

ABSTRACT
The Spindle Assembly Checkpoint (SAC) is part of a complex feedback system designed to ensure that cells do not proceed through mitosis unless all chromosomal kinetochores have attached to spindle microtubules. The formation of the kinetochore complex and the implementation of the SAC are regulated by multiple kinases and phosphatases. BubR1 is a phosphoprotein that is part of the Cdc20 containing mitotic checkpoint complex that inhibits the APC/C so that Cyclin B1 and Securin are not degraded, thus preventing cells going into anaphase. In this study, we found that PP2A in association with its B56γ regulatory subunit, are needed for the stability of BubR1 during nocodazole induced cell cycle arrest. In primary cells that lack B56γ, BubR1 is prematurely degraded and the cells proceed through mitosis. The reduced SAC efficiency results in cells with abnormal chromosomal segregation, a hallmark of transformed cells. Previous studies on PP2A's role in the SAC and kinetochore formation were done using siRNAs to all 5 of the B56 family members. In our study we show that inactivation of only the PP2A-B56γ subunit can affect the efficiency of the SAC. We also provide data that show the intracellular locations of the B56 subunits varies between family members, which is consistent with the hypothesis that they are not completely functionally redundant.
KEYWORDS: PP2A, PP2A-B56, B56, B56gamma, spindle assembly checkpoint, BUBR1, SAC
Introduction
Genomic stability after DNA replication and cell division depends on the spindle assembly checkpoint (SAC) to delay mitosis until chromosomes become attached to the microtubule spindle apparatus through their kinetochores.1-3 Deregulation of the spindle assembly checkpoint can transform a normal cell to a malignant cell through the accumulation of errors during chromosomal segregation.4 Because of the importance of safeguarding genomic integrity during mitosis, it is not surprising that the proteins involved in the SAC are very highly conserved and present in all eukaryotes, from yeast to humans.5
Kinetochores, the attachment site for the spindle microtubules, consist of more than 80 proteins assembled at the centromere. The outer kinetochore proteins Knl1, Mis12 and Ndc80, have been shown to bind to the spindle assembly checkpoint proteins that include Mps1, Mad1, Mad2, Bub1, Bub2, and BubR1. The binding of these SAC proteins to the kinetochore relies on the phosphorylation of Knl1 by the Mps1 kinase.6,7 While the SAC proteins are localized to unattached chromosomes they produce the mitotic checkpoint complex (MCC) that consists of Cdc20, Mad2, Bub3 and BubR1. The MCC maintains the APC/C (Anaphase Promoting Complex) in an inactive state, thus preventing the degradation of Securin and Cyclin B and the progression into anaphase. After microtubule binding to the kinetochore, the SAC proteins are removed from the kinetochore and the MCC stops interacting with the APC/C. The APC/C then proceeds to ubiquinate Securin and Cyclin B targeting them for degradation, allowing the cells to progress through mitosis.8
Kinetochore assembly and the spindle assembly checkpoint are regulated by protein phosphorylation involving interactions between multiple kinases and phosphatases, including Protein Phosphatase 2A (PP2A).9,10 PP2A exists primarily as a heterotrimeric complex consisting of the catalytic subunit C, a scaffold subunit A and a regulatory B subunit.11 There are 3 major families of B subunits, with multiple genes coding for related proteins; B/B55/PR55 has 4 isoforms (α, β, δ, γ), B’/B56/PR61 has 5 isoforms (α, β, δ, γ, ε), B”/PR72/130 is coded by one gene that has multiple splice variants. In addition to the B subunits there are multiple PP2A inhibitors and activators that can modulate the catalytic activity of PP2A. The exact functions that each of these regulatory B subunits possess is not completely understood. There has been structural evidence obtained by X-ray crystallography that the B subunits directly bind to the PP2A catalytic subunit and thus could influence substrate specificity.12 There have also been studies showing that the regulatory B subunits can direct PP2A to specific cellular locations depending on which subunit is incorporated into the PP2A heterotrimer.13,14
PP2A containing B56 regulatory subunits (PP2A-B56) have been identified as having significant roles in the control of the chromosome congression, chromosome distribution, and the SAC. PP2A-B56 has been detected at the kinetochore during mitosis using GFP-A subunit and GFP-B56 fusion proteins.15 Subsequent B56 siRNA treatment resulted in misaligned chromosomes, implicating PP2A-B56 as being required for proper chromosome distribution. Aurora B kinase is needed to phosphorylate outer kinetochore proteins to destabilize microtubule interactions to reset them to allow new connections to form. In the region of the kinetochore, PP2A-B56 has been shown to act to counterbalance the Aurora B kinase activity toward the KMN complex to stabilize the kinetochore microtubule interaction.16 The localization of PP2A to the kinetochores has been shown to depend on its interactions with BubR117 and Shugoshin-1 (Sgo1).18 In addition, Bub1 has also been shown to be required for PP2A-B56 to bind with Sgo1 at the centromeres and counterbalance Plk1 activity as needed for proper chromosomal segregation.19 PP2A-B56 BubR1 interaction has also been proposed to promote motor-driven chromosome movement toward the metaphase plate to establish stable and functional kinetochore-microtubule attachments.20 In the absence of the PP2A-B56 BubR1 interaction, aneuploidy is observed that is similar to that in Mosaic Variegated Aneuploidy syndrome cell lines.21
We have previously shown that inactivation of the B56γ gene in mice causes heart development and behavior defects which result in about 50% neonatal lethality.22 However, no high incidence of tumor formation or early embryonic lethality was observed, suggesting that there is not an inherent cell growth or chromosomal distribution abnormality present in these animals. Because of prior studies suggesting that B56γ may be a tumor suppressor,23,24 mouse embryonic fibroblasts (MEFs) were isolated from B56γ- mice to further investigate potential roles in cell cycle control and to identify more subtle requirements for PP2A-B56γ activity. We did not observe any growth differences in B56γ- MEFs under normal culture conditions but when treated with nocodazole, the B56γ- MEFs did not arrest in metaphase as expected. We found that this escape from the SAC correlates with a reduction in BubR1 localized to the kinetochore in the B56γ- cells. Previous studies involving PP2A-B56 and its role in chromosomal distribution have been done using siRNA that inactivated all B56 subunits and have been performed in continuous cell lines.15-17 In our study, we have used primary cells and genetic inactivation of only one B56 family member to detect a novel SAC function for PP2A-B56γ.
Materials and methods
B56γ mice
A mouse strain that expresses no functional B56γ was used as the cell source for all the experiments.22 PCR was used to genotype B56γ mice as described previously. Mouse embryonic fibroblasts (MEFs) were harvested at gestation day 14.5 from intercrosses of B56γ+/− mice. Only MEFs from homozygous wild type (+/+, B56γ+) or homozygous gene trapped (−/−, B56γ-) fetuses were used for experiments. MEFs were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum along with streptomycin and penicillin and cultured in the presence of 5% CO2. MEFs between passages 3 and 6 were used for all experiments.
Cell cycle analysis
106 MEFs from B56γ+ and B56γ- mice were allowed to adhere to T75 flasks for 24 hours and then grown in medium with, or without 200 ng/ml nocodazole for 18 hours. The cells were trypsinized and washed in 0.5 ml phosphate-buffered saline (PBS). Cells were permeabilized with addition of 100% methanol and incubated on ice for 30 min. Then the cells were resuspended in 0.5 ml of Propidium Iodide (PI)/RNase Staining Solution (Cell Signaling Technology) and incubated for 30 min at room temperature. Samples were analyzed for DNA content on a FACSCanto II (Becton Dickinson).
Immunohistochemistry
For all immunohistochemistry experiments, approximately 2 × 104 MEFs were plated in Nunc Lab-Tek™ II Chamber Slides. The cells were allowed to adhere overnight and then incubated for 18 hours with or without 200 ng/ml of nocodazole. MEFs were washed and then fixed in 4% paraformaldehyde/PBS for 10 minutes. Blocking was performed with 10% serum in TTBS buffer.
Identification of mitotic cells by immunohistochemistry and identification of lagging chromosomes
B56γ+and B56γ- MEFs were incubated with mitosis specific marker antibody, Phospho-Histone H3 (Ser10) conjugated with Alexa Fluor 488 (Cell Signaling Technology, 3465, 1:50) and α-Tubulin conjugated with Alexa Fluor 555 (Sigma, 05–829-AF555, 1:500). DNA was counterstained with DAPI. All the images were captured at 60X magnification. To determine the percentage of Phospho-Histone H3 (Ser10) positive mitotic cells and SEM, 300 total cells from both B56γ+and B56γ- MEFs were analyzed, in groups of approximately 50 cells.
Live cell imaging
3 × 105 B56γ+and B56γ- MEFs were plated into 2 well chambered systems (Nunc, Rochester, NY) and then synchronized using serum starvation (0.1% FBS) for 24 hours. The medium was then changed to 10% FCS medium along with 200 ng/ml nocodazole and CellLight Histone 2B-GFP, BacMam 2.0 (Thermo Fisher Scientific) and was incubated for 18 hours. The cells were visualized by a Zeiss Cell Observer Spinning Disk Confocal Microscope system (Carl Zeiss Microscopy, Thornwood, NY) in a PECON environmental chamber. Optical sections of confocal images were acquired every 3 min for 20–50 minutes and movie files were created using Zeiss Axiovision software (ver. 4.8.2). Quantitative analysis was performed using the Image J software (National Institutes of Health). The images were obtained from multiple independent experiments.
Apoptosis enumeration
B56γ+and B56γ- MEFs were incubated with cleaved Caspase-3 (ASP175) antibody (Cell Signaling Technology, 9661, 1:400) and secondary antibody Fluorescein isothiocyanate (FITC) anti-rabbit (Jackson Immunoresearch, 1:200). Nuclei were visualized using DAPI. To determine the percentage of apoptotic cells and SEM, 300 total cells from both B56γ+and B56γ- MEFs were analyzed, in groups of approximately 50 cells.
B56, BubR1, and cyclin B1 immunohistochemistry
B56γ+and B56γ- MEFs were incubated with primary antibodies to B56α (NOVUS Biologicals, NB100–41412, 5 μl/ml), B56β (Sigma, SAB2500140, 2 μl/ml), B56δ (Sigma, HPA029046, 1:100), B56ε (Sigma, HPA006034, 1:100), BUBR1 (BD Biosciences, 612502, 1 μg/ml), and Cyclin B1 (Cell Signaling Technology, 4138, 1:200). B56γ antibody was custom made as described previously.22 Secondary antibodies, Fluorescein isothiocyanate (FITC) anti-rabbit, Fluorescein isothiocyanate (FITC) anti-goat, Tetrarhodamine isothiocyanate (TRITC) anti-mouse, Tetrarhodamine isothiocyanate (TRITC) anti-rabbit from (Jackson Immunoresearch, 1:200) were used. Vectashield mounting media containing DAPI was used to visualize nuclei. Images were captured using a Zeiss LSM 710 Confocal microscope.
RT-PCR
About 106 B56γ+ MEFs were detached using 0.25% trypsin, washed and re-suspended in phosphate-buffered saline (PBS). Total RNA was isolated using Trizol reagent (Life Technologies) according to the manufacturer's protocol. B56α mRNA was reverse transcribed using B56a-1 (AATCAGGACTCTCCAAGAATCTC, mRNA bp 1148–1126) and PCR amplified using primers B56a-1 and B56a-3 (CGGAGAAAGTGGACGGCTTC, mRNA bp 678–697). B56β mRNA was reversed transcribed using B56b-3 (TACACCCGATGCAAGATGGTC, mRNA bp 1050–1030) and PCR amplified using primers B56b-1 (CTTCTCAGTTCCGCTATCAGAG, mRNA bp 528–549) and B56b-2 (CTGGAAATCTGGACTCTCCAAG, mRNA bp 934–913). B56γ mRNA was reversed transcribed using B56g-5 (ATCTTGTGTTCCTCCTTCAGTGG, mRNA bp 704–682) and PCR amplified using primers B56g-1 (CAGAAGCTACGCCAGTGTTGTG, mRNA bp 100–120) and B56g-4 (GTAACTCCGCTATGCCATTGTG, mRNA bp 643–622). B56δ mRNA was reversed transcribed using B56d-2 (AGTGACAACATCACGGCTGTG, mRNA bp 556 - 536) and PCR amplified using primers B56d-3 (TGATATACTCCACCATCTCATTG, mRNA bp 532–511) and B56d-1 (AGATGTCCTATAAACTGAAGAAGG, mRNA bp 87–110). B56ε mRNA was reversed transcribed using B56e-3 (AGGGTCTTCACTGTCAAATAGC, mRNA bp 1152–1131) and PCR amplified using primers B56e-2 (CTACTTCAGGGTAAGTCTGCTC, mRNA bp 919–898) and B56e-1 (GCCACAGATCATCTTCCGATTC, mRNA bp 527–548). All the primers were custom ordered from Integrated DNA Technologies.
Western blot
106 B56γ+and B56γ- MEFs were plated into a T75 flask. MEFs were allowed to adhere to flasks for 24 hours and then synchronized using serum starvation (0.1% FBS) for 24 hours. Then the medium was changed to 10% FCS medium with or without 200 ng/ml Nocodazole and incubated for 18 h. Total protein extracts were obtained from these MEFs and equal amounts of protein were separated by SDS-PAGE (4–12% gels; Invitrogen) and then transferred onto polyvinylidene difluoride membranes (Millipore). The membranes were blocked with 10% Blocking solution (Odyssey-Li-COR) and sequentially incubated with antibodies against BubR1 (BD Biosciences, 612502, 1 μg/ml), Cyclin B1 (Cell Signaling Technology, 4138, 1:200), PP2A catalytic subunit (Cell Signaling Technology, 2038, 1:1000), and then with anti-rabbit and anti-mouse secondary antibody IRDye 800 CW (Odyssey-Li-COR), respectively. Western blots were imaged and analyzed by Odyssey Li- COR infrared System.
Results
To investigate the role of PP2A-B56γ in controlling cell growth, mouse embryonic fibroblasts (MEFs) were harvested at gestation day 14.5 from intercrosses of B56γ+/− mice. Only MEFs from homozygous wild type (+/+, B56γ+) or homozygous B56γ- (−/−, B56γ-) fetuses were used for experiments.
B56γ−/− MEFs arrest less efficiently in G2/M following nocodazole treatment
Flow cytometry was used to investigate any potential cell cycle differences in MEFs that lack PP2A-B56γ. Under normal culture conditions, no significant differences in the cell cycle distribution of B56γ- MEFs were seen compared with wild type MEFs (Fig. 1A, B). However, when the cells were treated with nocodazole for 18 hours, only 45% of B56γ- MEFs arrested in G2/M as compared with 68% of the wild type cells (Fig. 1C–E). Nocodazole interferes with the polymerization of microtubules and will normally arrest cells with G2/M DNA content due to the SAC. The lower percentage of B56γ- MEFs in G2/M is consistent with a requirement for PP2A-B56γ activity to efficiently implement the SAC.
Figure 1.
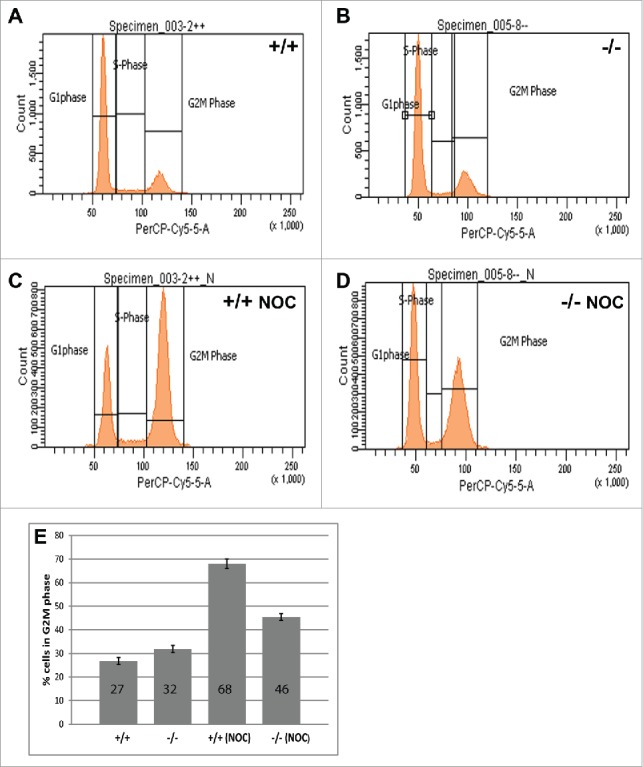
B56γ- MEFs arrest less efficiently in G2/(M)following nocodazole treatment. (A-D) DNA content was measured by flow cytometry using propidium iodide in untreated wild type (+/+) and B56γ- (−/−) MEFs. No difference in DNA content was seen between cell populations in untreated cells (A, B). DNA content analysis following 18 hour incubation in 200 ng/ml nocodazole (NOC) shows less G2/M B56γ- MEFs compared with wild type cells (C, D). (E) Percentage of nocodazole treated cells with G2/M DNA content, B56γ- 45%, wild type 68%. The mean and SEM were calculated from 10 experiments.
Nocodazole treated B56γ- MEFs have fewer M phase cells compared with wild type
To identify cells in M phase, an antibody specific for phosphorylation of histone H3 at Ser10 (pH3) that occurs during chromatin condensation was used.25 Following nocodazole treatment, B56γ- MEFs (Fig. 2C, D) had a lower number of pH3 positive cells compared with wild type (Fig. 2A, B), indicating fewer cells in M phase. Image analysis showed that there are just 10% cells in M phase in nocodazole treated B56γ- MEFs as compared with 21% in the wild type (Fig. 2E). No difference was observed in the number of cells in M phase in untreated wild type and B56γ- MEFs (2E, images not shown).
Figure 2.
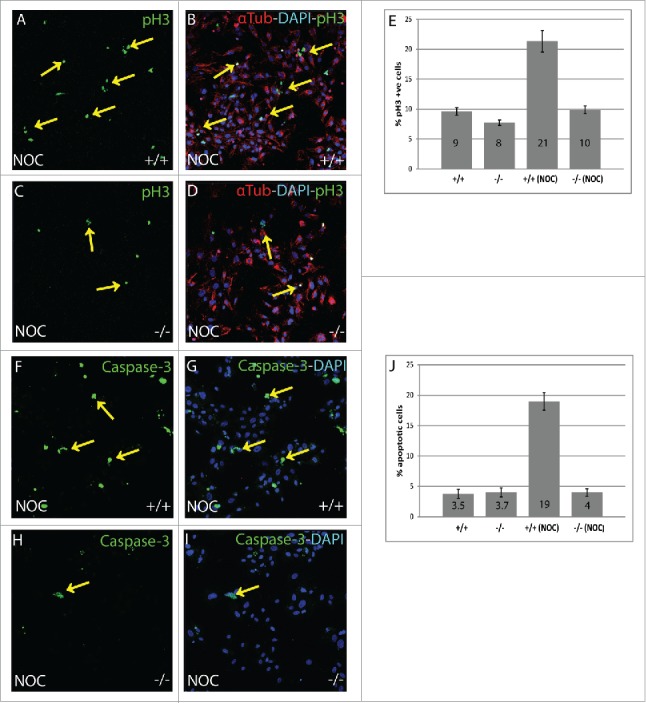
Nocodazole treated B56γ- MEFs have fewer (M)phase cells compared with wild type. (A-D) Immunostaining showing Histone H3 pSER10 (pH3, green, arrows), DAPI (Blue) labeling in conjunction with α-Tubulin (red) in wild type (+/+) and B56γ- (−/−) MEFs. Lower number of cells in M phase in B56γ- MEFs (C, D) were observed as compared with wild type (A, B), following 18 hours of nocodazole treatment. Images are representative of 3 independent experiments from each genotype group. All the images were captured at 60X magnification. (E) Percent pH3+ cells and SEM represent data from 300 MEFs of each genotype. (F-I) Caspase-3 labeling (green) and DAPI (blue), showing apoptotic cells in wild type (+/+) and B56γ- (−/−) MEFs. Higher numbers of apoptotic cells were observed in wild type MEFs (F, G) as compared with the B56γ- MEFs (H, I) following 18 hours of nocodazole treatment (NOC). Images are representative of 3 independent experiments from each genotype group. (J) Percent apoptotic cells and SEM represent data from 300 MEFs of each genotype.
A portion of B56γ- MEFs are able to overcome nocodazole induced apoptosis
The lower number of M phase cells in B56γ- MEFs following nocodazole treatment could be due to a higher level of cell death. Therefore, an apoptosis assay was performed to detect apoptotic cells using a Caspase-3, apoptosis specific antibody.26 Nocodazole treated B56γ- MEFs (Fig. 2H, I) had fewer apoptotic cells as compared with the wild type (Fig. 2F–G). There were only 4% apoptotic cells in B56γ- MEFs as compared with 19% in wild type (Fig. 2J) so the reduction in G2/M B56γ- MEFs is not due to increased apoptosis. Untreated wild type and B56γ- MEFs showed fairly equal amount of apoptotic cells (2J, images not shown). This result coupled with the reduction in the number of mitotic B56γ- MEFs after nocodazole treatment is consistent with a portion of the B56γ- cells bypassing the mitotic checkpoint and undergoing cell division.
Chromosomal abnormalities are detected in B56γ- MEFs
If nocodazole treated B56γ- MEFs proceed through the SAC then it would be expected that some of the cells would have difficulties in chromosomal segregation. Indeed, lagging or misaligned chromosomes were detected by immunohistochemistry at the rate of about 2% per 1000 total cells in B56γ- MEFs following nocodazole treatment (Fig. 3A–D). Live cell imaging was then used to visualize the fate of individual cells as they progressed from nuclear envelop breakdown (NEBD) through mitosis. Abnormal chromosomal segregation was detected in 62% (13 out of 21) of mitotic B56γ- MEFs treated with nocodazole (Fig. 3E and supplemental data). Five of the B56γ- MEFs arrested at NEBD and 3 went through mitosis without observable defects. In contrast, all 21 B56γ+ cells that were imaged did not proceed through mitosis.
Figure 3.

Chromosomal abnormalities are detected in B56γ- MEFs. (A-D) Immunohistochemical analysis with antibodies specific for Histone H3 pSER10 (pH3, green), DAPI (blue) and α-Tubulin (red), showing lagging or misaligned chromosomes (shown with yellow arrows) in the B56γ- MEFs treated with nocodazole. Representative images were obtained using a 100X objective. Chromosomal lagging was recorded at approximately 2% per 1000 total cells in nocodazole treated B56γ- MEFs. Data are representative of 3 independent experiments, using 3 B56γ- and B56γ+ MEF samples. E) Time lapse microscopy live images captured at the indicated time points from NEBD of nocodazole treated B56γ- MEFs. Of the 21 B56γ- MEFs, 13 displayed abnormal chromosomal segregation (shown with yellow arrows).
B56 subunits are present in different intracellular locations
Most prior PP2A-B56 studies have been done by inactivating all 5 B56 genes simultaneously under the assumption that they are functionally redundant. However, we found that inactivation of only the B56γ gene was enough to have an effect on the SAC. To help determine if our B56γ phenotype was due to the lack of expression of other B56 family members in MEFs, or was due to a unique B56γ requirement, we decided to study the native expression of all B56 subunits in MEFs. First, RT-PCR analysis was performed on the mRNA samples obtained from wild type E14.5 MEFs. Low levels of expression were seen for the B56α, B56β and B56δ subunits, whereas B56γ and B56ε were more highly expressed (Fig. 4A). Next, immunostaining was performed to study the cellular localization of these B56 subunits. B56α was found solely in the cytoplasm, primarily in the perinuclear region (Fig. 4B). No B56β protein was detected (Fig. 4C). B56δ was found primarily distributed throughout the cytoplasm, but only in a portion of the MEFs (Fig. 4D). B56γ was seen primarily localized to the nucleus as previously reported22 (Fig. 4E). B56ε was observed primarily in the cytoplasm, but there was also some B56ε detected in the nucleus (Fig. 4F). The localization of B56 subunits other than B56γ were found to be unaltered in the B56γ- MEFs (data not shown). The variable location of the B56 subunits is consistent with them directing PP2A to distinct substrates and functions within the cell.
Figure 4.
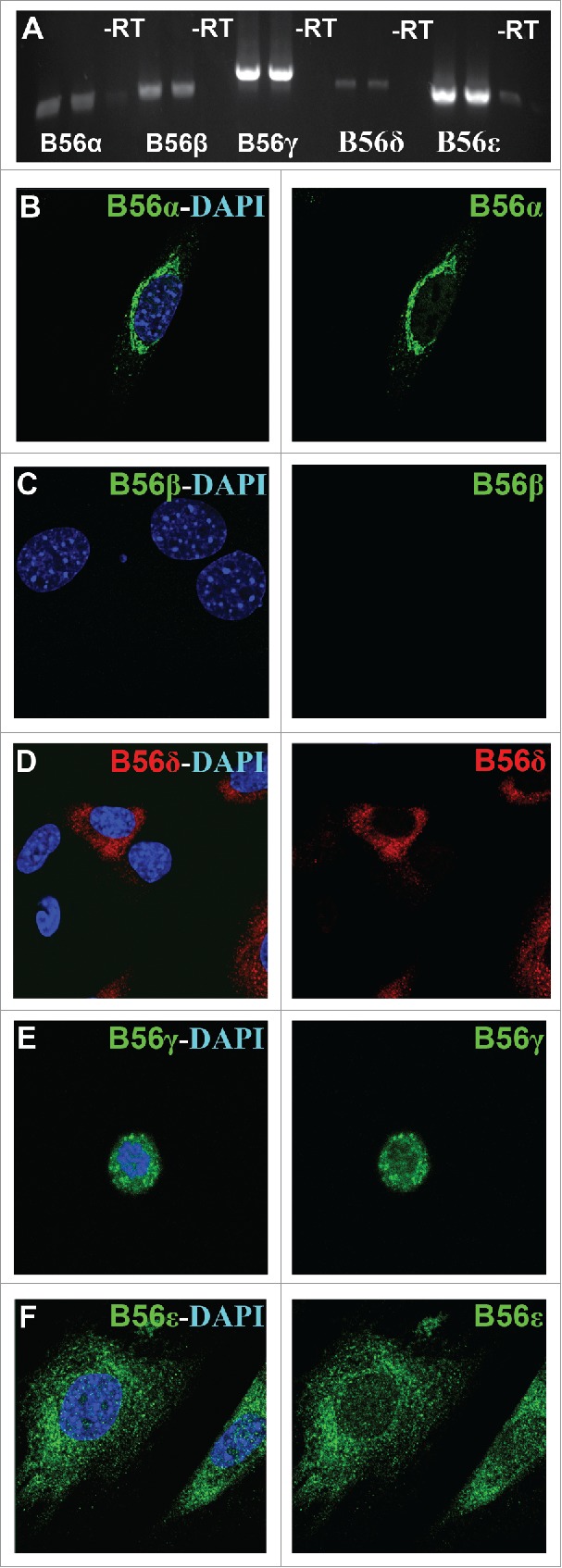
Endogenous expression of B56 subunits in MEFs. A) RT-PCR was used to detect native B56α, B56β, B56γ, B56δ and B56ε mRNA in wild type MEFs, -RT = no transcriptase. (B - F) Immunohistochemical analysis using antibodies to B56α, B56β, B56γ, B56ε (green) and B56δ (red) with DAPI (blue) in wild type MEFs. Representative immunofluorescence images from MEFs at E14.5. Representative images from 3 independent experiments were obtained using a 60X objective.
B56γ and B56ε localization during mitosis
B56γ and B56ε are both found concentrated around condensed chromosomes during prophase (Fig. 5A, E). In metaphase, B56γ and B56ε are similarly localized around the mitotic spindle (Fig. 5B, F). However, in anaphase and telophase, B56γ and B56ε localization differ. In anaphase, B56γ is found at the polar microtubules and mitotic poles (Fig. 5C) while B56ε is found predominately at the polar microtubules (Fig. 5G). In telophase, B56γ is found at the aster and around decondensing chromosomes (Fig. 5D), while B56ε expression in telophase was found to be concentrated in the mid-body (Fig. 5H).
Figure 5.
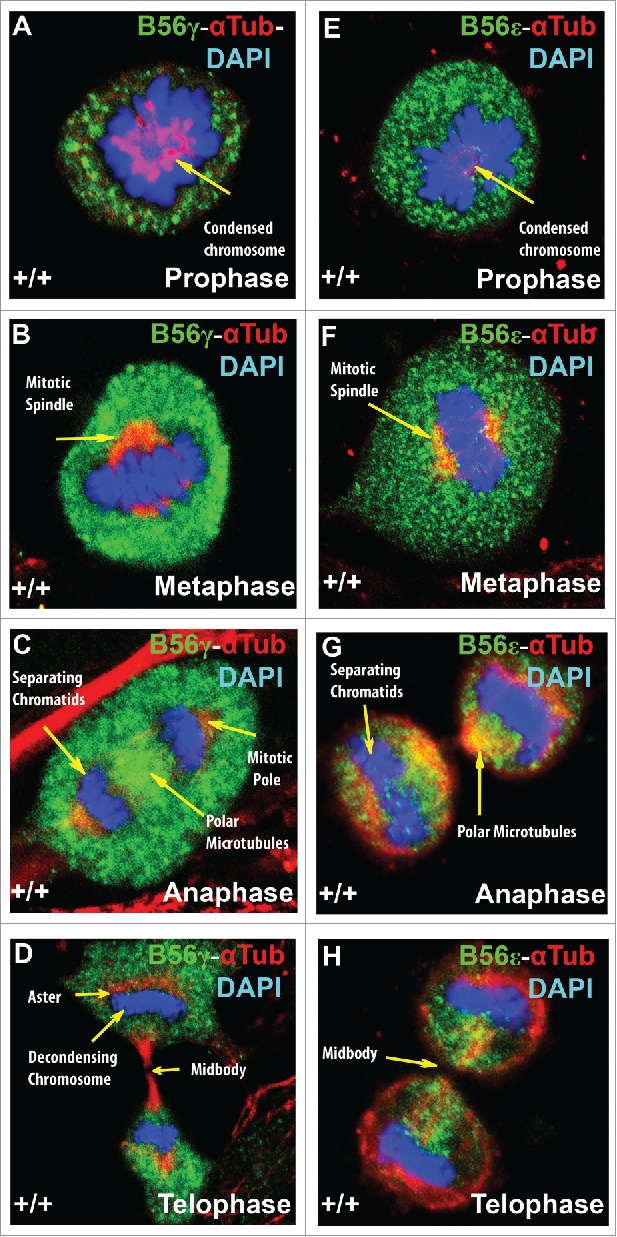
B56γ and B56ε localization during mitosis. Immunostaining of MEFs was done using antibodies against B56γ (green, A - D) or B56ε (green, E- H) along with α Tubulin (red) and DAPI (blue) in prophase, metaphase, anaphase and telophase respectively. Representative immunofluorescence images from MEFs at E14.5. Representative images from 3 independent experiments were obtained using a 60X objective.
B56γ co-localizes with checkpoint complex protein BubR1 during mitosis
Data from other investigators have found that B56γ is a binding partner of BubR1.17,20,21 To understand this interaction in more depth, we performed immunohistochemical studies of B56γ and BubR1 in mitotic MEFs. In prophase, BubR1 and B56γ were seen to co-localize in and around condensing chromosomes (Fig. 6A, B). BubR1 and B56γ are located on the chromosomal circle, as it opens to mobilize the chromosomes to form the metaphase plate (Fig. 6C, D). In metaphase, BubR1 and B56γ are both found at the metaphase plate (Fig. 6E, F). BubR1 and B56γ co-localization continues in anaphase and was observed in separating daughter chromosomes (Fig. 6G, H). BubR1 expression diminishes by telophase and is barely detectable around the decondensing chromosomes (Fig. 6I, J). Thus, B56γ is in proximity of BubR1 throughout mitosis, making it plausible that it could directly affect BubR1s SAC associated functions.
Figure 6.
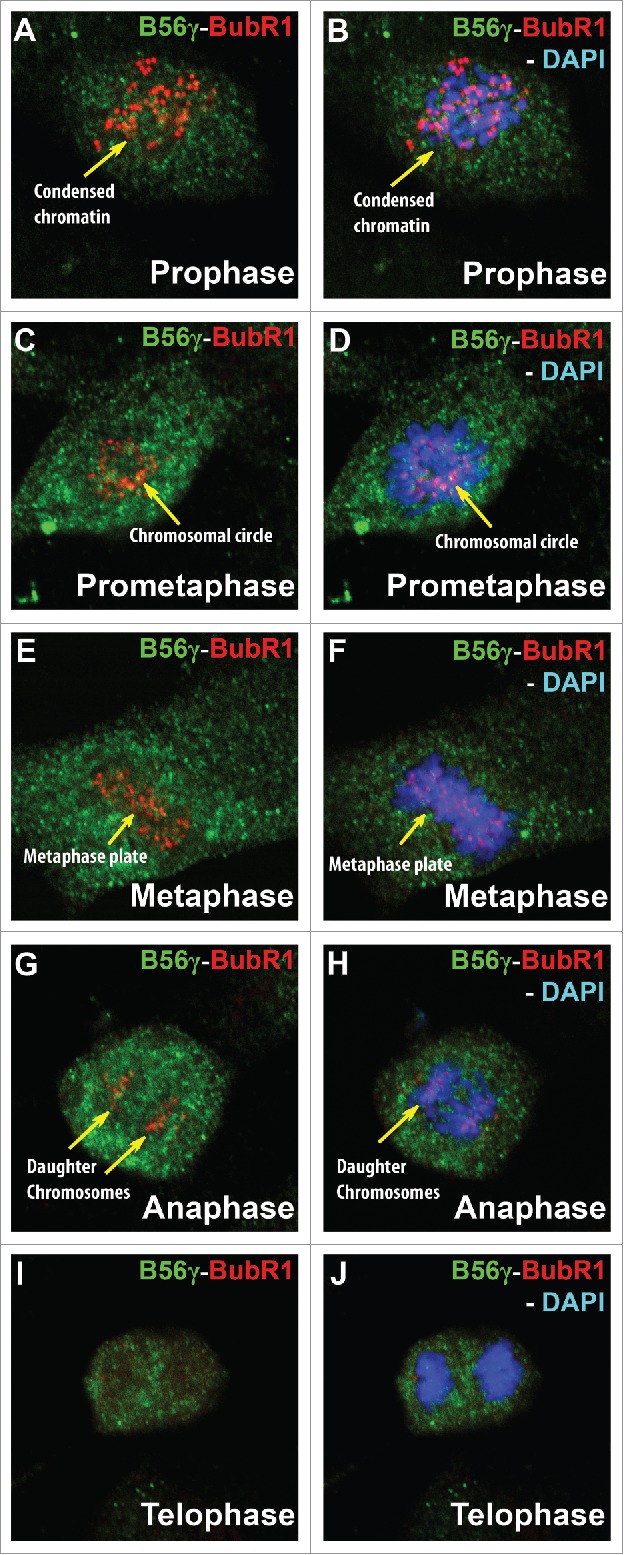
B56γ is present near or co-localizes with BubR1 throughout mitosis. (A, B) Merge images show B56γ (green) and BubR1 (red) in close proximity during prophase in condensed chromatin (yellow arrow). At prometaphase, co-localization was observed at the chromosomal circle (C, D). B56γ and BubR1 were present at the metaphase plate (E, F) and in the separating daughter chromosomes in anaphase (G, H). BubR1 expression diminishes by telophase (I, J). Representative immunofluorescence images from 3 independent experiments were obtained using a 60X objective.
Nocodazole treatment of B56γ- MEFs depletes BubR1 and cyclin B
Western blot analysis was performed using the extracts from synchronized E14.5 MEFs treated with nocodazole. BubR1 expression was found to be reduced in untreated synchronized B56γ- MEFs and nearly absent in the nocodazole treated B56γ- MEFs (Fig. 7). Cyclin B1 was also found to be absent in the nocodazole treated B56γ- MEFs (Fig. 7). Since there is no BubR1 present, the mitotic checkpoint complex formation (MCC) would not be able to form and inhibit the APC/C and thus the lack of Cyclin B1 correlates with the reduction of BubR1. No change was observed in the expression of the catalytic subunit of PP2A.
Figure 7.
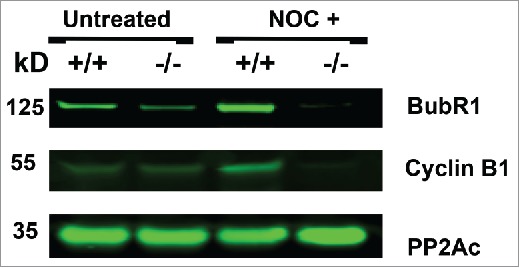
Nocodazole treatment of B56γ- MEFs depletes BubR1 and Cyclin B. Western blot analyses were performed on protein extracts from synchronized wild type (+/+) and B56γ- (−/−) MEFs. Less BubR1 is present in untreated B56γ- MEFs than in wild type MEFs. BubR1 and Cyclin B1 were not detected in the nocodazole treated B56γ- MEFs. No change was observed in the expression of the catalytic subunit of PP2A. Data are representative of 3 experiments, using 3 B56γ- and B56γ+ MEF extracts.
Immunohistochemical analysis BubR1 and cyclin B1 during mitosis
To further study the timing of the BubR1 and Cyclin B1 degradation in B56γ- cells, we performed immunohistochemical analysis during mitosis. B56γ- MEFs (Fig. 8E–H) showed an absence of BubR1 localized to the kinetochore of mitotic cells treated with nocodazole, even in early prophase, when it is normally present there as seen in wild type MEFs (Fig. 8A–D). Less Cyclin B1 was observed in the nocodazole treated B56γ- MEFs (Fig. 8M, N) at early prophase compared with wild type MEFs (Fig. 8I, J). By late prophase, Cyclin B1 was not detectable (Fig. 8O, P) but present in the wild type MEFs (Fig. 8K, L). Premature degradation of Cyclin B1 and BubR1 is consistent with, and could be the cause of the inefficient SAC seen in B56γ- cells treated with nocodazole. No differences were observed in Cyclin B1 or BubR1 localization in B56- mitotic MEFs without the nocodazole treatment (images not shown).
Figure 8.
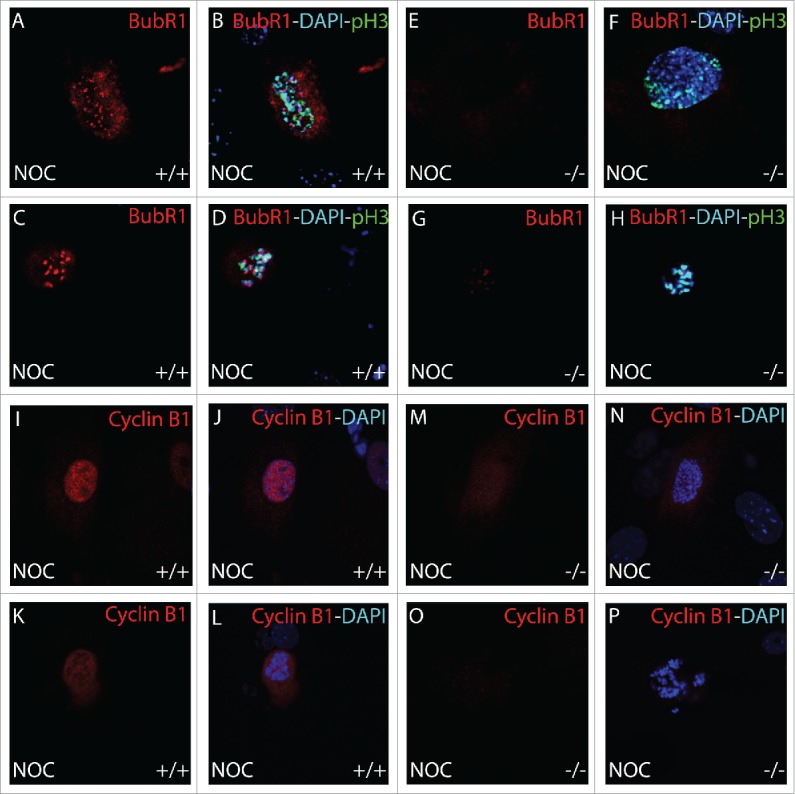
Immunohistochemical analysis of BubR1 and Cyclin B1 during mitosis. BubR1 (red, A-H) or Cyclin B1 (red, I-P), Histone H3 pSer10 (pH3, green) and DAPI (blue) were detected by immunohistochemistry. Following 18 hour nocodazole (NOC), BubR1 expression was found to be absent throughout prophase in B56γ- (−/−) MEFs (E-H) but present in the wild type (+/+) MEFs (A-D). Less Cyclin B1 was observed in the nocodazole treated B56γ- MEFs (M, N) at early prophase compared with wild type MEFs (I, J). By late prophase, Cyclin B1 was not detectable (O, P) but present in the wild type MEFs (K, L). All images were captured at 60X magnification. Data are representative of 3 independent experiments from approximately 50 different cells from each genotype.
Discussion
BubR1 has previously been shown to be required for chromosomal congression and SAC function.16,21,27 These functions rely on the localization of BubR1 to the kinetochore where it acts as a sensor for stable microtubule attachment. Under SAC activating conditions, BubR1 is incorporated into the mitotic checkpoint complex which binds to Cdc20 and inhibits the APC/C so that Cyclin B1 and Securin are not degraded, thus preventing cells from going into anaphase. We found that in B56γ- nocodazole treated cells, BubR1 was not detected in prophase/metaphase cells by immunohistochemistry or Western blot (Figs. 7 and 8). Since BubR1 was not present, apparently due to dysregulation of BubR1 stability, it could not inhibit the APC/C which resulted in the premature proteolysis of Cyclin B1 (Figs. 7 and 8) allowing B56γ- MEFs to escape from the SAC. The mechanism behind the reduced amount of BubR1 in the nocodazole treated B56γ- cells is unknown, but BubR1 stability and its role in the SAC has previously been shown to be regulated by acetylation.28 Since PP2A-B56γ has been shown to directly bind to BubR1, it could disrupt the acetylation or the binding of BubR1 to Cdc20, either scenario could result in decreased SAC activity.29 Alternatively, PP2A-B56γ could also affect BubR1 by either directly dephosphorylating it, or by participation in a phosphorylation signaling pathway that causes the early release of BubR1 from the kinetochore.
When cells were grown under normal conditions, BubR1 was detected at the kinetochore in B56γ- cells as in the wild type cells. This result shows that B56γ is not normally required for BubR1 localization and stability, or that the other remaining B56 family members can compensate for lack of B56γ under normal growing conditions.
Prior studies that examined the interaction between BubR1 and PP2A regulatory units have found that the B56 family was required for chromosome congregation and regulation of the SAC. These studies have mostly assumed that the 5 highly related B56 subunits are all functionally redundant and have been performed with siRNA inhibitors that inactivated all the B56 subunits.15,21 However, for the B56 subunits to be functionally redundant they would have to not only have the same effect on PP2A activity, they would also need to localize to the same regions within the cell. In this study we report that the B56 subunits do not localize to the same intracellular regions (Fig. 4) and therefore cannot be completely functionally redundant. B56α and B56δ were primarily found in the cytoplasm, B56ε was detected in both the nucleus and the cytoplasm, while B56γ was located mostly in the nucleus. In our study, primary cells were used and native protein was detected. In contrast, other studies have used overexpressed, epitope tagged subunits and immortalized cell lines to conclude that all the B56 family members were functionally redundant. Antibody selection is also very important since the B56 subunits have large regions of conserved peptide sequence that may result in antibodies that detect multiple B56 family members. For our experiments, all the antibodies used were made to recognize regions where there is no amino acid conservation between B56 subunits.
In our studies we observed that the inactivation of a single B56 subunit resulted in the inability to efficiently arrest at the SAC (Figs. 1–3). This was true even though both B56ε and B56γ were expressed in similar locations at pre-metaphase, the time when the SAC would be activated by the nocodazole treatment. One possible explanation for this observation would be that B56ε and B56γ are both performing the same function and that removal of the B56γ protein results in reduction in SAC efficiency due to a dosage effect. The other explanation is that the 2 subunits are performing different functions, which is consistent with their different patterns of expression at anaphase and telophase as well as during interphase. Additional studies using cells that have combinations of genetically inactivated B56 subunits will be needed to further define the in vivo functions of B56 subunits.
Treatment of B56γ- MEFs with nocodazole resulted in less cells arrested in mitosis and less apoptotic cells than observed in wildtype MEFs grown under the same conditions. These results are consistent with B56γ- MEFs bypassing the SAC prometaphase arrest that nocodazole induces due to the inhibition of microtubule formation and stable spindle attachment to the kinetochores. If the SAC is bypassed, it can result in cells with aneuploidy and abnormal chromosome distribution, which are hallmarks of cellular transformation. The function of PP2A-B56γ in the efficient implementation of the SAC could be important to the ability of the B56 regulatory subunits to behave as putative anti-oncogenes.24,30 B subunit inactivating mutations found in tumor samples and the ability of SV40 small T antigen to transform cells when it replaces PP2A regulatory B subunits in PP2A heterotrimers,23 further demonstrate the importance that the B56 regulatory subunits play in maintaining stable cell growth.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Steven Bauer and Deborah Hursh for valuable comments that improved the manuscript. J. J. Dyson was supported by an appointment to the Research Participation Program at the Center for Biologics Evaluation and Research administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration.
References
- [1].Etemad B, Kops GJ. Attachment issues: kinetochore transformations and spindle checkpoint silencing. Curr Opin Cell Biol 2016; 39:101-8; PMID:26947988; https://doi.org/ 10.1016/j.ceb.2016.02.016 [DOI] [PubMed] [Google Scholar]
- [2].Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol 2012; 22:R966-80; PMID:23174302; https://doi.org/ 10.1016/j.cub.2012.10.006 [DOI] [PubMed] [Google Scholar]
- [3].Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol 2013; 14:25-37; PMID:23258294; https://doi.org/ 10.1038/nrm3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Funk LC, Zasadil LM, Weaver BA. Living in CIN: Mitotic Infidelity and Its Consequences for Tumor Promotion and Suppression. Dev Cell 2016; 39:638-52; PMID:27997823; https://doi.org/ 10.1016/j.devcel.2016.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 2007; 8:379-93; PMID:17426725; https://doi.org/ 10.1038/nrm2163 [DOI] [PubMed] [Google Scholar]
- [6].Yamagishi Y, Yang CH, Tanno Y, Watanabe Y. MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat Cell Biol 2012; 14:746-52; PMID:22660415; https://doi.org/ 10.1038/ncb2515 [DOI] [PubMed] [Google Scholar]
- [7].Hiruma Y, Sacristan C, Pachis ST, Adamopoulos A, Kuijt T, Ubbink M, von Castelmur E, Perrakis A, Kops GJ. CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science 2015; 348:1264-7. [DOI] [PubMed] [Google Scholar]
- [8].Sacristan C, Kops GJ. Joined at the hip: kinetochores, microtubules, and spindle assembly checkpoint signaling. Trends Cell Biol 2015; 25:21-8; PMID:25220181; https://doi.org/ 10.1016/j.tcb.2014.08.006 [DOI] [PubMed] [Google Scholar]
- [9].Godek KM, Kabeche L, Compton DA. Regulation of kinetochore-microtubule attachments through homeostatic control during mitosis. Nat Rev Mol Cell Biol 2015; 16:57-64; PMID:25466864; https://doi.org/ 10.1038/nrm3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Funabiki H, Wynne DJ. Making an effective switch at the kinetochore by phosphorylation and dephosphorylation. Chromosoma 2013; 122:135-58; PMID:23512483; https://doi.org/ 10.1007/s00412-013-0401-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Molecular cell 2009; 33:537-45; PMID:19285938; https://doi.org/ 10.1016/j.molcel.2009.02.015 [DOI] [PubMed] [Google Scholar]
- [12].Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, Yu JW, Strack S, Jeffrey PD, Shi Y. Structure of the protein phosphatase 2A holoenzyme. Cell 2006; 127:1239-51; PMID:17174897; https://doi.org/ 10.1016/j.cell.2006.11.033 [DOI] [PubMed] [Google Scholar]
- [13].McCright B, Rivers AM, Audlin S, Virshup DM. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem 1996; 271:22081-9; PMID:8703017; https://doi.org/ 10.1074/jbc.271.36.22081 [DOI] [PubMed] [Google Scholar]
- [14].Flegg CP, Sharma M, Medina-Palazon C, Jamieson C, Galea M, Brocardo MG, Mills K, Henderson BR. Nuclear export and centrosome targeting of the protein phosphatase 2A subunit B56alpha: role of B56alpha in nuclear export of the catalytic subunit. J Biol Chem 2010; 285:18144-54; PMID:20378546; https://doi.org/ 10.1074/jbc.M109.093294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Foley EA, Maldonado M, Kapoor TM. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol 2011; 13:1265-71; PMID:21874008; https://doi.org/ 10.1038/ncb2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Suijkerbuijk SJ, Vleugel M, Teixeira A, Kops GJ. Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore-microtubule attachments. Dev Cell 2012; 23:745-55; PMID:23079597; https://doi.org/ 10.1016/j.devcel.2012.09.005 [DOI] [PubMed] [Google Scholar]
- [17].Kruse T, Zhang G, Larsen MS, Lischetti T, Streicher W, Kragh Nielsen T, Bjorn SP, Nilsson J. Direct binding between BubR1 and B56-PP2A phosphatase complexes regulate mitotic progression. J Cell Sci 2013; 126:1086-92; PMID:23345399; https://doi.org/ 10.1242/jcs.122481 [DOI] [PubMed] [Google Scholar]
- [18].Meppelink A, Kabeche L, Vromans MJ, Compton DA, Lens SM. Shugoshin-1 balances Aurora B kinase activity via PP2A to promote chromosome bi-orientation. Cell Rep 2015; 11:508-15; PMID:25892238; https://doi.org/ 10.1016/j.celrep.2015.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tang Z, Shu H, Qi W, Mahmood NA, Mumby MC, Yu H. PP2A is required for centromeric localization of Sgo1 and proper chromosome segregation 1. DevCell 2006; 10:575-85. [DOI] [PubMed] [Google Scholar]
- [20].Xu P, Virshup DM, Lee SH. B56-PP2A regulates motor dynamics for mitotic chromosome alignment. J Cell Sci 2014; 127:4567-73; PMID:25179604; https://doi.org/ 10.1242/jcs.154609 [DOI] [PubMed] [Google Scholar]
- [21].Xu P, Raetz EA, Kitagawa M, Virshup DM, Lee SH. BUBR1 recruits PP2A via the B56 family of targeting subunits to promote chromosome congression. Biol Open 2013; 2:479-86; PMID:23789096; https://doi.org/ 10.1242/bio.20134051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Varadkar P, Despres D, Kraman M, Lozier J, Phadke A, Nagaraju K, McCright B. The protein phosphatase 2A B56gamma regulatory subunit is required for heart development. Dev Dyn 2014; 243:778-90; PMID:24425002; https://doi.org/ 10.1002/dvdy.24111 [DOI] [PubMed] [Google Scholar]
- [23].Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene 2005; 24:7746-55; PMID:16299534; https://doi.org/ 10.1038/sj.onc.1209038 [DOI] [PubMed] [Google Scholar]
- [24].Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor suppressor. CurrOpinGenetDev 2005; 15:34-41. [DOI] [PubMed] [Google Scholar]
- [25].Juan G, Traganos F, James WM, Ray JM, Roberge M, Sauve DM, Anderson H, Darzynkiewicz Z. Histone H3 phosphorylation and expression of cyclins A and B1 measured in individual cells during their progression through G2 and mitosis. Cytometry 1998; 32:71-7; PMID:9627219; https://doi.org/ 10.1002/(SICI)1097-0320(19980601)32:2%3c71::AID-CYTO1%3e3.0.CO;2-H [DOI] [PubMed] [Google Scholar]
- [26].Mamada H, Sato T, Ota M, Sasaki H. Cell competition in mouse NIH3T3 embryonic fibroblasts is controlled by the activity of Tead family proteins and Myc. J Cell Sci 2015; 128:790-803; PMID:25588835; https://doi.org/ 10.1242/jcs.163675 [DOI] [PubMed] [Google Scholar]
- [27].Kapanidou M, Lee S, Bolanos-Garcia VM. BubR1 kinase: protection against aneuploidy and premature aging. Trends Mol Med 2015; 21:364-72; PMID:25964054; https://doi.org/ 10.1016/j.molmed.2015.04.003 [DOI] [PubMed] [Google Scholar]
- [28].Park I, Lee HO, Choi E, Lee YK, Kwon MS, Min J, Park PG, Lee S, Kong YY, Gong G, et al.. Loss of BubR1 acetylation causes defects in spindle assembly checkpoint signaling and promotes tumor formation. J Cell Biol 2013; 202:295-309; PMID:23878276; https://doi.org/ 10.1083/jcb.201210099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Malureanu LA, Jeganathan KB, Hamada M, Wasilewski L, Davenport J, van Deursen JM. BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev Cell 2009; 16:118-31; PMID:19154723; https://doi.org/ 10.1016/j.devcel.2008.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J 2016; 283:1004-24; PMID:26507691; https://doi.org/ 10.1111/febs.13573 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.