

Abstract
Megalencephaly and macrocephaly present with a head circumference measurement 2 standard deviations above the age-related mean. However, even if pathologic events resulting in both megalencephaly and macrocephaly may coexist, a distinction between these two entities is appropriate, as they represent clinical expression of different disorders with a different approach in clinical work-up, overall prognosis, and treatment. Megalencephaly defines an increased growth of cerebral structures related to dysfunctional anomalies during the various steps of brain development in the neuronal proliferation and/or migration phases or as a consequence of postnatal abnormal events. The disorders associated with megalencephaly are classically defined into 3 groups: idiopathic or benign, metabolic, and anatomic. In this article, we seek to underline the clinical aspect of megalencephaly, emphasizing the main disorders that manifest with this anomaly in an attempt to properly categorize these disorders within the megalencephaly group.
Keywords: brain dysfunction, large head, macrocephaly, megalencephaly, metabolic disorders
1. Introduction
Measuring the head circumference is an essential component of the physical examination in pediatric practice and in particular in neuropediatric assessment. The measurement of the head circumference is a challenge for pediatricians, as it is not easy to carry out in young children. In some cases, a correct reading of the head measurement may be particularly unfeasible, as the child may be too much restless or the tape measure may be not placed properly and the patient's hair may be too thick to give a correct result. A proper measure of the head circumference should be performed by putting the tape measure along the most prominent diameter of the occiput and the mid forehead; then, the results of the measurement must be checked with the head circumference growth charts, according to the age, gender, and height parameters.[1] Correlation with the maternal and paternal head circumference is useful. The newborn brain is reported to weigh about 370 g and increases about 4-fold from infancy to childhood till reaching an adult's weight of about 1500 g.[2]
2. Distinguishing between megalencephaly and macrocephaly
By definition, both in megalencephaly and macrocephaly, the measurement of head circumference is reported to be 2 standard deviations (SDs) above the age-related mean. Distinction between these two abnormally large head circumferences is useful because they represent clinical expression of different disorders with different approaches in clinical work-up, overall prognosis, and treatment.[3,4] Sometimes, this distinction is not performed, and the terms are not appropriately referred in scientific papers but instead are used interchangeably. Megalencephaly defines an increased growth of cerebral structures related to dysfunctional anomalies during the various steps of brain development in the neuronal proliferation and/or migration phases or as a consequence of postnatal abnormal events that cause excessive cerebral growth. In contrast, in macrocephaly, the increased head circumference is linked to various events that can result in an increase of orbito-frontal head circumference for age, including anomalies of bone skull structures, subdural fluid collections, hydrocephalus, intracranial masses, and arteriovenous malformations.
However, sometimes, adverse events resulting in both megalencephaly and macrocephaly may coexist in the same individual.
Megalencephaly is maintained to be prognostically more severe than macrocephaly and has been reported more frequently in the group of patients with intellective delay, epilepsy, and drug-resistant epilepsy.
In a clinical scenario, distinction between megalencephaly and macrocephaly is useful. In this article, we seek to underline the clinical aspects of megalencephaly, emphasizing the main disorders that manifest with this anomaly, in an attempt to properly distinguish these disorders within the category of megalencephaly.
3. Megalencephaly classification
Fletcher [5] is credited to have introduced the term megalencephaly while reporting a patient with this anomaly. The same author and successively Wilson [6] backed up that this term should be limited to patients with cerebral overgrowth in association with neurological dysfunction.[7] Megalencephaly may involve all the encephalon or only a localized portion. Ono et al[8] reported 3 neurologically normal adolescents who showed enlargement of the right frontal lobe with increased volume of subcortical and deep white matter and thickening of the ipsilateral genu of the corpus callosum. At present, megalencephaly is indicated by an oversized and overweight brain, aside from any neurological dysfunction. Megalencephaly is classically defined into 3 groups, according to the etiology: idiopathic or benign, metabolic, and anatomic (Fig. 1).
Figure 1.
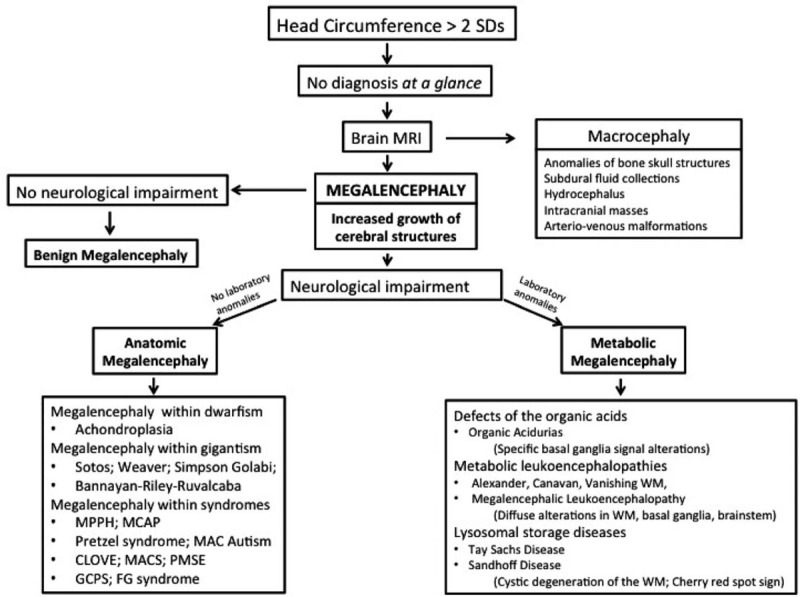
Diagnostic flowchart for increased head circumference in children.
Benign or idiopathic megalencephaly refers to children who have an abnormally large head with no neurological impairment. An increased head circumference is often reported in one or both the parents. In these individuals, the head circumference gradually increases from infancy to become stable at around 18 months of age or more with normal cerebral development.[4]
4. Metabolic megalencephaly
Some inborn errors of metabolism may manifest with megalencephaly. Diagnosis of this disorder is based on specific neurological features associated with the megalencephaly. Familial history of similar disorders with a recessive hereditary pattern, either autosomal or sex-linked, or a history of marriages among relatives may be suggestive for a metabolic disorder. The clinical course is clearly progressive with a more or less rapid neurological impairment, which may also involve other organs, including the eyes, heart, spleen and liver, skin, and muscles. In these patients, increased intracranial pressure may also be present.
Physical examination in pediatric patients with megalencephaly and metabolic disorders show signs of neurological impairment, such as large or tense fontanels, enlarged sutures, the sun-setting sign, hypotonia, irritability, or, in older children, neurodevelopment delay, lethargy, and/or seizures. In these patients, the physical examination should be extended to the other organs; hepatosplenomegaly and/or other anomalous manifestations may indicate the diagnosis of neurometabolic disease. Laboratory findings are mandatory for a diagnosis of metabolic impairment through the research of enzyme or chemical defect, which can be observed in serum, urine, cerebrospinal fluid, and tissue culture. Some of these patients may die before the head circumference ever reaches the maximum level.
Three groups of metabolic disorders have been suggested to be associated with megalencephaly: defects of the organic acids, metabolic leukoencephalopathies or “metabolic encephalopathies”, and lysosomal storage diseases.[4,9]
4.1. Defects of organic acids
Defects of organic acids include glutaric aciduria type 1 (gene defect GCDH) and L–2-hydroxyglutaric aciduria (gene defect L2HG). Glutaric aciduria type 1 is caused by deficiency of glutaryl-CoA dehydrogenase involved in the pathway of lysine to hydroxylysine and L-tryptophan. Abnormal head circumference is present since birth and is the earliest and most distinctive sign of this disorder. The enzymatic defect causes an accumulation of glutaric acid and 3-hydroxyglutaric acid that interferes in the energetic metabolism and oxidative stress, provoking neuronal impairment.[10] Untreated patients present with dystonic movement disorders in infancy. Brain MRI presents with widening of Sylvian fissure (batwing appearance), diffuse white matter signal abnormalities, and bilateral high signal in the basal ganglia. Fronto-temporal atrophy has also been reported.[11] The pathological features are characterized by striatal injury consequent to encephalopathic crises associated with frequent infectious episodes.[12]
L-2-hydroxyglutaric aciduria includes a group of neurometabolic disorders in which the metabolic defect causes an elevated elimination of hydroxyglutaric acid in the urine. L-2-hydroxyglutaric aciduria is the most common and severe form that manifests with neurological impairment, large head, cerebellar sign, and epileptic seizures.[13] The MRI images reported by Steenweg et al [14] displayed a cerebral white matter involvement that mainly affected the frontal and subcortical regions. The abnormal subcortical white matter anomalies show a mildly swollen appearance with an initial aspect partially multifocal. Neuropathologic examination shows mild cortical neuronal loss with intense gliosis, spongiosis, and vacualitation of the neuronal cells. Subcortical white matter contains numerous hyperplastic astrocytes with severe demyelinization and cystic cavities.[15]
D-2-hydroxyglutaric aciduria is caused by recessive mutation in D2HGDH (type 1) or by dominant gain-of-function mutations in IDH2 (Type 2). In this form, the megalencephaly is often present and is associated with cardiomyopathy, hypotonia, and intellectual delay. Brain MRI shows confluent subcortical white matter lesions that spread centrifugally with atrophy of the cerebellar vermis and involvement of dentate nuclei.[16]
4.2. Metabolic encephalopathies
Canavan disease is caused by deficiency of aspartoacylase that leads to the accumulation of N-acetylaspartic acid (NAA) in the brain and in white matter. In this disorder, there is a degeneration of myelin in the phospholipid layer that isolates the axon. The NAA in a high concentration results in myelin vacuolization and astrocyte swelling. The clinical features in the infantile form are characterized by a rapidly increasing head circumference, severe hypotonia, and irritability with an onset at around 3 to 6 months of age. Subsequently, feeding difficulties with poor growth become more evident as the delayed milestones. Hypertonia, joint stiffness, and seizures develop rapidly, and most of the patients die in the first decade of life. MRI reveals diffuse white matter degeneration mainly involving cerebral hemispheres with extensive thickening of the white matter. Diagnosis is made in the presence of high levels of urinary NAA.[17] Pathologic studies show spongy degeneration of white matter with no specific morphologic changes.
Alexander disease has been the first identified primary genetic disorder of astrocytes and is caused by mutations in the gene encoding glial fibrillary acidic protein (GFAP). Progressive neurological impairment, megalencephaly, and a typical MRI pattern are classically recognized as diagnostic. The brain MRI shows characteristic symmetric and extensive abnormalities with frontal predominance and relative sparing of occipital and temporal white matter.[18]
Van der Knaap et al,[19] in a report concerning the brain MRI of patients with Alexander disease, have identified 5 criteria, 4 of which are remarkable, for the diagnosis of the disease: an extensive cerebral white matter change with frontal predominance; a periventricular rim with high signal on T1-weighted images and low signal on T2-weighted images; abnormalities of basal ganglia and thalami; brain stem abnormalities; and contrast enhancement of particular gray and white matter structures. The pathological findings show a brain that is typically too large and too heavy; extensive paucity of myelin is found in the hemispheres in which frontal lobes are more affected than the others. The presence of Rosenthal fibers throughout the central nervous system (CNS) is the pathological hallmark of the disorder. A notable increase of astrocytes filled with Rosenthal fibers is reported to mainly affect the subpial, perivascular, and subependymal pattern. Rosenthal fibers are mainly found in the outer, subpial layers of the cerebral cortex, in frontal white matter, and near the periventricular area. [20,21]
Megalencephalic leukoencephalopathy (MLC) with subcortical cysts associated with MLC1 and HEPACAM gene mutations and leukoencephalopathy with vanishing white matter, which is linked to mutations in EIFB1, EIFB2, EIFB3, EIFB4, and EIFB5 (eukaryote translational initiation factor B1-B5) genes, have been reported in patients presenting an abnormal head circumference. In MLC, the large head may be present at birth, but more often it appears during the first year of life. The degree of abnormal head circumference may reach 4 to 6 SDs above the average. After the first year, the head growth tends to normalize and to stabilize within the 98th percentile. Patients present with hypotonia, cognitive delay, and seizures.[22] The brain MRI shows diffusely abnormal and mildly swollen white matter. Subcortical cysts are almost invariably present in the anterior temporal region and often in the frontal-parietal region. The subcortical cysts tend to increase in size and number.[23]
In leukoencephalopathy with vanishing white matter, the clinical signs begin to appear during early childhood with spasticity and motor incoordination. The pathological findings show a gray matter of normal structure, while the white matter appears rarefied with a small number of axons and U-fibers and presence of small cavities in the white matter. The presence of foamy oligodendrocytes is typical of this disorder. The brain MRI displays reversal of signal intensity of the white matter. Recovery sequences and holes in the white matter are found.[23,24]
4.3. Lysosomal storage diseases
Lysosomal storage diseases are also associated with an abnormally large brain. Tay–Sachs disease (TSD) is a disorder due to mutations in the HEXA gene located on chromosome 15q23–24. Two forms of β-hexosaminidase are recognized: β-hexosaminidase A [HEXA] is a heterodimer comprised of an α-acid and a β subunit, while β-hexosaminidase B [HEXB] is formed by two β subunits. The clinical signs begin at the age of 6 months with a progressive delay of the developmental milestones and hypotonia. A large head becomes apparent by 1 year of age due to an abnormal content of the ganglioside GM2, which accumulates in the brain. Short stature, progressive spasticity, and seizures with visual degeneration and deafness are the presenting clinical signs. Cherry red spots at the fundoscopic examination are useful for diagnosis. Pathologic examination displays massively increased weight and volume of the brain, which may weigh more than twice than a normal brain. Cystic degeneration of the cerebral white matter (status spongiosus) and atrophy of the cerebellar hemispheres are frequently observed.[25]
Sandhoff disease is linked to an inherited deficiency of HEXA and HEXB that are needed for the degradation of the neuronal membrane components (ganglioside GM2, its derivate GA2, the glycolipid globoside). The HEXB gene is located on chromosome 5q13. As for TSD, signs begin at the age of 6 months with large head, seizures, cherry red spots, and doll-like facies.[26] In the brain, an amount of GM2 asialoganglioside that is 300 times more than normal is found in affected patients.[25]
Large head circumference above the normal has been reported in patients with other metabolic disorders, but they do not have all the characteristics to be included in the group of megalencephaly, for example, Mucopolisaccharidoses (MPS) type 1-Hurler (gene mutation IDLIA), MPS II-Hunter syndrome (IDS gene), and MPS III-Sanfilippo syndrome (SGSH, HAGLN, HGSNAF, GNS genes). In these patients, the head circumference is usually normal at birth, and it gradually increases in the first years of life (usually between 1 and 3 years). The increased head circumference in Type I-Hurler and type II-Hunter is linked to chronic communicating hydrocephalus, progressive cerebral storage of glycosaminoglycans, enlarged cerebral perivascular spaces, and neuroinflammation. In MPS III, the increase of head circumference is noticed in the first decade, but there is a progressive decrease, and in adulthood, the head circumference, due to progressive brain atrophy, returns to a normal range.
Volumetric analysis of cortical gray matter, cortical white matter, corpus callosum, and frontal lobes in MPS II, as reported by Yund et al,[27] notes that the mean volumes are larger, but they are not statistically significant different than controls. In a large study of 118 patients with MPS III by de Ruijter et al,[28] mean head circumference SDs according to age was 0.88 for all patients (boy 0.71, girl 1.1, P < .001). The data reported by Yund et al[27] confirmed our experience of several patients followed in the metabolic center of the University of Catania.
Large head has been anecdotally described in patients with Krabbe disease, but as reported by Barone et al [29] in 11 patients with classic infantile and late-onset Krabbe disease, no patients with an abnormally large head have been found.
5. Anatomic megalencephaly
This group of disorders manifests with developmental megalencephaly linked to a single gene mutation involving early brain cellular growth, migration, or replication. Mutation in the mammalia Target of Rapamyicin (mTOR), mitogen-activated protein kinase, originally called “extracellular signal-regulated kinases” (MAPK/ERK), and Sonic hedgehog (SHH) pathways have been frequently reported as pathogenetic events causing megalencephaly as a single anomaly or in association with other body structural anomalies.[4]
5.1. Megalencephaly within dwarfism
Achondroplasic patients have a mutation at the FGF3 gene, codon 380. In these patients, the cranium is disproportionately large relative to height, but it is unusual to reach level above 2 SDs. The patients present with a prominent forehead, the nasal bridge is moderately flat, and the chest is narrow. Most of these signs may be present at birth. The cognitive aspect is normal, but cerebral complication may be frequent.[30]
5.2. Megalencephaly within gigantism (more often associated with ventricular megalencephaly)
Sotos syndrome (also known as cerebral gigantism) is a prenatal and postnatal overgrowth syndrome linked to a mutation of the gene encoding the nuclear receptor set domain containing protease 1 (NSD1) on chromosome 5q35. Patients present with tall stature, large head, distinctive craniofacial features, gait dyspraxia, seizures, and developmental delay. Some of these patients also present an auto- and hetero-aggressivity causing severe damage. Height tends to normalize after puberty, whereas the head circumference persists to be large.[31]
Brain neuroimaging shows abnormalities involving the cavum septum pellucidum and cavum vergae, hypoplasia of corpus callosum, enlarged subarachnoidal spaces, ventricle dilatation, slight hypodensity of the white matter, and cerebral atrophy.[31] Schaefer and Buehler [32] reported a neuroradiologic study performed in 40 patients affected by Sotos syndrome. Most of the anomalies were found in the ventricular system with ventricles enlarged, and prominence of the trigone and occipital horns, extracerebral supratentorial, and increased posterior fossa fluid spaces; gray matter heterotopias, periventricular leukomalacia, periventricular leukomalacia, cavum septum pellucidum, and corpus callosum anomalies were also reported.
Phenotypic overlap between Sotos and Weaver syndrome has been widely reported. In Weaver syndrome, tall stature has been reported as the most common feature observed in 90% and intellectual disability in 80% of patients.[33] Additional clinical features include camptodactyly, soft and doughy skin, umbilical hernia, and a low hoarse cry.
Simpson–Golabi–Behmal syndrome (SGBS) types I and II show clinical signs of multiple congenital abnormalities, including pre-post natal overgrowth, distinctive craniofacial features, large head, and organomegaly. Other anomalies may be observed involving the skeletal system, heart, CNS, kidney, and gastrointestinal tract.[34] Genomic rearrangements and point mutations involving the glycan-3 gene (GPC3) at Xq26 are the causal events associated with SGBS. Large head has been reported in about 70% of children with SGBS. MRI shows midline defect, such as abnormal corpus callosum, central lipomata, and hydrocephalia.[34] Craniosynostosis may be frequently present.
Bannayan–Riley–Ruvalcaba syndrome (BRRS) is a well-known disorder linked to a germline mutation of PTEN. This syndrome, with its allelic disorders including the Cowden syndrome, manifests with an overgrowth of connective tissue and multiple benign hamartomatous lesions and megalencephaly. The Cowden syndrome is associated with a high risk for thyroid, breast, and endometrial cancers. Patients with BRRS present a large head, intestinal polyposis, lipomas, and pigmented penile macules.[35] Bhargava et al [36] reported a brain neuroradiologic study in 7 patients affected by BRRS. They found the presence of white matter cysts localized in the parietal lobe in all the patients, whereas cysts were also reported in the frontal lobe in 3 and in 1 in the temporal lobe, respectively. The cysts were predominantly surrounded by white matter t 2 hyperintensities. Both these syndromes are enclosed in the spectrum of PTEN hamartoma tumor syndromes linked to the germline mutations in the tumor suppressor PTEN gene located in chromosome 10q23.3.[37]
5.3. Megalencephaly within syndromes
Recently, megalencephaly as a presenting sign has been reported in two syndromes, which represent a typical example of anatomic brain overgrowth: megalencephaly–polymicrogyria–polydactily–hydrocephalus (MPPH) and megalencephaly-capillary malformation (MCAP). De novo germline mutations in APT3 and PIK3R2 have been reported in patients with MPPH. This syndrome is characterized by abnormally increased head sizes, reaching levels above 10 SDs. Ventriculomegaly that may progress to hydrocephalus, cerebelllar tonsillar ectopia, and polymicrogyria are other associated anomalies. Cutaneous lesions are accompanying features consisting of capillary malformations and variable connective tissue dysplasia. Mild focal or segmental body overgrowth, together with finger or toe syndactily and postaxial polydactily may be present.[9] MCAP has many features overlapping the MPPH. It has been also linked to the PIK3CA gene. The genes PIK3R2, ATK3, and PIK3CA are members of the phosphatidyl inositol-3kinase of the AKT pathway and are present in all the developmental brain cellular activities, including apoptosis. MPPH shows many of the same features as the MCAP, including the severe megalencephaly, with the exception of the postaxial polydactyly, which is more common and more typical in the MPPA syndrome.[38–40]
5.4. Pretzel syndrome
Polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE), which is also called Pretzel syndrome, is characterized by infantile-onset epilepsy, neurocognitive delay, and craniofacial dysmorphism. PMSE is caused by homozygous deletion of exons 9 to 13 of the LYK5/STRADA gene that encodes the pseudokinase STRADA, an upstream inhibitor of mammalian target of rapamycin complex 1.[41] Polyhydramnios is frequently reported: around 80% of the cases reported present with this anomaly. The psychomotor disturbances are very impressive with cognitive delay, early seizures, and muscle involvement that are hypotrophic. The affected child lies in a particular position termed a “Pretzel-like posture.” Atrial septal defect is reported in one-third of the patients. The large head commonly present in this child as reported by brain MRI is linked to the presence of extracerebral fluid or hydrocephalus in association with excessive cerebral growth. Histopathologic evidence of heterotopic neurons in subcortical white matter and subependymal regions were reported by Puffenberger et al [42] wherein a single postmortem neuropathologic study showed megalencephaly, ventriculomegaly, cytomegaly, and extensive vacuolization and astrocytosis of white matter. A constitutive activation of mTORC1 signaling pathway was reported by the authors in the frontal cortex, basal ganglia, hippocampus, and spinal cord.
5.5. Other syndromes
PTEN gene mutations have been observed in patients with increased head circumference and autistic behavior. Butler et al [43] reported 18 individuals who presented a germline PTEN mutation with X-linked intellectual disability, large head, and neurobehavioral features of autistic spectrum disorder. In these patients (13 males), the head circumerence increase ranged from 2.5 to 8.0 SDs. Large head and autism has been reported in patients with RAB1q mutation.[9]
Congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE) syndrome has been reported in association with CNS malformations and seizures.[44] Among the patients reported by Gucev et al,[44] a newborn girl presented with massive lymphatic truncal vascular malformation with cutaneous venous anomaly and overgrown feet and splayed toes. Cranial computed tomography (CT) showed encephalomalacia, widening of ventricles and the sulci, and hemimegalencephaly. Patients with CLOVE syndrome and megalencephaly have also been reported,[44] but megalencephaly is not a prominent sign in this syndrome.
Macrocephaly, alopecia, cutis laxa, and scoliosis is a rare condition with an autosomal recessive inheritance. Mutations in RIN2 (chromosome 20p11.23) have been linked to this disorder.[45]RIN2 gene encodes the RAS and RAB interactor protein 2, which is involved in cell trafficking. The presenting clinical features consist of progressive facial coarsening, gingival hypertrophy, severe scoliosis, sparse hair, cutis laxa, and joint hyperlaxity.[46]
PMSE [42] is linked to homozygous 7-kilobase deletion in LYK5, which encodes STE20-related adaptor protein, a pseudokinase functionally necessary for proper localization and function of serine/threonine kinase 11 (a.k.a. LKB1). The LYK5 deletion is associated with polyhydramionis, preterm labor, and distinctive craniofacial features in addition to megalencephaly and multifocal seizures. Puffenberger et al [42] reported 16 patients of whom 4 (38%) died during childhood due to status epilepticus, congestive heart failure, and hypernatremic dehydratation. A pathologic examination carried out in a single patient disclosed the presence of megalencephaly, ventriculomegaly, cytomegaly, and extensive vacuolization and astrocytosis in the white matter.
Greig cephalo-polysyndactyly syndrome (GCPS) is linked to a mutation involving the GLI3 protein, which is a zinc finger transcription factor that is expressive in early development. Patients present with preaxial or postaxial polydactily with or without syndactily and craniofacial features, including large head and hypertelorism. Abnormalities located in the corpus callosum have been reported.[47] Démurger et al [48] state that macrocephaly was found in 32 of 53 (60%) patients with GCPS, but in 7 of 18 patients (39%), a severe ventricular dilatation was present.
Acrocallosal syndrome is an autosomal recessive disorder that manifests with corpus callosum agenesis, facial dysmorphism, postaxial polydactily of the hands as well as preaxial polydactyly of the feet. The disorder is linked to a mutation of the KIF7 gene that encodes a molecule within the sonic hedgehog.[49] The reported large head has been related to enlarged ventricles [50] and therefore should not be included in the group of megalencephaly as with the GCPS.
In Opitz–Kaveggia syndrome, also known as “FG syndrome,” the mutation involves the MED12 gene, which is located on Xq13 and is a member of the large mediator complex; it has a relevant role in RNA polymerase II transcription. The mutations of MED12 can cause, in addition to the FG syndrome, the Lujan syndrome and Ohdo syndrome.[51,52] Affected male patients present distinctive facial appearance, mental retardation, large head, imperforate anus, and hypotonia. In the classical report of Opitz and Kaveggia,[53] the syndrome was designed as a multiple congenital anomaly syndrome with signs of relative macrocephaly, broad and flat thumbs, emperforate anus, hypotonia, and moderately severe cognitive delay.
Nguyen et al [54] reported a new autosomal-recessive neurological condition characterized by megalencephaly, thick corpus callosum, and severe intellectual disability. The boy presented with neonatal overgrowth of the occipital-frontal circumference at the 90th percentile, +3 SDs at the age of 14 years and 66.5 cm at 18 years of age. Facial features included a long face with high forehead, prognatism, and long and thin feet and hands. Severe myopia, thick corpus callosum, enlarged white matter, and small cerebellum were also reported. The syndrome seems to be caused by a homozygous nonsense variant in the HERC1 protein that is an ubiquitin ligase that interacts with tuberous sclerosis complex 2, an upstream negative regulator of the mTOR pathway.
Grotto et al [55] reported 5 patients affected by severe intellectual disability linked to BRWD3 nonsense mutation, p.Tyr131, and compared the clinical presentations to those of 4 patients previously reported. The main symptoms consisted of intellectual disability that ranged from mild to moderate (9/9) and speech delay (8/9); also, a large head was present in 7 of 9 patients. Among these, one patient presented with neonatal overgrowth and large head; at the age of 12 years, the weight, length, and head circumference measurements were +3.5 to 4.5 SDs. In this group of patients, dysmorphic features were also present, including high forehead, hypertelorism, short palpebral fissures, anteverted nares, pointed chin, broad hands and feet, joint laxity, genu valgum, and small penis.
Developmental delay labeled macrocephaly syndrome in a large Amish kindred with an autosomal recessive inheritance was described and related to homozygous or compound heterozygous mutations in the KPTN gene encoding kaptin.[56]
6. Conclusions
Megalencephaly is a congenital condition characterized by severe overdeveloped brain size. Several congenital conditions and several molecular mutations may cause megalencephaly, and in most of the cases they are associated with other cerebral and clinical anomalies. The condition has to be distinguished by the macrocephaly that presents with different clinical situations. Among the different gene mutations that cause cerebral overgrowth, particular attention should be given to those involving RTK/P13K/mTOR and MAPK/ERT and SHH pathways.[38–40]
Footnotes
Abbreviations: BRRS = Bannayan–Riley–Ruvalcaba syndrome, CLOVE = Congenital lipomatous overgrowth, vascular malformations, and epidermal nevi, GCPS = Greig cephalo-polysyndactyly syndrome, HEXA = β-hexosaminidase A, HEXB = β-hexosaminidase B, MCAP = Megalencephaly-capillary malformation, MLC = Megalencephalic leukoencephalopathy, MPPH = Megalencephaly-polymicrogyria-polydactily-hydrocephalus, MPS = Mucopolisaccharidoses, MRI = Magnetic Resonance Imaging, NAA = N-acetylaspartatic acid, PMSE = Polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome, SDs = Standard Deviations, SGBS = Simpson–Golabi–Behmal syndrome, TSD = Tay-Sachs disease.
The Authors of the present article do not have any conflict of interest in the subject treated. Ethical committee approval was not necessary.
References
- [1].Sniderman A. Abnormal head growth. Pediatr Rev 2010;31:382–4. [DOI] [PubMed] [Google Scholar]
- [2].Blinkov SM, Glazer II. The Human Brain in Figures and Tables. A Quantitative Handbook. New York: Plenum Press; 1968. [Google Scholar]
- [3].DeMyer W. Megalencephaly: types, clinical syndromes, and management. Pediatr Neurol 1986;2:321–8. [DOI] [PubMed] [Google Scholar]
- [4].Winden KD, Yuskaitis CJ, Poduri A. Megalencephaly and macrocephaly. Semin Neurol 2015;35:277–87. [DOI] [PubMed] [Google Scholar]
- [5].Fletcher HM. A Case of Megalencephaly. Vol 51. 1900;London: Pathological Society of London, 230–232. [Google Scholar]
- [6].Wilson SAK. Megalencephaly. J Neurol 1937;14:193. [Google Scholar]
- [7].DeMeyer W. Megalencephaly in children: clinical syndromes, genetic patterns, and differential diagnosis from other causes of megalocephaly. Neurology 1972;22:634–43. [DOI] [PubMed] [Google Scholar]
- [8].Ono Y, Saito Y, Maegaki Y, et al. Three cases of right frontal megalencephaly: clinical characteristics and long-term outcome. Brain Dev 2016;38:302–9. [DOI] [PubMed] [Google Scholar]
- [9].Mirzaa GM, Poduri A. Megalencephaly and hemimegalencephaly: breakthroughs in molecular etiology. Am J Med Genet C Semin Med Genet 2014;166C:156–72. [DOI] [PubMed] [Google Scholar]
- [10].Jafari P, Braissant O, Bonafé L, et al. The unsolved puzzle of neuropathogenesis in glutaric aciduria type I. Mol Genet Metab 2011;104:425–37. [DOI] [PubMed] [Google Scholar]
- [11].Strauss KA, Puffenberger EG, Robinson DL, et al. Type I glutaric aciduria, part 1: natural history of 77 patients. Am J Med Genet C Semin Med Genet 2003;121C:38–52. [DOI] [PubMed] [Google Scholar]
- [12].Kölker S, Christensen E, Leonard JV, et al. Diagnosis and management of glutaric aciduria type I: revised recommendations. J Inherit Metab Dis 2011;34:677–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Steenweg ME, Jakobs C, Errami A, et al. An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a genotype-phenotype study. Hum Mutat 2010;31:380–90. [DOI] [PubMed] [Google Scholar]
- [14].Steenweg ME, Salomons GS, Yapici Z, et al. L-2-Hydroxyglutaric aciduria: pattern of MR imaging abnormalities in 56 patients. Radiology 2009;251:856–65. [DOI] [PubMed] [Google Scholar]
- [15].Seijo-Martínez M, Navarro C, Castro del Río M, et al. L-2-hydroxyglutaric aciduria: clinical, neuroimaging, and neuropathological findings. Arch Neurol 2005;62:666–70. [DOI] [PubMed] [Google Scholar]
- [16].Nyhan WL, Shelton GD, Jakobs C, et al. D-2-hydroxyglutaric aciduria. J Child Neurol 1995;10:137–42. [DOI] [PubMed] [Google Scholar]
- [17].Matalon R, Michals-Matalon K. Canavan disease. In: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington; 1993–2015. September 16, 1999. [Google Scholar]
- [18].Rodriguez D. Leukodystrophies with astrocytic dysfunction. Handb Clin Neurol 2013;113:1619–28. [DOI] [PubMed] [Google Scholar]
- [19].van der Knaap MS, Naidu S, Breiter SN, et al. Alexander disease: diagnosis with MR imaging. AJNR Am J Neuroradiol 2001;22:541–52. [PMC free article] [PubMed] [Google Scholar]
- [20].Sawaishi Y. Review of Alexander disease: beyond the classical concept of leukodystrophy. Brain Dev 2009;31:493–8. [DOI] [PubMed] [Google Scholar]
- [21].Johnson AB. Alexander disease: a review and the gene. Int J Dev Neurosci 2002;20:391–4. [DOI] [PubMed] [Google Scholar]
- [22].van der Knaap MS, Scheper GC. Megalencephalic leukoencephalopathy with subcortical cysts. In: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington; 1993–2015. August 11, 2003. [updated November 03, 2011]. [Google Scholar]
- [23].van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol 2006;5:413–23. [DOI] [PubMed] [Google Scholar]
- [24].Pronk JC, van Kollenburg B, Scheper GC, et al. Vanishing white matter disease: a review with focus on its genetics. Ment Retard Dev Disabil Res Rev 2006;12:123–8. [DOI] [PubMed] [Google Scholar]
- [25].Swaiman KF, Menkes JH, Devivo DC, Prensky AL. Swaiman KF, Wright FS. Metabolic disorders of the central nervous system. The Practice of Paediatric Neurology 2nd ed. Vol. ILondon: The C.V. Mosby Co; 1982. 575. [Google Scholar]
- [26].Krivit W, Desnick RJ, Lee J, et al. Generalized accumulation of neutral glycosphingolipids with GM2 ganglioside accumulation in the brain. Sandhoff's disease (variant of Tay-Sachs disease). Am J Med 1972;52:763–70. [DOI] [PubMed] [Google Scholar]
- [27].Yund B, Rudser K, Ahmed A, et al. Cognitive, medical,;1; and neuroimaging characteristics of attenuated mucopolysaccharidosis type II. Mol Genet Metab 2015;114:170–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].de Ruijter J, Broere L, Mulder MF, et al. Growth in patients with mucopolysaccharidosis type III (Sanfilippo disease). J Inherit Metab Dis 2014;37:447–54. [DOI] [PubMed] [Google Scholar]
- [29].Barone R, Brühl K, Stoeter P, et al. Clinical and neuroradiological findings in classic infantile and late-onset globoid-cell leukodystrophy (Krabbe disease). Am J Med Genet 1996;63:209–17. [DOI] [PubMed] [Google Scholar]
- [30].Bouali H, Latrech H. Achondroplasia: current options and future perspective. Pediatr Endocrinol Rev 2015;12:388–95. [PubMed] [Google Scholar]
- [31].Mauceri L, Sorge G, Baieli S, et al. Aggressive behavior in patients with Sotos syndrome. Pediatr Neurol 2000;22:64–7. [DOI] [PubMed] [Google Scholar]
- [32].Schaefer CB, Buehler BA. Saul RA, Phelan MC. Neuroanatomic Features of Sotos Syndrome. Proceedings of the Greenwood Genetic Center. Greenwood, SC: Greenwood Genetic Center; 1993. 36. [Google Scholar]
- [33].Tatton-Brown K, Murray A, Hanks S, et al. Childhood Overgrowth Consortium, Rahman N. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A 2013;161A:2972–80. [DOI] [PubMed] [Google Scholar]
- [34].Tenorio J, Arias P, Martínez-Glez V, et al. Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis 2014;9:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pilarski R, Stephens JA, Noss R, et al. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet 2011;48:505–12. [DOI] [PubMed] [Google Scholar]
- [36].Bhargava R, Au Yong KJ, Leonard N. Bannayan-Riley-Ruvalcaba syndrome: MRI neuroimaging features in a series of 7 patients. AJNR Am J Neuroradiol 2014;35:402–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Piccione M, Fragapane T, Antona V, et al. PTEN hamartoma tumor syndromes in childhood: description of two cases and a proposal for follow-up protocol. Am J Med Genet A 2013;161A:2902–8. [DOI] [PubMed] [Google Scholar]
- [38].Mirzaa G, Conway R, Graham JM, Jr, Dobyns WB. PIK3CA-related segmental overgrowth. In: Pagon RA, Adam MP, Ardinger HH, et al, editors. SourceGeneReviews® [Internet]. Seattle, WA: University of Washington; 1993–2015.
- [39].Conway RL, Pressman BD, Dobyns WB, et al. Neuroimaging findings in macrocephaly-capillary malformation: a longitudinal study of 17 patients. Am J Med Genet A 2007;143A:2981–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mirzaa GM, Conway RL, Gripp KW, et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A 2012;158A:269–91. [DOI] [PubMed] [Google Scholar]
- [41].Parker WE, Orlova KA, Parker WH, et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med 2013;5: 182ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Puffenberger EG, Strauss KA, Ramsey KE, et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain 2007;130:1929–41. [DOI] [PubMed] [Google Scholar]
- [43].Butler MG, Dasouki MJ, Zhou XP, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005;42:318–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gucev ZS, Tasic V, Jancevska A, et al. Congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE) syndrome: CNS malformations and seizures may be a component of this disorder. Am J Med Genet A 2008;146A:2688–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aslanger AD, Altunoglu U, Aslanger E, et al. Newly described clinical features in two siblings with MACS syndrome and a novel mutation in RIN2. Am J Med Genet A 2014;164A:484–9. [DOI] [PubMed] [Google Scholar]
- [46].Syx D, Malfait F, Van Laer L, et al. The RIN2 syndrome: a new autosomal recessive connective tissue disorder caused by deficiency of Ras and Rab interactor 2 (RIN2). Hum Genet 2010;128:79–88. [DOI] [PubMed] [Google Scholar]
- [47].Biesecker LG. The Greig cephalopolysyndactyly syndrome. Orphanet J Rare Dis 2008;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Démurger F, Ichkou A, Mougou-Zerelli S, et al. New insights into genotype-phenotype correlation for GLI3 mutations. Eur J Hum Genet 2015;23:92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ibisler A, Hehr U, Barth A, et al. Novel KIF7 mutation in a Tunisian boy with acrocallosal syndrome: case report and review of the literature. Mol Syndromol 2015;6:173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Speksnijder L, Cohen-Overbeek TE, Knapen MF, et al. A de novo GLI3 mutation in a patient with acrocallosal syndrome. Am J Med Genet A 2013;161A:1394–400. [DOI] [PubMed] [Google Scholar]
- [51].Graham JM, Jr, Schwartz CE. MED12 related disorders. Am J Med Genet A 2013;161A:2734–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Graham JM, Jr, Clark RD, Moeschler JB, et al. Behavioral features in young adults with FG syndrome (Opitz-Kaveggia syndrome). Am J Med Genet C Semin Med Genet 2010;154C:477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Opitz JM, Kaveggia EG. Studies of malformation syndromes of man 33: the FG syndrome. An X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z Kinderheilkd 1974;117:1–8. [DOI] [PubMed] [Google Scholar]
- [54].Nguyen LS, Schneider T, Rio M, et al. A nonsense variant in HERC1 is associated with intellectual disability, megalencephaly, thick corpus callosum and cerebellar atrophy. Eur J Hum Genet 2016;24:455–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Grotto S, Drouin-Garraud V, Ounap K, et al. Clinical assessment of five patients with BRWD3 mutation at Xq21.1 gives further evidence for mild to moderate intellectual disability and macrocephaly. Eur J Med Genet 2014;57:200–6. [DOI] [PubMed] [Google Scholar]
- [56].Pajusalu S, Reimand T, Õunap K. Novel homozygous mutation in KPTN gene causing a familial intellectual disability-macrocephaly syndrome. Am J Med Genet A 2015;167A:1913–5. [DOI] [PubMed] [Google Scholar]