size and genome size. In this study, we examined body size and repetitive DNA elements in relationship to the evolution of genome size in North American representatives of a single beetle family, the Lampyridae (fireflies). The 23 species considered represent an excellent study system because of the greater than 5-fold range of genome sizes, documented here using flow cytometry, and the 3-fold range in body size, measured using pronotum width. We also identified common genomic repetitive elements using low-coverage sequencing. We found a positive relationship between genome size and repetitive DNA, particularly retrotransposons. Both genome size and these elements were evolving as expected given phylogenetic relatedness. We also tested whether genome size varied with body size and found no relationship. Together, our results suggest that genome size is evolving neutrally in fireflies.
Keywords: C-value paradox, flow cytometry, Coleoptera, Lampyridae, transposable elements, 454
Introduction
Eukaryotic genome sizes vary widely, from 2.19 Mb in the microsporidian fungus Encephalitozoon romaleae to 148,851.60 Mb in the angiosperm plant Paris japonica (Kullman et al. 2005; Bennet and Leitch 2012), begging the question, “How and why is this variation generated and maintained?” It is now established that much of genome size variation is due to differences in repetitive DNA content, at the genome (whole genome duplication), chromosome (aneuploidy, supernumerary chromosomes), and/or segmental levels (indels, gene duplications/deletions, transposable elements [TEs], satellite DNA) with the relative importance of each varying across taxa (reviewed in Petrov 2001; Gregory 2005). Some general trends have emerged—for example, there is a positive correlation between genome size and TE content across eukaryotes (Elliott and Gregory 2015). However, the specific TEs involved differ across organisms—some genomes show proliferation of a single TE family (i.e. transposon release, e.g. legumes: Macas et al. 2015), while others show expansion of several TE families (e.g. in the grasses: Estep et al. 2013). While TE mobilization is expected to be deleterious due to insertional disruption of genes, the degree to which selection drives genome-size evolution remains a mystery in most taxa.
In addition to natural selection acting on TEs and on genome size, the degree of mutational bias (i.e. the relative rates and sizes of mutations that increase versus decrease genome size), and genetic drift will play a role in genome size evolution. For example, if large genome size has deleterious but small fitness consequences, and/or if mutational bias is weak and in the direction of reduced genome size, then we expect the observed negative correlation between effective population size and genome size across broad taxonomic scales (Lynch and Conery 2003; but see Charlesworth and Barton 2004; Whitney and Garland 2010). In contrast, if the fitness consequences of large genome size are substantial, then selection will be the primary determinant of genome size evolution. For example, there is a strong positive correlation between cell size and genome size in most taxa (reviewed in Gregory 2001). Cell size can influence body size (Arendt 2007), which is one of the most ecologically important traits of an organism (Peters 1986). Thus, genome size variation may be a target of strong selection through its indirect effects on body size, itself correlated with effective population size. Furthermore, metabolic and developmental correlates of genome size have also been described (reviewed in Gregory 2005; Ellis et al. 2014; Arnqvist et al. 2015). For example, genome size in birds is negatively correlated with both flight muscle size and heart size, perhaps causally through effects on flight metabolism (Wright et al. 2014). Genome size also correlates with developmental rate and complexity in many taxa (e.g. Drosophila: Gregory and Johnston 2008; mosquitos: Ferrari and Rai 1989; ladybird beetles: Gregory et al. 2003), although such correlations are not always present (Juan and Petitpierre 1991; Gregory et al. 2003). In addition, if mutational bias is strong, then it will be a major determinant of genome size evolution (Petrov 2002).
Given the many forces that can play a role in genome size evolution, a complete understanding requires documentation of genome sizes, measurements of likely selective correlates such as body size, estimates of the strength of genetic drift, measurement of mutational biases, and characterization of genomic content across species in a phylogenetic framework. There are relatively few animal systems, particularly insects, in which all of these factors have been estimated. To begin to address this gap, we present results on genome size evolution in the beetle family Lampyridae, the fireflies, in which we investigate three of these factors. Worldwide, there are over 2,000 firefly species perhaps best known for the production of nocturnal lighted mating displays. In addition to large variation in body size, fireflies also vary in physiology, which allows testing if metabolic rates are correlated with genome sizes: a negative relationship has been seen in birds (Wright et al. 2014). In lighted species, flying males emit flash signals to females in the vegetation (Lloyd 1966). In contrast to lighted species, some fireflies are unlighted—they have lost light, are diurnal, and use long-distance pheromones to find mates (e.g. Phosphaenus hemipterus:De Cock and Matthysen 2005). If the energetic costs of signal production or searching for mates are consistently different for lighted versus dark fireflies, then lighted species may have a higher metabolic rate and smaller genome size than dark species. Flying insects have higher resting metabolic rates and are thus expected to have relatively small genome sizes (Reinhold 1999). In some firefly species, females lack or have reduced wings, eliminating their ability to fly (reviewed in South et al. 2011), therefore firefly species in which both sexes fly might be under stronger selection for smaller genomes as compared to species with flightless females. Importantly, a molecular phylogeny is available to investigate the relationship between these potential selective correlates and genome size in a comparative approach (Stanger-Hall et al. 2007; Sander and Hall 2015; Stanger-Hall and Lloyd 2015).
In this study, we used flow cytometry to determine the genome size for over 20 North American firefly species and found that genome size varies substantially, over 5-fold across species. We used comparative methods to test for neutral evolution of genome size. In addition, we performed low-coverage sequencing to identify common repetitive DNA sequences that most contribute to genome size variation (e.g. Macas et al. 2007; Tenaillon et al. 2011; supplementary file 1 S6.2). Finally, we quantified body size, allowing us to test for selection on genome size acting through body size, light production, and female wing reduction. In all of these analyses we used a molecular phylogeny to correct for nonindependence of related species (Felsenstein 1985).
Materials and Methods
Specimen Collection and Species Identification for Flow Cytometry
Adult specimens were collected from natural populations (table 1) and kept alive in 50 ml plastic conical tubes containing a piece of damp paper towel to retain moisture. Upon return to the laboratory, specimens were flash-frozen in liquid nitrogen and individual heads were harvested for flow cytometry. Specimens were identified to species and sex in the field using flash pattern and morphology, and males verified in the laboratory using genital morphology (Fender 1966; Green 1956, 1957; Luk et al. 2011). Both male and female species identification were molecularly confirmed using 376 bp of the mitochondrial cytochrome oxidase I (COI) locus (primers HCO, LCO; Stanger-Hall et al. 2007). Sequences were aligned with MUSCLE (Edgar 2004) in Geneious R7 (Biomatters Ltd.) along with reference sequences from voucher specimens (Stanger-Hall et al. 2007; Stanger-Hall and Lloyd 2015; Sander and Hall 2015) and used to create a neighbor-joining phylogeny. Species identity was considered validated if the specimen grouped with its corresponding voucher in the phylogeny. Where species identification was ambiguous, an additional 895 bp of the COI locus (primers 2183, 3014) were sequenced as well as two nuclear loci: 594 bp of rudimentary (CAD; primers CD806F, CD1098R2), and 420 bp of wingless (WG; primers Wg550F, WgAbRZ; for additional primer information and PCR amplification parameters see Stanger-Hall and Lloyd 2015). For small specimens, whole bodies were retained for flow cytometry to reduce the chance of freeze-drying the neural tissue, which renders it unusable; in these cases, molecular data from proxy specimens were used to verify the field identification. Proxy specimens were caught at the same location as flow cytometry specimens, typically on the same night. Bodies of large specimens and all proxies are retained in the KSH collection at the University of Georgia.
Table 1.
Collection Dates and Localities of Specimens Used in Genome Size Estimation
| Genus | Species | N a | State(s) Collected | Date(s) |
|---|---|---|---|---|
| Ellychnia | corrusca | 1 | PA | June 2012 |
| Lucidota | atra | 5(5) | IL, OH, PA, TN | June 2012 |
| punctata | 2 | PA, TN | June 2012 | |
| Phausis | reticulata | 2 | TN | June 2011 |
| sp. WATb | 2(5) | GA | March 2012 | |
| Photinus | australis | 5 | GA | June 2011 |
| brimleyi | 2 | TN | June 2012 | |
| carolinus | 7(5) | GA, PA | June 2011, June 2012 | |
| cooki | (1) | TN | June 2012 | |
| curtatus | 5(2) | IL, OH | June 2012 | |
| indictus | 2 | PA | June 2012 | |
| macdermotti | 10(6) | GA, PA | June 2011, April 2012, June 2012 | |
| marginellus | 6(5) | TN, PA | June 2012, July 2012 | |
| obscurellus | 3 | PA | June 2012, July 2012 | |
| pyralis | 4(5) | GA, MO, MS, TN | June 2011, May 2012, June 2012 | |
| sabulosus | 3 | OH | June 2012 | |
| scintillans | 6(1) | PA | June 2012 | |
| Photuris | frontalis | 5 | GA | June 2011 |
| multiple sp. | 17(8)c | IL, IN, MO, MS, PA, TN | June 2012 | |
| Pyractomena | angulata | 5 | IN, MO | June 2012 |
| borealis | 5 | GA | March 2012 | |
| marginalis | 2 | PA | June 2012 | |
| Pyropyga | decipiens | 4(5) | PA | June 2012 |
Total number of males (females) per species.
These specimens were treated as a separate species from Phausis reticulata due to morphological and molecular differences (see supplementary material S3.4, Supplementary Material online for details).
A single unknown Photuris with the largest measured genome size was used in further analysis (details in supplementary material S3.6, Supplementary Material online).
Flow Cytometry
Genome size estimates were obtained by flow cytometric determination of relative fluorescence of propidium iodide stained nuclei after Hanrahan and Johnston (2011). In brief, one half of the head of a firefly was placed into 1 ml of Galbraith buffer and coprepared in a 2 ml Kontes Dounce tissue grinder along with two internal standards, the head of a female Drosophila virilis (1C = 328 Mb) and 1/3 of the head of a male Periplaneta americana (1C = 3,338 Mb). Nuclei from the sample and standards were released by 15 strokes of the “B” (loose) pestle at a rate of 1.5 strokes/second. The released nuclei in solution were filtered through a 42-µm nylon filter, and stained for at least one hour in the cold and dark with 25 µg/ml of propidium iodide (PI). The mean relative red (PI) fluorescence of the 2C nuclei of the two standards and of the sample were scored as channel numbers using a Partec CyFlow flow cytometer equipped with a solid-state laser emitting at 532 nm. To increase the precision of estimates, each individual was re-measured using an independent preparation of the remaining head tissue. At least 1,000 nuclei were scored under each 2C peak with the CV of all 2C peaks <2.5. Genome size was estimated as the ratio of the mean 2C channel number of the sample divided by the mean 2C channel number of the standard times the 1C amount of DNA in the standard. A total of four values were produced for each individual, with the two values based on the different standards and two independent sets of scores for each individual. The estimated 1C genome size for each sample was the average of these four measures. Between 1 and 17 individuals of each sex were scored for genome size in field-identified species (N >23 species; table 1). Mean estimates for each individual measured are reported in the Supplementary Material online.
Statistical Analyses of Genome Size
The variance in genome size estimates across taxonomic levels was analyzed using standard least squares in JMP Pro 10 (SAS Institute Inc. 2012) with restricted maximum likelihood to account for differences in sample size. The full model included the effects of genus, species nested within genus, and sex nested within species and genus. Subsequently, Student’s t-tests were used to test for sex differences in seven species that had estimates for at least two individuals of each sex (Lucidota atra, Phausis sp. WAT, Photinus carolinus, Photinus curtatus, Photinus macdermotti, Photinus marginellus, and Photinus pyralis). Significance levels were adjusted to control the false discovery rate (FDR) using the Benjamini–Hochberg correction for multiple comparisons (Benjamini and Hochberg 1995).
454 Sequencing
To identify abundant repetitive elements that might account for variation in genome size, low-coverage genomic sequencing was performed on 21 individuals, representing 20 species, and 7 genera (supplementary material S1.1, Supplementary Material online). The proportion of repetitive elements in the sample should reflect their abundance in the genome if sequencing is unbiased (Macas et al. 2007; Swaminathan et al. 2007). Genomic DNA was isolated from thorax or whole body of single specimens using phenol–chloroform extraction with RNAse digestion. Sequencing libraries for each specimen were uniquely barcoded and then all libraries pooled into two lanes of 454 FLX Titanium XLR70 (Roche Diagnostics Corporation). Library preparation, sample barcoding, sequencing, and demultiplexing were performed at the Georgia Genomics Facility (Athens, GA).
Sequences were assessed for quality using fastqc v. 0.11.2 (Babraham Bioinformatics 2012) and subsequently trimmed for adapters and low-quality regions using the fastq-mcf program in ea-utils v. 1.1.2 (parameters: -q 20 -p 10 –D1 5 -x 0.01 -w 20; Aronesty 2013). Seqtk v. 1.0 (https://github.com/lh3/seqtk) was used to trim 19 bases from the beginning of each read due to skewed base distributions and PCR duplicates were collapsed using the fastx toolkit v. 0.0.13.2 (http://hannonlab.cshl.edu/fastx_toolkit). Mitochondrial sequences were identified using BLASTn (e-value = 1e−6; Altschul et al. 1990) of collapsed reads against a database of complete mitochondrial genomes from Elateroid beetles, including fireflies, and removed (supplementary material S1.2, Supplementary Material online). Prokaryotic contaminants were identified and removed using kraken v. 0.10.5 using a minikraken kmer library constructed from all RefSeq bacteria, archaea, plasmids, and virus sequences filtered for repetitive sequences using the BLAST + dustmasker (Wood and Salzberg 2014). Finally, all reads less than 80 bp were removed to increase the efficiency of repetitive element identification and assembly.
Repetitive Element Identification and Classification
Repetitive elements were identified using the RepeatExplorer Galaxy server with default parameters (Novák et al. 2013). RepeatExplorer identifies repetitive elements de novo using a graph-based method to group reads into discrete clusters based on all-by-all blast similarity. It then annotates clusters using RepeatMasker (Smit et al. 2013–2015) using all or a subset of RepeatMasker databases and then assembles contigs from the reads belonging to each cluster using CAP3 (parameters: -O -p 80 -o 40; Huang and Madan 1999; Novák et al. 2010). To be inclusive, we used all of the RepeatMasker databases during annotation. We only annotated clusters consisting of at least 20 reads, which we term “top” clusters. This cut-off was low enough to fully capture highly abundant repeats in all species (supplementary material S1.3, Supplementary Material online), while remaining computationally tractable. Remaining clusters are “bottom” clusters and were not annotated using RepeatMasker (supplementary material S1.4, Supplementary Material online). All sequences across all species were pooled for clustering analysis to effectively increase our sequencing coverage across shared repeats.
Both top and bottom clusters were screened for contaminants by blasting assembled contigs against the NCBI nucleotide database (e-value: 1e−5) and excluding clusters with contigs that had high quality hits to mitochondrial, microbial, or human sequences (high quality = hits over 100 bp that were also over 50% of either the query or subject length). At least 60% identity was required to exclude mitochondrial and microbial contaminants, while at least 90% was required to exclude human sequences. Finally, the top clusters that cumulatively accounted for at least 50% of top cluster abundance within each species were manually curated using visual inspection of the RepeatExplorer assembled contigs, tblastx (default parameters) of contigs against the NCBI nt/nr database to identify conserved domains, and Tandem Repeats Finder (default parameters; Benson 1999). For the smallest data set, Photinus pyralis, all clusters were manually curated following the above procedure.
Top clusters were assigned to one of 10 repeat categories based on RepeatExplorer and manual annotations: 1) long terminal repeat (LTR), 2) long interspersed nuclear element (LINE), 3) DNA TE, 4) rolling circle TE, 5) low complexity repeat, 6) simple repeat (short repeats of less than 20 bp), 7) tandem repeat (large repeats of more than 20 bp), 8) histone gene, 9) ribosomal gene, and 10) unknown repeat (no annotation) (Kapitonov and Jurka 2008; Wicker et al. 2007). An 11th category comprised the sum of the bottom clusters. To examine patterns on a broader scale, we also performed analyses after grouping some categories into three groups: Class I TEs, or retrotransposons (categories 1 and 2), Class II DNA TEs (categories 3 and 4), and repeats (categories 5–9).
Validation of Low-Coverage Sequencing Approach
We investigated the effect of low-coverage sampling on our estimates of genomic repetitiveness for the seven species with the most data by randomly subsampling sequences to 0.01, 0.03, and 0.05× (without replacement) and then performing RepeatExplorer analysis on the individual data sets. Five replicates per species per coverage level were generated to examine the repeatability of estimates.
Low-coverage sequencing with blast-based de novo repeat identification is expected to underestimate true genomic repetitiveness due to not detecting low-copy number repeats. To assess how much true repetitiveness is underestimated, we simulated 454 sequences from the T. castaneum reference genome v 5.2 (NCBI: GCA_000002335.3) using our empirical read length distribution in ART v 2.6.0 (Huang et al. 2012). Data sets were generated at 0.02, 0.04, 0.06, 0.08, and 0.1× coverage (five replicates each) and used in RepeatExplorer analysis. Resulting repetitiveness estimates were compared with expected estimates developed using the distribution of copy numbers of annotated repetitive elements in the T. castaneum genome (Wang et al. 2008) at 0.01× to 0.1× sequencing depths. For a nucleotide position in an element to be identified as part of a repeat, we assumed that it must have 95% or greater probability of being detected in two or more sequencing reads and used the Poisson distribution to calculate the repeat abundance at which an element would be detected under this criterion (see supplementary material S6.1, Supplementary Material online for details).
Morphological Measurements
Body size measurements were obtained from ethanol-preserved adult specimens in the KSH collection at the University of Georgia. Individuals were first photographed on 1 mm grids, and then five morphological size characters (pronotum length, width, area, and elytron length and body length; supplementary material S2, Supplementary Material online) were measured from the images using ImageJ v. 1.42 (Schneider et al. 2012). Specimens used in size measurements were not the same as those used in flow cytometry due to differences in storage requirements for downstream processes. All photographed specimens were identified to species using morphology, flash behavior, and molecular methods (when necessary). All five size measurements were highly correlated (Pearson’s r > 0.8) and exhibited essentially identical loadings on the first principal component axis that accounted for 90% of the variance. For this reason, a single measure could be used for size. We chose pronotum width because it is robust to variation in adult nutrition and has been used in previous work (Vencl 2004). Where possible, size measures were obtained from at least three males and three females per species. Across species, there were significant differences in pronotum width between the sexes (two-tailed t-test: P = 0.017), though male and female measurements were highly correlated (b = 0.67, R2 = 0.88, P = <0.0001). Subsequent analyses were performed on male measurements only.
Data on presence/absence of adult light (scored as presence versus absence of adult light organ) and female wing reduction (full-size versus reduced elytra) were gathered from specimens in the KSH collection, the literature (Green 1956, 1957), and field observations. Presence versus absence of adult light and reduced elytra in females were coded as binary variables to test for correlations with genome size.
Phylogeny
Evolutionary relationships among species were reconstructed by extending the Photinus phylogeny of Stanger-Hall and Lloyd (2015) to include 10 additional taxa in 5 genera. For this purpose, representative specimens from each species were sequenced at the three loci cited above, aligned with the Stanger-Hall and Lloyd data set using MUSCLE (Edgar 2004) in Geneious R7 (Biomatters Ltd.), and manually reviewed. jModeltest2 (Darriba et al. 2012) was used to select an appropriate model of evolution for each locus (WG: K80 + I + G, CAD: TIM3 + I + G, COI: GTR + I+G). Phylogenies were constructed in BEAST v. 1.8 (Drummond et al. 2012) using an uncorrelated lognormal clock model to account for rate variation among lineages. BEAST was run twice for 30 million generations each with 25% burn-in, until the estimated sample size for all parameters was over 200. Independent runs were assessed for convergence using Tracer and the majority-rule consensus tree produced in TreeAnnotater. The final tree was trimmed to include only those taxa used in this study.
Phylogenetic Analysis
Patterns in genome size evolution were examined by estimating Pagel’s three parameters (Pagel 1999) using the pgls function in the caper package (Orme 2013) in R 3.0.2 (R Core Team 2013). The first parameter, λ, is a measure of phylogenetic dependence of trait covariances. A λ of 0 implies no phylogenetic dependence, while a λ of 1 indicates complete dependence based on a Brownian motion model. The second parameter, δ, ranges from 0 to 3 and measures the rate of evolution along shared branches in the phylogeny, with values below 1 suggesting that changes early in the phylogeny contribute more to trait evolution, whereas values above 1 suggest changes later in the phylogeny, towards tips. The third parameter, κ, ranges from 0 to 3 and measures where on average changes occur on individual branches, with values close to 0 indicating that changes happen early, i.e. immediately following speciation events (punctuated evolution). Measures of both δ and κ equal to 1 are consistent with gradual evolution. λ was also estimated for body size and repeat category/group abundance to examine their degree of phylogenetic dependence. To identify where in the phylogeny genome size changed we reconstructed ancestral states using the ape package v. 3.0-11 (Paradis et al. 2004) in R.
Subsequent analyses of correlations between genome size and explanatory variables accounted for relatedness between species using phylogenetic generalized least squares (PGLS) in the R package nlme (Pinheiro et al. 2016). The mean genome sizes of males and females were averaged to obtain an estimate for the species. Mean genome size was then log transformed for statistical adequacy and to conform to assumptions of Brownian motion (Quader et al. 2004). If a repeat category/group is a cause of genome size variation then the proportion of that category/group should increase with genome size. As sequencing coverage is expected to affect the estimated abundance of repeats in our samples, we performed comparative analysis on a RepeatExplorer dataset generated from a subset of taxa (N = 18), subsampled to 0.01× coverage for each taxon, with clusters annotated by transferring annotations from the curated dataset (N = 21 taxa, supplementary materials S1.5–S1.9, Supplementary Material online) and adding any new annotations from the reduced, uniform coverage analysis. We also performed analysis with and without an outlier taxon (Photinus obscurellus). A complete table of all traits measured is included in additional files available via figshare (https://doi.org/10.6084/m9.figshare.5077843.v1).
Results
Genome Size Varies over 5-Fold across North American Firefly Species
Estimates obtained from a total of 151 specimens of 23 species across seven genera showed that genome size varies over 5-fold across North American lampyrid species (range: 433–2,572 Mb; fig. 1; supplementary material S3.1, Supplementary Material online). Approximately 72% of the genome size variation occurred at the genus level, 28% at the species level, and there was a significant effect of sex (P < 0.0001, supplementary material S3.2, Supplementary Material online). Females had significantly larger genomes than males in four of the seven species for which we had at least two replicates of each sex: Pn.curtatus (P = 0.0017), Pn.macdermotti (P = 0.0002), Pn.marginellus (P = 0.0003), and Phausis sp. WAT (P = 0.0084) and marginally significant in one additional species, Pn.pyralis (P = 0.023, Benjamini-Hochberg FDR correction P = 0.017; supplementary material S3.3, Supplementary Material online). In these four species female genomes are approximately 5% larger than males. As fireflies have X0 sex determination (Dias et al. 2007), these data suggest that the X chromosome is ∼5% of the genome.
Fig. 1.

—Genome size varies over 5-fold across 23 North American firefly species. Genome size ranges from 433 Mb (Photinus pyralis) to 2572 Mb (Photuris sp.). Left: a molecular phylogeny generated from one mitochondrial and two nuclear loci with branch lengths proportional to relative time. Middle: means and standard deviations for nuclear genome size estimates (Mb) of males (filled circles) and females (empty circles) of each species. M, F: the sample sizes for males and females, respectively. Where bars are not visible and multiple individuals were measured, the standard deviations are entirely covered by the mean circles. L: lighted (empty squares) and unlighted (filled squares) species; WF: species with winged females (empty triangles) and females with reduced wings (filled triangles).
While most within-species genome size variation could be attributed to sex, there was a large difference in genome size among specimens of Pyropyga decipiens, with individuals clustering into one of two genome sizes, small, 699 Mb, and large, 1,079 Mb. Because there were no observable morphological or genetic differences between genome size types, it was not possible to distinguish them (supplementary materials S3.1 and S3.6, Supplementary Material online). Accordingly, we excluded Pg. decipiens from the comparative analyses involving morphological measurements and treated individuals of each genome size type as separate lineages in the repeat analysis. In addition, most of the Photuris specimens could not be identified to species morphologically or genetically (supplementary material S3.6, Supplementary Material online). Thus, we only used data for the single distinguishable Photuris species, Pt. frontalis, in the comparative analysis of morphology. For the repeat analysis using single specimens, both Photuris frontalis and the Photuris individual with the largest genome size estimate were included.
Evolutionary History and Genome Size
Pagel’s parameter estimates for genome size supported a Brownian motion model of evolution and complete phylogenetic dependence (λ = 1.00, 95% CI = 0.98–1.00, N = 21) supporting a neutral model. The other two parameters suggested gradual evolution in genome size across the entire phylogeny and along branches (δ = 1.35, 95% CI = 0.13–3.00, κ = 1.13, 95% CI = 0.64–1.50).
Ancestral state reconstruction indicated that the most recent common ancestor of North American fireflies had a genome size of ∼1,200 Mb (supplementary material S3.7, Supplementary Material online). There was a dramatic ∼1 Gb expansion in Photuris lineages and large expansions and contractions of several hundred Mb (up to 2.6-fold) within five of the six genera sampled. The exception was Pyractomena, in which all three species have very similar genome sizes (768.04–789.57 Mb, a difference of less than 3%).
Repetitive DNA and Genome Size
To determine whether the repeat abundance data explains genome size variation, we performed PGLS analysis at three different levels: Total repetitiveness, repeat groups, and repeat categories. As sequencing effort (coverage) is expected to affect estimates of repeat abundance, we generated a data set with sequences from each taxon sampled to uniform 0.01× coverage (N = 18 species). RepeatExplorer analysis on the 258,348 reads in this uniform data set resulted in 14,034 clusters after removal of putative contaminants, of which 209 were “top” clusters (at least 20 reads) and annotated by RepeatExplorer. Top cluster annotations for the uniform data set were confirmed and extended using annotations from an analysis that included all of the sequencing data (N = 21 species, supplementary materials S1.5–S1.9, Supplementary Material online).
Across samples, 10.3% (Photinus sabulosus) to 56.6% (Photinus obscurellus) of reads were assigned to clusters, and thus represent repetitive sequences (table 2). Clusters were generally limited in their phylogenetic occurrence—on average, top clusters were shared among 1.8 ± 1.0 species, whereas bottom clusters were shared among 1.2 ± 0.6 species. This suggests that many of the shared clusters identified in this analysis represent young (shared among a few close relatives) repeats, though there was no significant relationship between branch length (divergence) between species and number of shared clusters (Spearman’s ρ, corrected for ties = −0.13, P = 0.11). Our analysis also identified expected evolutionarily conserved repeats, including a ribosomal (rDNA) gene sequence that is shared among 15 of the 18 species (supplementary material S1.10, Supplementary Material online).
Table 2.
RepeatExplorer Metrics Uniform 0.01× Data Set
| Genus | Species | Sex | Genome Size (Mb)a | Mean Read Length (bp) | Total Repb | Groupsc |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Class I | Class II | Repeats | Unknown | Bottom | ||||||
| Ellychnia | corrusca | M | 781.56 | 627 | 13.5 | 1.69 | 0.45 | 0.02 | 0.60 | 10.71 |
| Lucidota | atra | F | 491.16 | 624 | 13.5 | 0.76 | 0.89 | 1.80 | 0.81 | 9.22 |
| punctata | M | 1300.06 | 654 | 17.8 | 2.81 | 0.74 | 0.84 | 1.46 | 11.89 | |
| Phausis | reticulata | M | 835.56 | 633 | 12.4 | 0.41 | 0.14 | 1.48 | 1.18 | 9.03 |
| sp. WAT | M | 1114.58 | 617 | 19 | 0.63 | 1.66 | 4.31 | 0.91 | 11.46 | |
| Photinus | australis | M | 1615.15 | 643 | 31.9 | 1.46 | 1.61 | 1.17 | 1.56 | 23.81 |
| brimleyi | M | 1180.76 | 631 | 20.9 | 1.70 | 1.18 | 0.87 | 0.95 | 16.15 | |
| cooki | F | 700.98 | 617 | 14.8 | 1.59 | 0.43 | 1.20 | 0.07 | 11.49 | |
| indictus | M | 433.19 | 606 | 13.3 | 1.08 | 0.62 | 1.07 | 0.48 | 10.04 | |
| macdermotti | F | 508.99 | 590 | 18.1 | 0.83 | 1.27 | 2.36 | 1.15 | 12.38 | |
| obscurellus | M | 674.34 | 686 | 54.6 | 0.11 | 0.03 | 0.38 | 0.96 | 52.6 | |
| sabulosus | M | 617.52 | 657 | 10.3 | 0.89 | 0.73 | 0.70 | 0.07 | 7.86 | |
| scintillans | F | 1032.40 | 664 | 24.8 | 1.51 | 0.63 | 0.22 | 0.77 | 21.62 | |
| Photuris | frontalis | M | 2154.37 | 679 | 18.2 | 2.93 | 0.78 | 0.48 | 0.17 | 13.80 |
| Pyractomena | angulata | M | 798.43 | 601 | 20.2 | 2.11 | 0.76 | 5.56 | 1.60 | 10.13 |
| marginalis | M | 768.04 | 663 | 15.4 | 2.50 | 1.20 | 0.01 | 1.02 | 10.22 | |
| Pyropyga | decipiens L | F | 1079.10 | 618 | 21.8 | 3.02 | 0.45 | 0.23 | 0.48 | 17.60 |
| decipiens S | F | 700.92 | 668 | 17.9 | 3.23 | 0.44 | 0.25 | 0.56 | 13.45 | |
Mean of average genome size estimates for the specified sex.
Total percent repetitive using all 14,034 clusters.
Percent repetitive due to each of the listed repeat groups.
We then assigned clusters to categories and larger groups based on their annotations to examine patterns of variation across the phylogeny (fig. 2). Across all 18 taxa, the category that contributed the most to total repetitiveness was composed of the summed Bottom (unannotated) clusters (mean 15 ± 10%). Of the repeat groups, Class I repeat abundance showed phylogenetic signal consistent with complete phylogenetic dependence (Estimated λ = 1.00, 95% CI = 0.95–1.00; supplementary material S4.1, Supplementary Material online). Among categories, LTR and LINE showed a similar pattern.
Fig. 2.

—Repetitive element content across species. (a) Left: three-locus phylogeny with branch lengths in units of relative time. Middle: mean genome size (Mb) per species (GS). Right: the repetitive fraction of each sample, color-coded by repeat group classification: retrotransposons (Class I), DNA transposons (Class II), repeats, unknown top clusters, and bottom clusters. Total bar length is equal to the total repetitiveness of each sample. All taxa have been sampled to 0.01× and Photinus obscurellus removed as an outlier. (b–e) the contribution of 4 repeat orders: long-terminal repeats (LTR), long interspersed nuclear elements (LINE), DNA transposons (DNA), and ribosomal repeats. Horizontal and vertical axes as in (a).
There was a significant relationship between genome size and total percent repetitive sequence after removing an outlier, Photinus obscurellus (fig. 3, PGLS: Estimated λ = 0.93, P = 0.0015), though this relationship was not significant if the outlier was included (PGLS: Estimated λ = 0.90, P = 0.37, supplementary material S4.2, Supplementary Material online). Within repeat groups, Class I TEs and Bottom clusters were significantly correlated with genome size (fig. 3; PGLS: Estimated λ = 1.00, Class I: P = 0.008; Bottom: P = 0.016). These significant group-level relationships were robust to both data transformation and inclusion/exclusion of the outlier. Among categories, none were significantly correlated with genome size.
Fig. 3.
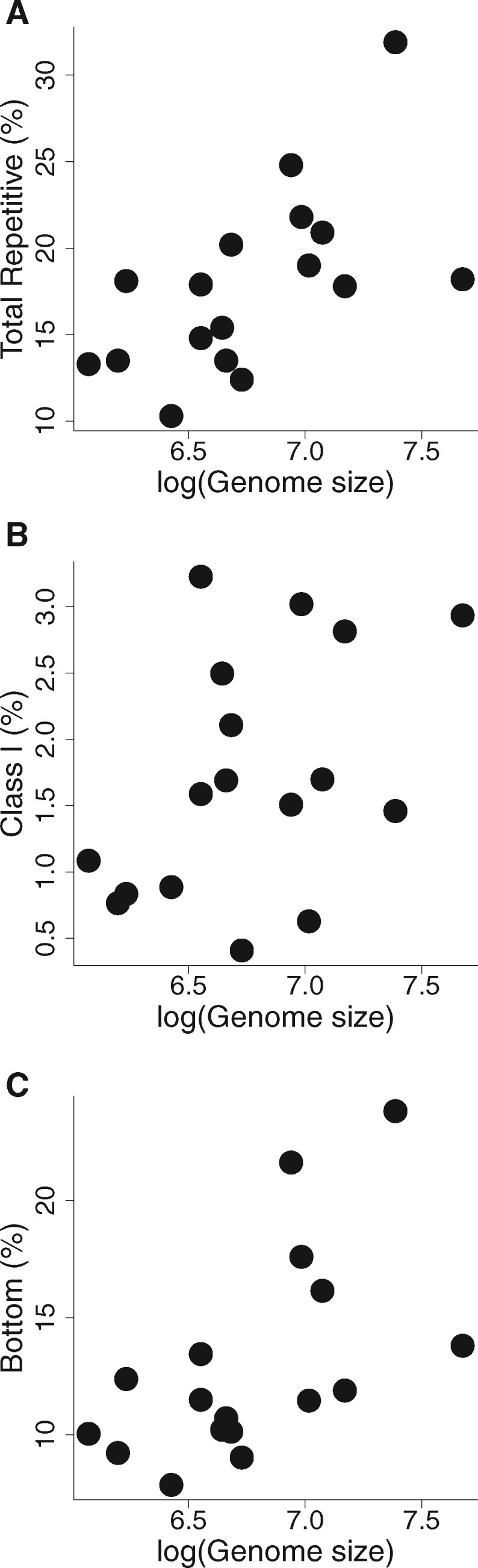
—Correlation of repeat content with genome size. Graphs show the relationships between genome size and repetitive elements, as classified by RepeatExplorer analysis, using the uniform 0.01x data set and excluding an outlier, Photinus obscurellus (N = 17 taxa). (a) Total percent of the low coverage sample classified as repetitive, (b) percent classified as Class I repetitive elements, (c) percent classified as Bottom clusters. Genome size was log-transformed prior to analysis to conform to assumptions of PGLS analysis (Quader et al. 2004).
Effects of Low-Coverage Sequencing
As expected, our low coverage simulated data substantially underestimated total repetitiveness (fig. 4a). Part of this underestimation was due to the fact that low genome coverage is unable to detect low-copy number repeats because two copies will not be sequenced at low depth, which is required for detection using this method. The highest repeat estimates possible in the simulated data given the distribution of repeats in the genome are 50–80% of the actual value (fig. 4a, comparing black diamonds to red squares). The simulated data are also well below the expectation based on repeat abundance, presumably due to RepeatExplorer not assigning reads to the same repeat when they were in fact from the same repeat. This error could be caused by some combination of sequence divergence and/or insufficient overlap between reads, or possibly some other factor. Within the firefly data set, estimates of total repetitiveness increased with increasing coverage when we resampled species at different depths (fig. 4b). Encouragingly, the relative relationships among the taxa remained the same across coverage levels, thus validating our comparative approach.
Fig. 4.

—The effects of low coverage sampling on repetitiveness estimates. (a) Observed and expected estimates of total repetitiveness in Tribolium castaneum. Estimates of total repetitiveness were obtained from RepeatExplorer analysis of simulated 454 reads from the T. castaneum genome (gray triangles; average of five replicates per coverage). Expected estimates based on the probability of detecting a repeat due to its copy number in the genome are shown in black diamonds. The red squares show the estimate of genomic repetitiveness obtained from the genome sequence on which simulations are based (Wang et al. 2008). (b) Estimates of repetitiveness across different coverage levels for seven firefly species (average of five replicates per coverage). Lines connect points from the same species across coverages. Lines do not cross, indicating that the ranking among samples remains the same. Species abbreviations: Latra: Lucidota atra, Lpunc: Lucidota punctata, PdeAS: Pyropyga decipiens small genome size type, Pindi: Photinus indictus, Pobsc: Photinus obscurellus, PretG: Phausis reticulata, Psabu: Photinus sabulosus.
Morphology and Genome Size
We tested whether large genome sizes are associated with large body sizes, the presence/absence of adult light (scored as presence versus absence of adult light organ), and female wing reduction (full-size versus reduced elytra) by performing PGLS analysis with data from 21 species (supplementary material S2.3, Supplementary Material online). None of these morphological traits were correlated with genome size (full model: log[genome size] −male pronotum width + reduced-winged females + lighted/unlighted; Estimated λ = 1.00, P > 0.2).
Discussion
Genome Size Variation within Fireflies
Genome size varies over 5-fold across the North American firefly species measured in this study. We are confident that genome size estimates are accurate because, for four species, the estimates of genome size obtained in this study correctly predicted the depth of coverage we obtained from genomic sequencing in another study (Sander and Hall 2015). Given our sample of 23 species, the within-family genome size variation in fireflies is on par with that seen in other beetle families: 11-fold in Chrysomelidae (N = 64), 9-fold in Coccinellidae (N = 31), and 5-fold in Tenebrionidae (N = 69) (Gregory 2015: Animal Genome Size Database; last accessed February 24, 2015). Across all Coleoptera, genomes generally are in the 154–2,650 Mb range (Hanrahan and Johnston 2011) thus, firefly genome sizes appear to be representative of Coleoptera.
Unexpectedly, we discovered substantial genome size variation within a single population of Pg.decipiens, with some individuals having genomes of ∼700 Mb and others of ∼1,080 Mb, a 1.5-fold difference. Both types seemed to be common; three individuals had large and six had small genomes. Individuals were collected on the same day on the same plants in the same field, and the genetic data does not distinguish the two types, suggesting that they may represent a single species. This polymorphism in genome size within a population, combined with the lack of karotypic variation in fireflies (supplementary material S5.1, Supplementary Material online), suggests that either supernumerary B chromosomes or triploidy may play a role.
B chromosomes are selfish, unessential chromosomes, generally made of repetitive DNA, that are polymorphic across and within populations (reviewed in Houben et al. 2014), and can contribute substantially to genome size. For example, they are responsible for a 155% increase in the DNA content in rice (Jones and Rees 1982) and cause up to a 20% difference in genome size in grasshoppers (Rees et al. 1978). Some firefly species are known to have B chromosomes (Photinus pyralis, Pyractomena angulata, Aspisoma laterale, reviewed in Dias et al. 2007), though the extent of variation in B chromosome size and number across populations and species remains largely unknown. If the ∼50% difference in genome size between the two genome size types was entirely due to high-copy number repetitive sequences, as are often found on B chromosomes (Camacho et al. 2000), then we would expect repeat content to increase from 17.9% in small- to 45.3% in large-genomed individuals (supplementary material S6.1, Supplementary Material online), a 27% difference between size types. However, we observed only a 3.9% difference, thus arguing against B chromosomes as an explanation for the genome size variation in this species.
An alternative explanation is that individuals with large genomes represent recent triploids. Support for recent triploidy includes: 1) genome size estimates that are approximately 50% different (i.e. 2n diploids versus 3n triploids), 2) a modest difference in estimated repeat content between the two genome size types (fig. 2), and 3) all three putative triploid individuals were females, which is consistent with the association between triploidy and parthenogenesis in another beetle group, the weevils (Suomalainen et al. 1976). However, to date there are no reports of parthenogenesis in fireflies. Future collections of Pg. decipiens from this population and others are needed to determine the extent of genome size variation within this species, and provide karyotypic evidence for polymorphic B chromosomes or triploidy.
Genome Size and Repetitive DNA
Given the greater than 5-fold variation in genome size in our samples, we expected to find evidence of repetitive elements substantially contributing to nuclear DNA content, particularly in species where ancestral state reconstruction suggests there has been an increase in genome size (e.g. Photuris species). We did find that the total repetitive proportion increases with genome size across the 17 species in the uniform 0.01× sequencing coverage data set (fig. 3). Class I elements were responsible for part of this significant relationship, which is not surprising given their copy-and-paste mechanism of proliferation in genomes. Unlike in Drosophila, which show some evidence for selective optima in genome size in some clades (Sessegolo et al. 2016), neither genome size nor significant repeat classes showed evidence of non-neutral evolution in fireflies (i.e. departure from Brownian motion).
Despite the correlation between genome size and repeat content, the 3-fold variation, from 10% to 32% (57% including the outlier, Pn. obscurellus; table 2), in repetitive DNA across species is lower than that predicted based on a model of genome size expansion due solely to repeats, indicating that we have underestimated true genomic repeat content (supplementary material S6, Supplementary Material online). This is not surprising given our low-coverage sequencing scheme. Repeat detection requires that at least two copies of an element are sequenced in order to be identified as a repeat, thus low coverage genomic sequencing misses many of the repeats that are present in low-copy numbers in the genome. Furthermore, even if two or more copies of a repeat are present in the sequencing reads, they may not be identified as being from the same repeat because they do not overlap sufficiently, or they are too diverged. Simulated low-coverage 454 reads from the Tribolium castaneum genome (GCA_000002335.3; Wang et al. 2008) show that both genomic copy number and sequence divergence can have substantial impacts on repeat content estimation at low coverage (fig. 4; supplementary material S6, Supplementary Material online). However, this underestimation of total repeat content does not impact our comparative analyses as every species suffers from the same underestimation bias: Subsampling from 0.01 to 0.05× across seven species for which we had enough data preserved the relative relationships among species across coverages (fig. 4).
Prior to the study, we hypothesized that species with large genomes would have relatively more high-copy repeats, indicative of one or two transposons contributing to large genomes. If so, we should have detected a large proportion of repeats in species with large genome size estimates. For example, if the entire difference in genome size between the smallest and largest firefly genomes were due entirely to high copy number repeats, then repeat content should have varied from 10% to 86%, rather than our observed range of 10–35% (excluding Pn. obscurellus; supplementary material S6.1, Supplementary Material online). Thus, we can conclude that either high copy repeats are not the primary determinant of genome-size variation across firefly species, or they are being underestimated due to 1) the age of repeat expansions, 2) the dynamics of repeat-mediated gain/loss, or both. 1) If repeat expansions occurred in the distant past and there has been a long period of time for repeats to diverge from the consensus, these repeats will essentially be low-copy number in the genome. As a result, low-coverage sequencing will either miss them or RepeatExplorer will classify them as low-abundance (Bottom) elements. Indeed, we find that, unlike in some plant species (e.g. Estep et al. 2013), the repetitive landscape in fireflies is not dominated by a high-copy single repetitive element (cluster), even in species with extremely large genome sizes. Instead, it is dominated by “Bottom clusters” that are below the abundance threshold we used for annotation, and therefore represent low-copy number sequences. 2) Another explanation is that firefly genome size variation is governed by loss of elements rather than by gain, such that, in the species being examined, there have been few increases in genome size. This is a definite possibility given the ∼1,200 Mb genome size estimate for the common ancestor of lampyrids. This ancestral genome estimate is larger than over half the species in our study, indicating that much of the evolution in the family could represent loss of repetitive DNA sequences. Further genome size estimates and sequencing across the phylogeny of fireflies and sister taxa will help elucidate these dynamics.
Genome Size and Morphology
The observation that genome size often varies with organism physiology suggested several specific expectations in fireflies. In particular, we expected to find the commonly observed positive correlation between genome size and body size, which has been hypothesized to be caused by the nucleotypic effects of cell size/volume (reviewed in Cavalier-Smith 1978; Gregory 2005). However, we found no evidence for a positive correlation between genome size and body size. This is not unusual for beetles- while a positive correlation between body size and genome size has been noted across a diversity of animal taxa, including other insects (reviewed in Gregory 2001), previous studies within beetles have documented either a negative or no correlation (e.g. Tenebrionidae: Juan and Petitpierre 1991; Palmer et al. 2003; Coccinellidae: Gregory et al. 2003; Chrysomelidae: Petitpierre and Juan 1994). Our negative finding, incorporating phylogenetic correction, suggests this previous beetle work is robust to assumptions about relatedness of species.
In addition, we tested for a relationship between genome size and energetic costs imposed by light production and/or flight, as indicated by the presence/absence of an adult light organ and/or female wing reduction. We found no evidence for a correlation between genome size and light production (light organ presence/absence). This may be due to the small magnitude of difference in resting metabolic rate between lighted and dark fireflies; i.e. there was no significant difference in carbon dioxide production between individuals of three lighted and two unlighted species during intervals where they were not flashing or moving (Woods et al. 2007), however, metabolic rates during flight and mate signaling remain to be investigated across species.
We also found no relationship between genome size and the presence/absence of reduced wings in females. It is possible that, because males must still fly to search for females in all species, resting metabolic rates may not significantly differ between species with and without flightless females. However, it is also possible that male flight effort is smaller in species with flightless females, which are more restricted in dispersal than their winged counterparts. Our sample size is small (only four species with flightless females) and more species are needed to rigorously test this hypothesis. In addition, there are presently no data on resting metabolic differences between species with flighted versus flightless females. Flightless females have evolved several times independently in fireflies (Branham and Wenzel 2003) and so a sample size increase is feasible in future studies.
The Evolution of Genome Size in Fireflies
We find no evidence to support strong selection acting on genome size in fireflies. Specifically, 1) genome size evolution is gradual, 2) exhibits complete phylogenetic dependence, and 3) there is no relationship with measured morphological variables. To determine if there is weak selection favoring reduced genome size, it would be useful to examine genome size versus effective population size in fireflies. Unfortunately, we do not have sufficient data from nuclear markers across taxa to perform this analysis. When we looked at mitochondrial sequence data, we did not find a relationship between genome size and COI sequence variation (supplementary material S5.2, Supplementary Material online), but this is difficult to interpret given the unique evolutionary history of mitochondria, presence of Wolbachia sequences in several of our low-coverage sequencing taxa, and large variance in sample size (Ballard and Whitlock 2004; Hurst and Jiggins 2005).
We do not find evidence that high copy repeats make a relatively larger contribution to large genome sizes. This suggests recent proliferation of a single TE family does not play a disproportionate role in genome size variation in fireflies. Instead, it seems that both low copy elements and high copy elements together shape genome size variation across fireflies. The dynamics are likely due small changes in the relative rates of gains versus losses of elements led to trends in genome size reduction or increase across different lineages.
It is possible that further investigations will identify relationships with other potential selective correlates (e.g. cell size, egg size, developmental time), as we did not exhaustively sample traits known to be associated with genome size. In addition, future comprehensive taxon sampling will provide more information about evolutionary patterns in genome size and content across the worldwide distribution of fireflies. Overall, our results support a dynamic picture of genome size and content variation in this family of beetles.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors would like to thank Jim Lloyd and Lynn Faust for identification of many of the specimens used in body size measurements. The authors would also like to thank the following people and organizations for collection assistance and permission: Allegheny National Forest, Ashley Brown, Megan Behringer, Tom Brightman (Longwood Gardens), Lynn Faust, the David Fisk family, the Entomological Society of Pennsylvania, Great Smoky Mountains National Park (permit GRSM-2011-SCI-0049), Illinois Department of Natural Resources (permit NH12.5615), Indiana University, Michael Marsh (Whitehall Experimental Forest, University of Georgia), Jerry McCollum (Wharton Conservation Center), David McNaughton (Fort Indiantown Gap), Jenna Pallansch, David Queller, Willem Roosenberg, State of Tennessee Department of Environment and Conservation (permit 2012-16), Tonya Saint John, Kevin Smith (Tyson Research Center), Joan Strassman, and Dorset Trapnell. This work was supported by a National Science Foundation Graduate Research Fellowship (to S.S.L.), a National Science Foundation Dissertation Improvement Grant (DEB-1311315 to D.W.H. and S.S.L.), and an award from the National Institute of General Medical Sciences of the National Institute of Health (award number T32GM007103 to S.S.L.).
Literature Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Arendt J. 2007. Ecological correlates of body size in relation to cell size and cell number: patterns in flies, fish, fruits and foliage. Biol Rev Camb Philos Soc. 82:241–256. [DOI] [PubMed] [Google Scholar]
- Arnqvist G, et al. 2015. Genome size correlates with reproductive fitness in seed beetles. Proc R Soc Lond B. 282:20151421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronesty E. 2013. Comparison of sequencing utility programs. Open Bioinforma J. 7:1–8. [Google Scholar]
- Ballard JWO, Whitlock MC.. 2004. The incomplete natural history of mitochondria. Mol Ecol. 13:729–744. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 57:289–300. [Google Scholar]
- Bennet MD, Leitch IJ.. 2012. Plant DNA C-values database [Internet][cited 2015 February 27]. Available from: http://www.kew.org/cvalues/
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branham MA, Wenzel JW.. 2003. The origin of photic behavior and the evolution of sexual communication in fireflies (Coleoptera: Lampyridae). Cladistics. 19:1–22. [DOI] [PubMed] [Google Scholar]
- Camacho JP, Sharbel TF, Beukeboom LW.. 2000. B-chromosome evolution. Phil Trans R Soc B 355:163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. 1978. Nuclear volume control by nucleoskeletal DNA, selection for cell volume and cell growth rate, and the solution of the DNA C-value paradox. J Cell Sci. 34:247–278. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Barton N.. 2004. Genome size: does bigger mean worse? Curr Biol. 14:R233–R235. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D.. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Meth. 9:772–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cock R, Matthysen E.. 2005. Sexual communication by pheromones in a firefly, Phosphaenus hemipterus (Coleoptera: Lampyridae). Anim Behav. 70:807–818. [Google Scholar]
- Dias CM, Schneider MC, Rosa SP, Costa C, Cella DM.. 2007. The first cytogenetic report of fireflies (Coleoptera, Lampyridae) from Brazilian fauna. Acta Zool. 88:309–316. [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A.. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29:1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott TA, Gregory TR.. 2015. What's in a genome? The C-value enigma and the evolution of eukaryotic genome content. Phil Trans R Soc B. 370:20140331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis LL, et al. 2014. Intrapopulation genome size variation in D. melanogaster reflects life history variation and plasticity. PLoS Genet. 10:e1004522.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estep MC, DeBarry JD, Bennetzen JL.. 2013. The dynamics of LTR retrotransposon accumulation across 25 million years of panicoid grass evolution. Heredity. 110:194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 1985. Phylogenies and the comparative method. Am Nat. 125:1–15. [Google Scholar]
- Fender KM. 1966. The genus Phausis in America North of Mexico (Coleoptera-Lampyridae). Northwest Sci. 40:83–95. [Google Scholar]
- Ferrari JA, Rai KS.. 1989. Phenotypic correlates of genome size variation in Aedes albopictus. Evolution. 43:895–899. [DOI] [PubMed] [Google Scholar]
- Green J. 1956. Revision of the Nearctic species of Photinus (Lampyridae: Coleoptera). Proc Calif Acad Sci. 28:561–613. [Google Scholar]
- Green J. 1957. Revision of the nearctic species of Pyractomena (Coleoptera: Lampyridae). Wasmann J Biol. 15:237–284. [Google Scholar]
- Gregory TR. 2001. Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol Rev Camb Philos Soc. 76:65–101. [DOI] [PubMed] [Google Scholar]
- Gregory TR. 2005. Genome size evolution in animals In: Gregory TR, editor. The evolution of the genome. Burlington, MA: Elsevier Inc; p. 4–67. [Google Scholar]
- Gregory TR. 2015. Animal Genome Size Database. http://www.genomesize.com.
- Gregory TR, Johnston JS.. 2008. Genome size diversity in the family Drosophilidae. Heredity 101:228–238. [DOI] [PubMed] [Google Scholar]
- Gregory TR, Nedved O, Adamowicz SJ.. 2003. C-value estimates for 31 species of ladybird beetles (Coleoptera: Coccinellidae). Hereditas. 139:121–127. [DOI] [PubMed] [Google Scholar]
- Hanrahan S, Johnston JS.. 2011. New genome size estimates of 134 species of arthropods. Chromosome Res. 19:809–823. [DOI] [PubMed] [Google Scholar]
- Houben A, Banaei-Moghaddam A, Klemme S, Timmis J.. 2014. Evolution and biology of supernumerary B chromosomes. Cell Mol Life Sci. 71:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Li L, Myers JR, Marth GT.. 2012. ART: a next-generation sequencing read simulator. Bioinformatics. 28:593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A.. 1999. CAP3: a DNA sequence assembly program. Genome Res. 9:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst GD, Jiggins FM.. 2005. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc R Soc Lond B. 272:1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RN, Rees H.. 1982. B Chromosomes. Chicago, IL: Academic Press. [Google Scholar]
- Juan C, Petitpierre E.. 1991. Evolution of genome size in darkling beetles (Tenebrionidae, Coleoptera). Genome. 34:169–173. [Google Scholar]
- Kapitonov VV, Jurka J.. 2008. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 9:411–412. [DOI] [PubMed] [Google Scholar]
- Kullman B, Tamm H, Kullman K.. 2005. Fungal Genome Size Database [Internet] [cited 2015 February 27, 2015]. Available from: http://www.zbi.ee/fungal-genomesize
- Lloyd JE. 1966. Studies on the flash communication system in Photinus fireflies. Ann Arbor, Michigan: Miscellaneous Publications, Museum of Zoology, University of Michigan. [Google Scholar]
- Luk SPL, Marshall SA, Branham MA.. 2011. The fireflies of Ontario (Coleoptera: Lampyridae). Can J Arthropod Identif. 16:1–105. [Google Scholar]
- Lynch M, Conery JS.. 2003. The origins of genome complexity. Science. 302:1401–1404. [DOI] [PubMed] [Google Scholar]
- Macas J, Neumann P, Navrátilová A.. 2007. Repetitive DNA in the pea (Pisum sativum L.) genome: comprehensive characterization using 454 sequencing and comparison to soybean and Medicago truncatula. BMC Genomics. 8:427-427.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas J, et al. 2015. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe fabeae. PLoS One. 10:e0143424.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, Neumann P, Macas J.. 2010. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics. 11:378.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, Neumann P, Pech J, Steinhaisl J, Macas J.. 2013. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics. 29:792–793. [DOI] [PubMed] [Google Scholar]
- Orme D. 2013. The caper package: comparative analysis of phylogenetics and evolution in R. R package version 5.
- Pagel M. 1999. Inferring the historical patterns of biological evolution. Nature. 401:877–884. [DOI] [PubMed] [Google Scholar]
- Palmer M, Petitpierre E, Pons J.. 2003. Test of the correlation between body size and DNA content in Pimelia (Coleoptera: Tenebrionidae) from the Canary Islands. Eur J Entomol. 100:123–129. [Google Scholar]
- Paradis E, Claude J, Strimmer K.. 2004. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 20:289–290. [DOI] [PubMed] [Google Scholar]
- Peters RH. 1986. The ecological implications of body size. Cambridge: Cambridge University Press. [Google Scholar]
- Petitpierre E, Juan C.. 1994. Genome size, chromosomes and egg-chorion ultrastructure in the evolution of Chrysomelinae. Ser Entomol. 50:213–225. [Google Scholar]
- Petrov DA. 2001. Evolution of genome size: new approaches to an old problem. TRENDS Genet. 17:23–28. [DOI] [PubMed] [Google Scholar]
- Petrov DA. 2002. Mutational equilibrium model of genome size evolution. Theor Popul Biol. 61:531–544. [DOI] [PubMed] [Google Scholar]
- Pinheiro J, Bates D, DebRoy S, Sarkar D.. R Core Team. 2016. nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1-128, http://CRAN.R-project.org/package=nlme
- Quader S, Isvaran K, Hale RE, Miner BG, Seavy NE.. 2004. Nonlinear relationships and phylogenetically independent contrasts. J Evol Biol. 17(3):709–715. [DOI] [PubMed] [Google Scholar]
- Rees HF, Shaw DD, Wilkinson P.. 1978. Nuclear DNA variation among acridid grasshoppers. Proc R Soc Lond B. 202:517–525. [Google Scholar]
- Reinhold K. 1999. Energetically costly behaviour and the evolution of resting metabolic rate in insects. Funct Ecol. 13:217–224. [Google Scholar]
- Sander SE, Hall DW.. 2015. Variation in opsin genes correlates with signalling ecology in North American fireflies. Mol Ecol. 24:4679–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW.. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Meth. 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessegolo C, Burlet N, Haudry A.. 2016. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biol Lett. 12:20160407.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P.. 2013. –2015. RepeatMasker Open-4.0 [Internet]. Available from: http://www.repeatmasker.org
- South A, Stanger-Hall K, Jeng M-L, Lewis SM.. 2011. Correlated evolution of female neoteny and flightlessness with male spermatophore production in fireflies (Coleoptera: Lampyridae). Evolution. 65:1099–1113. [DOI] [PubMed] [Google Scholar]
- Stanger-Hall KF, Lloyd JE.. 2015. Flash signal evolution in Photinus fireflies: character displacement and signal exploitation in a visual communication system. Evolution. 69:666–682. [DOI] [PubMed] [Google Scholar]
- Stanger-Hall KF, Lloyd JE, Hillis DM.. 2007. Phylogeny of North American fireflies (Coleoptera: Lampyridae): implications for the evolution of light signals. Mol Phylogenet Evol. 45:33–49. [DOI] [PubMed] [Google Scholar]
- Suomalainen E, Saura A, Lokki J.. 1976. Evolution of parthenogenetic insects In Evolutionary biology. Springer US; p. 209–257. [Google Scholar]
- Swaminathan K, Varala K, Hudson ME.. 2007. Global repeat discovery and estimation of genomic copy number in a large, complex genome using a high-throughput 454 sequence survey. BMC Genomics. 8:132–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon MI, Hufford MB, Gaut BS, Ross-Ibarra J.. 2011. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol Evol. 3:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vencl FV. 2004. Allometry and proximate mechanisms of sexual selection in Photinus fireflies, and some other beetles. Integr Comp Biol. 44:242–249. [DOI] [PubMed] [Google Scholar]
- Wang S, Lorenzen MD, Beeman RW, Brown SJ.. 2008. Analysis of repetitive DNA distribution patterns in the Tribolium castaneum genome. Genome Biol. 9:R61–R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney KD, Garland T.. 2010. Did genetic drift drive increases in genome complexity? PLoS Genet. 6:e1001080.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, et al. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 8:973–982. [DOI] [PubMed] [Google Scholar]
- Wood DE, Salzberg SL.. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods WA Jr, Hendrickson H, Mason J, Lewis SM.. 2007. Energy and predation costs of firefly courtship signals. Am Nat. 170:702–708. [DOI] [PubMed] [Google Scholar]
- Wright NA, Gregory TR, Witt CC.. 2014. Metabolic ‘engines’ of flight drive genome size reduction in birds. Proc R Soc Lond B. 281:2013–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.