

Abstract
Rationale:
The carbamoyl phosphate synthetase I deficiency (CPS1D) was rare and hard to diagnose due to its atypical symptoms. Brain magnetic resonance imaging (MRI) was typically unavailable in other reports because most patients died before diagnosis was confirmed. Furthermore, it was found a new mutation that had not been described previously.
Patient concerns:
This is a case of neonatal-onset CPS1D with nonspecific clinical manifestations and deteriorating rapidly. Poor feeding, low activity, and tachypnoea were observed, with rapid progression on day 2 after birth. Severe systematic infection was considered first. However, blood culture and cerebrospinal fluid examination were negative. Symptoms were relief temporarily. Then seizure and tachypnoea reappeared as intravenous amino acids were provided. Further examination indicated severe hyperammonemia (serum ammonia level >500mmol/L). Brain MRI showed diffused white matter lesions.
Diagnoses:
Genetic analysis revealed 2 heterozygous mutations in the CPS1 gene: c.2407C>G (p.803, R>G) in exon 20 and C.323G>A (p.108, G>E) in exon 4. The diagnosis of CSP1D was confirmed.
Interventions:
Fasting, the withdrawal of amino acids and plans to treat hyperammonemia were immediately implemented.
Outcomes:
The parents decided to discontinue medical care.
Lessons:
Many CPS1D patients died before the diagnoses are confirmed due to its sudden onset, rapid deterioration, atypical symptoms, and low morbidity. Once hyperammonemia is confirmed, blood and urea amino acid analysis in combination with genetic examinations should be performed as early as possible, this approach would help establish diagnoses at an early stage and thus contribute to reducing mortality and improving prognosis.
Keywords: carbamoyl phosphate synthetase I deficiency, clinical features, CPS1 gene, neonatal onset
1. Introduction
Carbamoyl phosphate synthetase I deficiency (CPS1D), a congenital urea cycle disorder, is an autosomal recessive genetic disorder characterized by hyperammonemia. The morbidity rate is 1/50,000 to 1/300,000.[1,2] The CSP1D could be onset in different ages. The normal procedure of urea cycle is interrupted as a result of the lack of carbamoyl phosphate synthetase I (CPS1). Thus, urea could not be converted from ammonia. The accumulation of toxic ammonia in the blood would lead to neural function disturbance. Therefore, the severity of clinical manifestations of CPS1D mainly depends on the degree of enzymatic activity deficiency. Severe hyperammonemia is common in neonatal-onset patients, with severe clinical manifestations and a poor prognosis.[3–5] It is easy to be misdiagnosed because of atypical symptoms, sudden onset, rapid progression and low morbidity, especially the neonatal-onset types.[1,2] Thus, clinical data, especially the brain magnetic resonance imaging (MRI) was often limited. We present a case of neonatal-onset CSP1D, with nonspecific clinical manifestations and a rapid progression. However, brain MRI data were obtained after severe hyperammonemia was confirmed, as well as the genetic data. The management and diagnostic procedure of the case will be reported and discussed.
2. Case report
A 3-day-old girl was admitted due to progressive tachypnoea and recurrent seizures for 1 day.
The patient was delivered by caesarean section at a gestational age of 39 weeks and weighed 3500 g at birth. No abnormalities were documented during her birth. Her antenatal history was uneventful. Her mother was 29 years old. The parents did not have a consanguineous marriage, and no congenital diseases were documented in the family. She had a healthy 7-year-old brother.
Poor feeding, low activity, and tachypnoea were observed, with rapid progression on day 2 after birth. Irregular respiration and 3 seizures (involving hypertonia and spasms of the upper limbs), each of approximately 1 minutes in duration, were documented before admission. Noninvasive ventilation, antibiotics, and anticonvulsants were administered. Because the symptoms were not relieved, the baby was intubated and transferred to our hospital.
Upon admission, the patient's vital signs were a temperature of 38°C, a heart rate of 192 bpm, a respiratory rate of 70 breaths per minute, and a blood pressure of 94/52 mm Hg. Her body weight was 3040 g. She was unconscious, with moderate jaundice. Lung auscultation revealed coarse rales. Obvious hypotonia was observed, and the patient's primitive reflexes were absent. Bloody fluids from the gastric tube were found after admission.
Peripheral blood counts and blood gas were normal upon admission. Elevated levels of alanine aminotransferase (203 U/L) and unconjugated hyperbilirubinemia (234 μmol/L) as well as a mildly elevated serum creatine level (104 μmol/L) were found. Hypoglycemia (blood glucose of 2.01 mmol/L), hypernatremia (Na+ of 153.8 mmol/L) and hyperchloremia (Cl− of 114.2 mmol/L) suggested a potential disturbance in homeostasis. A coagulation function test indicated a mildly prolonged prothrombin time of 16.8 seconds. A chest X-ray revealed pneumonia with atelectasis of the superior lobe of the right lung. However, sputum culture, blood culture, and cerebrospinal fluid examination were negative.
Dysfunction of multiple organs and systems, neonatal sepsis, and upper digestive tract bleeding were considered, and intracranial infection was suspected upon admission. The patient was on mechanical ventilation at the time of admission. In addition, meropenem and anticonvulsants were administered, as were other supporting therapies, such as fasting for 3 days. Symptoms were relieved, and the patient was weaned off ventilation.
Oral feeding recommenced after extubation. Feeding difficulties became the main problem because the patient did not respond well to routine swallow training. Therefore, parenteral nutritional support was added. Seizure and tachypnoea reappeared as intravenous amino acids were provided. Vomiting, dyspnoea, and lethargy were present concurrently, with rapid deterioration. Blood gases suggested respiratory alkalosis with or without metabolic acidosis. Further examination indicated severe hyperammonemia (serum ammonia level >500 μmol/L). Brain MRI (Fig. 1) revealed extensive abnormalities in deep white matter of the bilateral cerebral hemisphere, subcortical white matter, caudate nuclei, the dorsal thalamus, and the cerebellar hemisphere, which suggested hereditary metabolic leukoencephalopathy. Blood tandem mass spectrometry revealed hypocitrullinemia (a serum citrulline level of 2.57 μmol/L), whereas urine gas mass spectrometry produced normal results. Urea cycle disorder was considered. Fasting, the withdrawal of amino acids and plans to treat hyperammonemia were immediately implemented.
Figure 1.
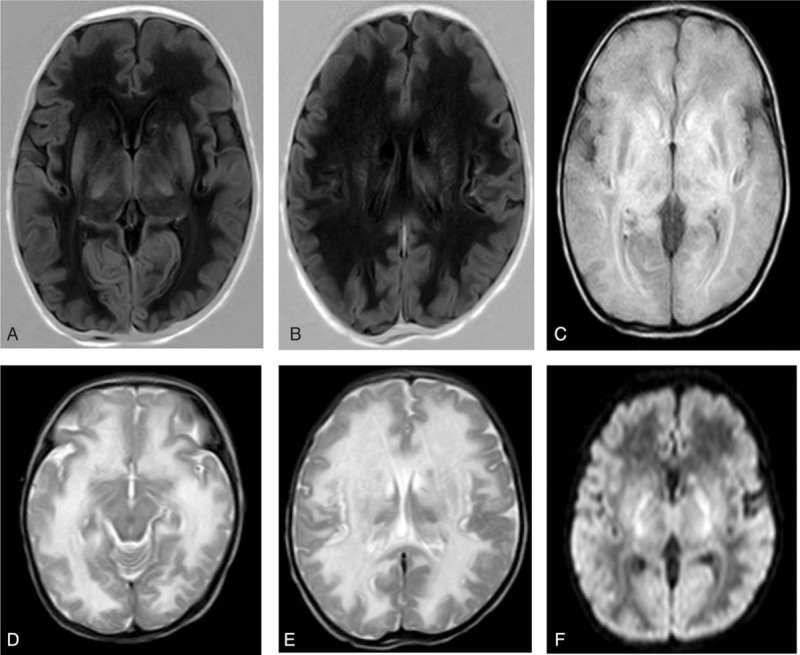
Brain magnetic resonance imaging (MRI) on day 18 of life. MRI revealed extensive abnormalities in the deep white matter of the bilateral cerebral hemisphere, subcortical white matter, caudate nuclei, the dorsal thalamus, and the cerebellar hemisphere, which suggested hereditary metabolic leukoencephalopathy. (A and B) Low signal intensity on T1 weighted image (T1WI); (C) high signal intensity on fluid attenuated inversion recovery (FLAIR); (D and E) high signal intensity on T2 weighted image (T2WI); (F) high signal intensity on diffusion weighted image (DWI).
An EDTA-anticoagulated blood sample was sent to Joy Orient Translational Medicine Research Center Co, Ltd, after informed consent was obtained from both parents. DNA was extracted and amplified via polymerase chain reaction (PCR). Sequencing and comparison with the NCBI RefSeq database revealed 2 heterozygous mutations in the CPS1 gene (Fig. 2): c.2407C > G (p.803, R > G) in exon 20 and C.323G > A (p.108, G > E) in exon 4. The diagnosis of CPS1D was confirmed. However, the parents decided to discontinue medical care and refused to allow further genetic examination of the family. Written informed consent was obtained from the parents of the patient for publication of this case report and any accompanying images.
Figure 2.
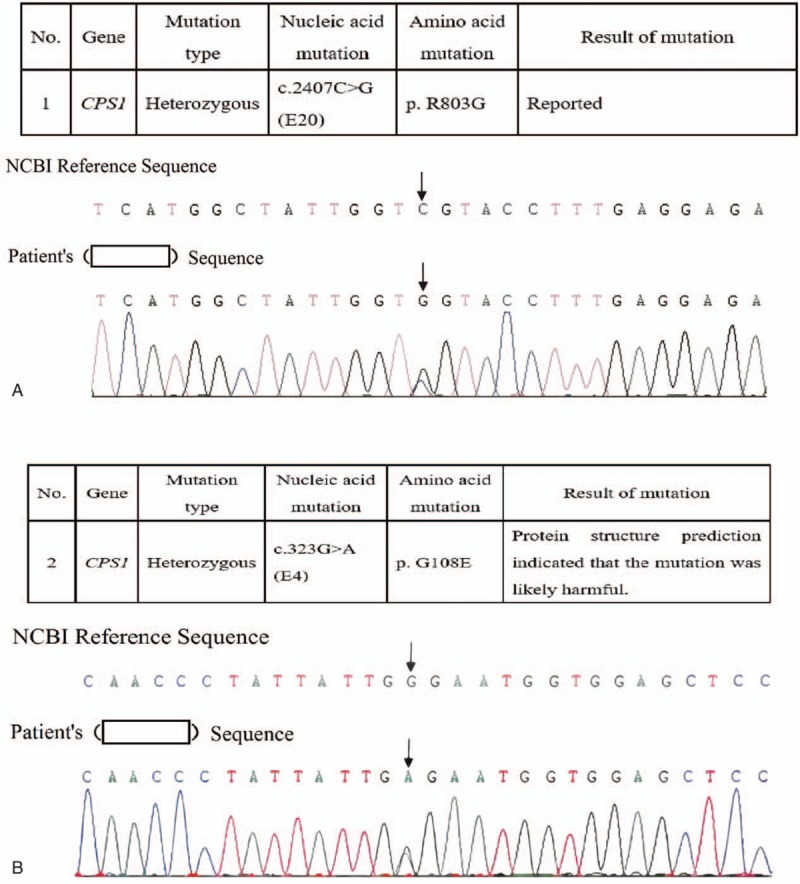
Genetic findings. Sequencing and comparison with the NCBI RefSeq database revealed 2 heterozygous mutations of the CPS1 gene, c.2407C > G (p.803, R > G) in exon 20 (A) and C.323G > A (p.108, G > E) in exon 4 (B).
3. Discussion
In this case, no family history was recorded. Just as other literatures showed that most cases of CPS1D are sporadic.[1–3] Brain MRI data, which were limited in other case reports, were recorded in this case. Meanwhile, a new mutation founded in this case had not previously been described.
Toxic ammonia can be converted to urea by the urea cycle, which is the main means by which urea is eliminated. The first step is the synthesis of carbamoyl phosphate from NH3, which is catalyzed by the key enzyme CPS1. A deficiency in CPS1 will lead to urea cycle disorder.[6] Hence, marked hyperammonemia and decreased downstream production are observed in CPS1D patients.
Clinical manifestations of CPS1D are mainly associated with neural function disturbances attributable to hyperammonemia. The severity of these manifestations depends on the degree of enzymatic activity deficiency. Two independent types of CPS1D, neonatal- and late-onset CPS1D, have been differentiated based on age of onset, clinical manifestations and degree of enzymatic activity deficiency.[4] Severe hyperammonemia and severe clinical manifestations are common in neonatal-onset CPS1D.[1] Neonatal-onset patients are often born normally. Symptoms appear as feeding is established and can include feeding difficulties, vomiting, somnolence, hypotonia, hypothermia, seizure, coma, and apnoea; rapid progression and high mortality are observed. In addition, mental retardation is common among survivors. Late-onset CPS1D patients can develop symptoms at different ages and can exhibit mild to severe clinical manifestations. CPS1D may have an intermittent course, whereas viral infection or a high-protein diet would induce deterioration.[4,5]
Brain MRI data for CPS1D are limited. A brain MRI of an 18-month-old boy with CPS1D revealed atrophy of cerebral white matter, the pallidus, lenticular nuclei, and the corpus callosum; in addition, high signal intensity was observed for periventricular white matter, subcortical white matter, basal ganglia, and caudate nuclei on T2 weighted image (T2WI).[3] In this case, white matter lesions were also apparent. Thus, white matter lesions might be the major imaging findings for CPS1D, and encephalatrophy might be a later finding.
Because it is a rare disease with atypical symptoms, CPS1D is easy to misdiagnose, particularly as a severe infection. A diagnosis of CPS1D depends on enzyme detection and genetic examination. The pathogenic gene CPS1 is located on chromosome 2q35. Hundreds of mutations in this gene have been reported, although only 10% of these mutations have been found in multiple unrelated patients.[7,8] In addition, new mutations continue to be reported. Therefore, the diagnosis of CPS1D is complicated. In the described case, genetic examination revealed the missense mutation c.2407C > G (p.803, R > G), which has previously been reported.[5] However, the C.323G > A (p.108, G > E) mutation was a new finding; this mutation might be a new pathogenic mutation because protein structure prediction indicated that it was likely to be harmful.
The aim of treatment is to manage hyperammonemia. Management approaches include a restricted or low-protein diet and drugs to decrease serum ammonia levels, such as oral l-arginine, natrium benzoicum, and sodium phenylacetate. In cases of severe hyperammonemia, hemodialysis or peritoneal dialysis should be considered.[6] Theoretically, N-carbamoyl-l-glutamate (NCG), an allosteric activator of CPS1, could help increase urea production and therefore decrease serum ammonia levels in patients with CPS1D. Potent effects of NCG have been reported, although insufficient clinical data have been collected.[9,10]
4. Conclusion
Many CPS1D patients die before their diagnoses are confirmed due to this disease's atypical symptoms, sudden onset, rapid progression, and low morbidity. The diagnosis process in the described case suggests that serum ammonia levels should be considered if clinical manifestations such as feeding difficulty, seizure, and disturbance of consciousness appear after feeding is established without indications of an infection or other possible causes of these symptoms. Once hyperammonemia is confirmed, blood and urea amino acid analysis in combination with genetic examinations should be performed early; this approach would help establish diagnoses at an early stage and thus contribute to reducing mortality and improving prognosis.
Acknowledgments
The authors thank Joy Orient Translational Medicine Research Center Co., Ltd, for genetic data analysis and Dr. Fumin Zhao and Dr. Haibo Qu for image acquisition.
Footnotes
Abbreviations: CPS1 = carbamoyl phosphate synthetase I, CPS1D = carbamoyl phosphate synthetase I deficiency, DWI = diffusion weighted image, FLAIR = fluid attenuated inversion recovery, MRI = magnetic resonance imaging, NCG = N-carbamoyl-l-glutamate, PCR = polymerase chain reaction, T1WI = T1 weighted image, T2WI = T2 weighted image.
Funding: The National Key Development Program of Clinical Specialist (neonatologist) (1311200003303); the National Science Foundation of China (81330016, 81630038); the Foundation of Health and Family Planning Commission of Sichuan Province (150104, 150107); the Science and Technology Bureau of Sichuan Province (2014SZ0149).
Ethical approval was obtained from the ethics committee of West China Second University Hospital, Sichuan University. Written informed consent was obtained from the parents of the patient for publication of this case report and any accompanying images.
The authors have no conflicts of interest to disclose.
References
- [1].Díez-Fernández C, Gallego J, Häberle J, et al. The study of carbamoyl phosphate synthetase 1 deficiency sheds light on the mechanism for switching on/off the urea cycle. J Genet Genomics 2015;42:249–60. [DOI] [PubMed] [Google Scholar]
- [2].Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab 2013;110:179–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Uchino T, Endo F, Matsuda I. Neurodevelopmental outcome of long-term therapy of urea cycle disorders in Japan. J Inherit Metab Dis 1998;21(suppl 1):151–9. [DOI] [PubMed] [Google Scholar]
- [4].Funghini S, Thusberg J, Spada M, et al. Phosphate synthetase 1 deficiency in Italy: clinical and genetic findings in a heterogeneous cohort. Gene 2012;493:228–34. [DOI] [PubMed] [Google Scholar]
- [5].Ali EZ, Khalid MK, Yunus ZM, et al. Carbamoylphosphate synthetase 1 (CPS1) deficiency: clinical, biochemical, and molecular characterization in Malaysian patients. Eur J Pediatr 2016;175:339–46. [DOI] [PubMed] [Google Scholar]
- [6].Häberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 2012;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Häberle J, Shchelochkov OA, Wang J, et al. Molecular defects in human carbamoy phosphate synthetase I: mutational spectrum, diagnostic and protein structure considerations. Hum Mutat 2011;32:579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Díez-Fernández C, Hu L, Cervera J, et al. Understanding carbamoyl phosphate synthetase (CPS1) deficiency by using the recombinantly purified human enzyme: effects of CPS1 mutations that concentrate in a central domain of unknown function. Mol Genet Metab 2014;112:123–32. [DOI] [PubMed] [Google Scholar]
- [9].Tuchman M, Caldovic L, Daikhin Y, et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res 2008;64:213–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ah Mew N, McCarter R, Daikhin Y, et al. Augmenting ureagenesis in patients with partial carbamyl phosphate synthetase 1 deficiency with N-carbamyl-L-glutamate. J Pediatr 2014;165:401–3. [DOI] [PMC free article] [PubMed] [Google Scholar]