

Replication of the DNA is a fundamental requirement for progression through the cell cycle and thereby for the development and maintenance of all tissues in the human body. Though all cells duplicate their DNA once before division, they do so in a manner dependent on cell type. Since replication tends to begin in regions with active genes, about half of the human genome is replicated in a cell type dependent temporal pattern, while the other half is cell type independent.1 Surprisingly, a functional significance of cell type specific DNA replication has not been established because of fundamental experimental challenges. It is not yet possible to manipulate all cell type specific origins and ask whether a cell can still progress through the cell cycle. Various observations, including feedback between replication progression and origin activity,2 as well as checkpoints monitoring the completion of S-phase before entry into mitosis, suggest that DNA replication is well prepared to duplicate the genetic information of any cell type. According to this interpretation, differences in DNA replication patterns are a mere adaptation to epigenetic changes required to establish cell type specific gene expression. Hence, most research to better understand different cell states has focused on the processes governing gene expression, while cell type specific DNA replication remains a little explored dimension of cellular biology.
The work of Chia and colleagues published in Nature Cell Biology supports a new model of the role of cell type specific DNA replication.3 The authors exchanged the genomes of human and mouse eggs with either the genome of another egg, of an embryonic cell, or of various differentiated somatic cell types. Replication of somatic chromatin in the egg resulted in DNA damage and chromosome missegregation at the first mitosis. Segregation errors were dependent on DNA replication, and independent of gene expression, as inhibition of transcription by α amanitin had no effect on cell cycle progression or error frequency. The incidence of missegregation depended on the origin of the transferred genome, with terminally differentiated T cell and fibroblast genomes resulting in the highest error rates. Transferring genomes from pluripotent stem cells led to intermediate error frequency, while transferring the genome of another oocyte did not increase error rates compared with unmanipulated controls. The authors also found that remodeling of somatic chromatin by chromosome condensation reduced replication-dependent segregation errors. Consistent with this observation, chromatin condensation is thought to remove most cell type specific aspects of nuclear architecture,4 and thereby mediate the transition between cell type specific replication patterns. Therefore, cell type-specific aspects of chromatin are required for normal progression through S phase.
A connection between genetic stability and cellular identity is also visible in other experimental systems. The conversion of somatic cells to induced pluripotent stem cells is associated with DNA damage and replication stress. In the absence of proteins involved in DNA damage repair, such as Brca1, Brca2 and Rad51, reprogramming fails.5 Furthermore, cancer cells are widely known to duplicate their genetic material in an aberrant manner, often associated with DNA damage and genomic instability. Since these experimental systems also involve profound changes in gene expression, it has not been possible to distinguish cause from consequence. Early embryonic cells are the only cell type that does not require gene expression for normal cell cycle progression. Therefore, the results by Chia and colleagues conclusively show that cell type specific DNA replication provides constraints on the ability to progress through the cell cycle, at least in this experimental context. Consequently, the pattern of DNA replication is not a mere adaption to a cell type specific gene expression program. Rather, it limits the number of possible cellular states compatible with genetic integrity during cell proliferation. The organization of the genome and the packaging of the DNA in chromatin determine the ability to replicate and also enable cell type specific gene expression patterns, providing a link between cell identity and genetic stability.
The significance of this model to cellular biology is profound. The model predicts that cells with abnormal gene expression states will also have a compromised replication program, resulting in the activation of checkpoints that prevent or slow cell cycle progression (Fig. 1). Thereby, checkpoints of genetic integrity are also checkpoints of cellular identity, enabling continuous selection for cellular fitness in proliferating cells. During growth and development, this ensures the optimization of cell and organ function. During aging, it results in cell cycle arrest of damaged cells. During terminal differentiation, the inactivation of a cell type specific replication program may be used in a physiological manner to permanently halt the proliferation of entire groups of cells. For instance, neurons do not proliferate, and forcing them into DNA replication results in cell death.6 Cell type specific DNA replication may also be the primary impediment to the growth of cells assuming an abnormal identity. Consistent with this observation, DNA replication stress has been identified as an early barrier to tumorigenesis.7 Proliferating cancer cells represent the rare gaps in this quality control system, allowing cell cycle progression of some abnormal cellular states, albeit with a compromised replication program and genetic instability. Most abnormal cell states are incompatible with DNA replication and cell cycle progression would result in lethal chromosomal abnormalities. Perhaps the most important implication of this model is that, if cell type specific DNA replication is a cellular mechanism preventing the proliferation of abnormal cells, it may also be therapeutically relevant.
Figure 1.
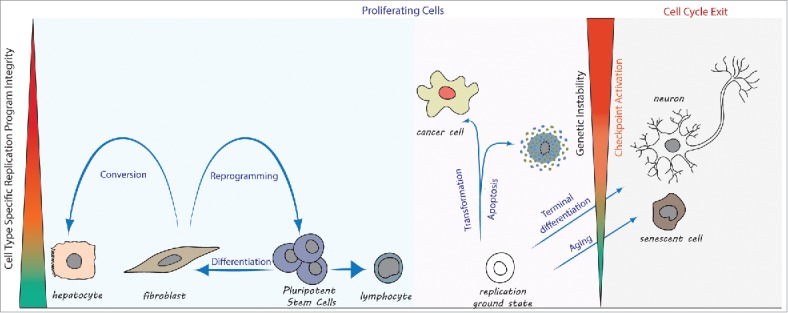
Cell type specific DNA replication programs limit changes in cell identity. Proliferating cells blue panel: Cell type transitions during normal development follow along cell type specific replication programs. In contrast, forced changes in cell identity during reprogramming to pluripotency (e.g. fibroblast to iPS cell) or direct conversion to another cell type (e.g., fibroblast to hepatocyte) undermine replication program integrity and activate cell cycle checkpoints. Proliferating cells pink panel: Deviation from a cell type specific replication program compatible with genetic stability (referred to as ‘replication ground state’) occurs during cell transformation. The outcome may be cell death, or genetic instability in proliferating cells. Cell cycle exit: The cell type specific replication program is compromised in a growing number of cells during aging, resulting in cellular senescence. Mature neurons lack a replication program compatible with genetic stability and permanently exit the cell cycle.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A 2010; 107:139-44; PMID:19966280; https://doi.org/ 10.1073/pnas.0912402107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 2004; 6:648-55; PMID:15220931; https://doi.org/ 10.1038/ncb1145 [DOI] [PubMed] [Google Scholar]
- [3].Chia G, Agudo J, Treff N, Sauer MV, Billing D, Brown BD, Baer R, Egli D. Genomic instability during reprogramming by nuclear transfer is DNA replication dependent. Nat Cell Biol 2017; 19(4):282-91; PMID:28263958; https://doi.org/ 10.1038/ncb3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, Dekker J. Organization of the mitotic chromosome. Science 2013; 342:948-53; PMID:24200812; https://doi.org/ 10.1126/science.1236083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gonzalez F, Georgieva D, Vanoli F, Shi ZD, Stadtfeld M, Ludwig T, Jasin M, Huangfu D. Homologous recombination DNA repair genes play a critical role in reprogramming to a pluripotent state. Cell Rep 2013; 3:651-60; PMID:23478019; https://doi.org/ 10.1016/j.celrep.2013.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci 2004; 24:9232-9; PMID:15496657; https://doi.org/ 10.1523/JNEUROSCI.3347-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al.. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434:864-70; PMID:15829956; https://doi.org/ 10.1038/nature03482 [DOI] [PubMed] [Google Scholar]