

Abstract
Symmetry is a common feature among natural systems, including protein structures. A strong propensity towards symmetric architectures has long been recognized for water-soluble proteins, and rationalized from an evolutionary standpoint. Proteins residing in cellular membranes, however, have traditionally been less amenable to structural studies, and thus the prevalence and significance of symmetry in this important class of molecules is not as well understood. In the past two decades great strides have been made in this area, providing exciting insights into the range of architectures adopted by membrane proteins. These structural studies have revealed a similarly strong bias toward symmetric arrangements, often unexpected, despite the restrictions imposed by the membrane environment on the possible symmetry groups. Moreover, membrane proteins disproportionately contain internal structural repeats resulting from duplication and fusion of smaller segments. Here, the types and origins of symmetry in membrane proteins are discussed, along with the implications for their function.
Keywords: oligomer, internal repeats, inverted-topology repeats, asymmetry, alternating access
1 INTRODUCTION
Symmetry, defined as the property of having the same appearance from two or more vantage points, is an aesthetically appealing and common feature of natural systems. In the structure of macromolecules, and in particular, of proteins, a multitude of symmetries and pseudo-symmetries have been identified, and these appear to have a range of functional advantages (37). As we reach the milestone of 500 unique structures of membrane proteins (38, 109), it seems timely to review the prevalence and mechanistic significance of symmetry in this special class of proteins.
After briefly introducing the major functional classes of integral membrane proteins, I discuss the emergence of symmetry in their structures as a result of gene duplication or oligomerization. I then describe the specific types of symmetry observed thus far, and their mechanistic implications. This discussion is focused on membrane proteins whose chains span the entire lipid bilayer one or more times (i.e. not monotopic, membrane-associated proteins (11)). I conclude with open questions and exciting future directions for the field.
2 FUNCTIONS OF MEMBRANE PROTEINS
Around 25–35% of the genes in a genome encode for integral membrane proteins (55, 82). These proteins serve a wide variety of functions, and can be grouped into four types: receptors, channels and transporters, enzymes, and co-factor scaffolds.
2.1 Receptors
Lipid bilayers serve as hydrophobic barriers that protect the interior of cells and organelles, but also impede numerous essential processes. So-called receptor proteins facilitate the transmission of information across membranes. In response to light or to chemical signals from the cell exterior, these proteins adopt a different state or conformation and thereby modulate their ability to interact with other proteins in the interior. The family of seven-transmembrane (TM) helix G-protein coupled receptors (GPCRs) is the most prominent example, and constitutes the largest functional class in eukarya (2). Receptor tyrosine kinases (RTK) comprise another large and important family of membrane proteins in this class (57).
2.2 Channels and transporters
The second most abundant membrane proteins, accounting for 2–15% of the genes in a genome (2, 4), are those that facilitate selective passage of chemicals across the lipid membrane.
In the simplest case, proteins called channels create pores through which ions and other molecules diffuse passively, i.e., along their concentration gradients. To regulate this process, many channels incorporate ‘gating’ mechanisms that respond to environmental stimuli, such as voltage.
Cells also need to expel toxic compounds, and to take up rare nutrients, which typically requires movement against a concentration gradient. So-called primary transporters derive the energy for such processes from ATP hydrolysis or light conversion. ATP hydrolysis is catalyzed by domains residing outside the membrane that are tightly coupled to the membrane-spanning domain through which the substrate passes.
Many primary transporters also serve as ion pumps, i.e. they accumulate e.g. H+ or Na+ ions on one side of the membrane and thereby generate an electrochemical gradient. Such concentration gradients are used as an energy source by membrane proteins known as secondary active transporters. Specifically, these proteins power the movement of one substrate against its gradient by harnessing the energy released from the dissipation of a gradient of a different substrate. The transport process may involve the substrates moving either in the same (symport) or opposite (antiport) directions. In all cases, these transporters function according to the so-called alternating-access mechanism (45), whereby the binding sites for the substrates are alternately exposed to one or other side of the membrane, but not both at the same time (reviewed in, e.g. (30)).
2.3 Membrane enzymes
A number of enzymatic reactions carried out by water-soluble proteins are also conducted by enzymes integrally embedded in the membrane. The membrane setting facilitates access to hydrophobic substrates, such as TM helices destined for proteolysis, but must also allow access to reactive water molecules. Indeed, recent structural studies have shown that membrane enzymes achieve this feat by creating an aqueous micro-environment within the membrane (24, 108).
2.4 Co-factor scaffolding proteins
The orientational confinement imposed by the lipid bilayer can also hold a functional advantage. During photosynthesis, for example, light is absorbed by co-factors in so-called light-harvesting complexes (LHCs) and, by way of resonance energy transfer, activates neighboring photosynthetic reaction centers (PRCs). By fixing the relative positions of their co-factors, LHCs and PRCs create optimal conditions for light absorption and transfer. Similarly, electron transfer reactions in the membranes of mitochondria and respiratory bacteria are facilitated by a series of scaffolding proteins. Ultimately, both photosynthesis and respiration result in a H+ electrochemical gradient across the membrane, which is harnessed by the ATP synthase to energize the production of ATP.
3 FUNCTIONAL DIVERSITY, PROTEIN SIZE AND SYMMETRY
The assortment of functions described above appears to have required a very diverse array of protein architectures. A possible evolutionary strategy to achieve this diversity is to create larger proteins and complexes from smaller structural units (37, 59). This strategy appears to have a number of advantages: the creation of new protein surfaces capable of binding to other molecules; enhanced stability, e.g. shielding of hydrophobic surfaces too small to match the membrane width (8, 77); the conformational stability of supra-molecular complexes; and, the potential for cooperative or other regulatory mechanisms, e.g., by tethering distinct functions within hetero-oligomers (16). As explained below, larger proteins are created by the assembly of multiple subunits, or by gene fusion of duplicated or dissimilar genes, and symmetry appears to be an intrinsic consequence of those processes (37, 59).
3.1 Oligomerization
The simplest mechanism by which larger proteins are formed is through assembly into homo- or hetero-oligomers. Oligomerization is remarkably common; Levy et al showed that between one-half and two-thirds of all proteins form obligate complexes (59). Unfortunately, the equivalent numbers for membrane protein structures have not been well documented, though a cursory analysis of the Protein Data Bank for TM proteins (PDB_TM (103), Figure 1) suggests that a similarly large fraction of membrane proteins (~65%) are obligate oligomers (59).
Figure 1.
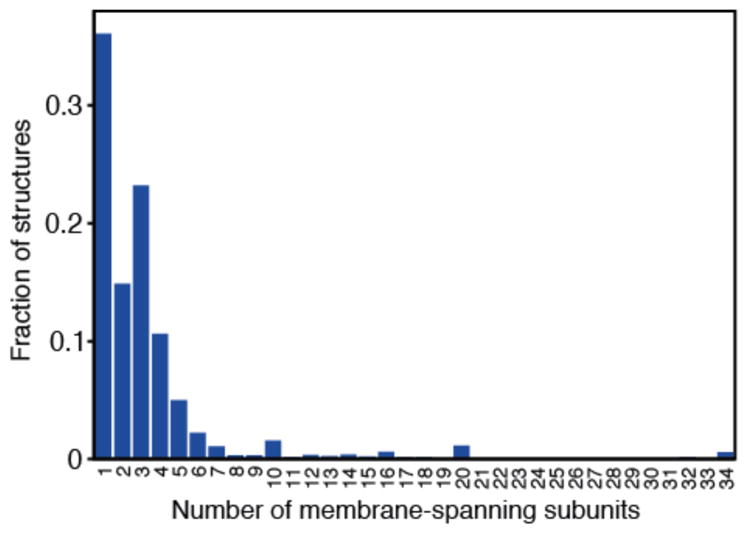
Degree of oligomer formation in membrane protein structures. Data was taken from the PDB_TM database (103) dated 2014.08.01 containing 2241 proteins. No filtering of low-resolution models or redundancy was applied. Thus, each data-point may contain a number of representatives from the same structural family. This analysis is necessarily biased toward those proteins that crystallize and are well studied. Oligomers are formed via interactions between membrane domains as well as between fused water-soluble domains.
Symmetry is found in ~85% of protein complexes, and is therefore the norm (59). It has been proposed that symmetry arises naturally from the fact that symmetric protein-protein interfaces contain duplications of all pair-wise contacts. Thus, the most favorable interactions are also duplicated, leading to more stable interfaces than those achievable by non-symmetric complexes (3, 69). A simple survey of available structures (see below), suggests that membrane protein oligomers are also predominantly symmetric, though a statistical analysis would be desirable. It will also be important to assess whether the above arguments regarding interface energies also apply, and whether the reduced degrees of freedom in the membrane either enhance or diminish this inherent predisposition toward symmetry.
3.2 Gene fusion
A second solution to the need for larger and/or more complex proteins is to combine pre-existing domains by gene fusion (37). Indeed, the majority of all proteins (55–67%) contain multiple detectable subdomains (72). However, when considering only membrane proteins, the opposite trend is observed, as only about 30% contain multiple, independently-functioning domains (65). Notably, this lack of fusion between membrane protein domains is not due to an inherent inability of their genes to fuse: indeed, as many as 90% of membrane proteins contain water-soluble domains (65). Thus, it may be that the reduced dimensionality of the membrane enhances the stability of protein-protein interactions, reducing the need for fusion (65).
If fusion of membrane-spanning segments is indeed less common than fusion of water-soluble domains, then one must assume that more complicated membrane protein functions, such as cooperativity, occur preferentially via oligomerization. However, the fraction of membrane protein oligomers in PDB_TM (Figure 1) is similar to that found for all proteins (59). A more systematic analysis of available membrane protein structures may help clarify this apparent discrepancy.
3.3 Internal repeats
The above discussion pertains to the fusion of domains with independent functionality. However, larger proteins can also be constructed by fusion of genes encoding small protein segments, for example, after duplication of secondary structure elements (70), which results in internal structural repeats. Of the proteins containing detectable internal repeats, about half are symmetric, suggesting that they originate from concurrent duplication and fusion of genes that encoded for homo-oligomeric complexes (1). In other cases, duplication and subsequent fusion of segments that did not previously form oligomers may have resulted in non-symmetric internal repeats (1). Either way, after fusion, internal repeat sequences are independently exposed to selective point mutations. Without a specific functional reason to maintain perfect internal symmetry, therefore, the primary sequences of the repeats are very likely to diverge, resulting in structures that are internally pseudo-symmetric, rather than symmetric.
The first studies of internal structural pseudo-symmetry in membrane proteins suggested that the proportion is as high as one-half (16, 40), while a recent study using more conservative repeat-detection strategy identified pseudo-symmetry in ~24% of membrane protein structures (80). This discrepancy suggests that a quarter of membrane proteins may contain harder-to-detect, highly divergent, internal repeats, although it is also possible that recently reported structures are less pseudo-symmetric. Either way, given that ~18% of all folds were found to be internally pseudo-symmetric, it is clear that available membrane protein structures are enriched in internal pseudo-symmetry relative to water-soluble proteins (80).
For a few membrane protein families, hints of these internal structural duplications were identified based on sequence analyses, long before structures were available, e.g. (84, 94). However, many other duplications are too distantly related (<10% identity) to be detected by sequence-based methods (16, 40, 51, 80).
4 SYMMETRY IN MEMBRANE PROTEIN STRUCTURES
Lipid bilayers themselves contain a planar symmetry that divides the hydrophobic core in half and reflects the two lipid head-group regions. One might therefore expect that some membrane proteins contain a similar structural symmetry, i.e., with an axis running along the mid-plane of the membrane. Nevertheless, the chemical environments at either side of the membrane are typically not equivalent.
4.1 Non-symmetric membrane proteins
By definition, the one-third of soluble and membrane proteins that are monomeric (37, 59)(Figure 1) cannot adopt oligomeric symmetry. Moreover, between one-half and four-fifths of individual domains also contain no internal repeats (16, 80) and therefore also lack any detectable symmetry. Such non-symmetric proteins ought to be well-suited to detecting differences between the environments at either side of the membrane (37). Indeed, none of the three classes of receptor with known structures, namely GPCRs, ligand-gated ion channels, and enzyme-linked receptors such as RTKs, exhibit any symmetry with respect to the membrane plane. Nevertheless, they all feature structural symmetry around an axis perpendicular to the membrane (see below).
Enzymatic reactions in the membrane appear to be accomplished readily by non-symmetric architectures. These include oligosaccharide transferase, OST (66), aspartate proteases (61), site-2-proteases (26), and rhomboid proteases (107, 110) (Table 1, Figure 2).
Table 1.
Membrane proteins of known structure with non-symmetrical or two-fold symmetrical folds
| Order | None | 2-fold | ||||
|---|---|---|---|---|---|---|
| Axis | Perpendicular | Planar | ||||
| Group | C2 | c pC2 | d aC2 | pC2 | 22 | |
| Receptor, Enzyme | YidC, TatC, OST, proteases, | RTK, sulfur-transferase, cyt c ox aa3, cyt c1, PS-II SR-II, class C GPCR, glycophorin A | BR, PRC, complex I, thiol oxidase, rhodopsin, UbiA prenyl-transferase | |||
| a Primary | P-type ATPase | ABC Export, ABC Import I, ABC Import II | ABC export, ABC Import I | ABC Import II1 | ||
| b Second-ary | CDF, MFS, APC, EIIC, DASS, CLC | MATE, MFS, RND1 | SMR | CLC, APC,NPA1, CaCA,EAAT CNT,DASS, MFS,NCS2 | Mrp1 | |
| Channel | BcsA, PorA1 | MgtE, SKT | MIP, Amt, SecY, FNT, UT, TMBIM | |||
Protein families (or one subunit of a larger complex) are organized by symmetry order, axis orientation (perpendicular or parallel to the membrane) and symmetry group. Bold indicates a regulatory role (e.g. stability, cooperativity).
Primary transporters,
secondary transporters.
pC2 indicates pseudo-C2 axis,
asymmetry. Italics indicates that the corresponding symmetry axis runs through the helices of the two repeats (interdigitating repeats). Blue text indicates asymmetry (synonymous with homo-oligomer, while pseudo-symmetry includes both hetero-oligomers or internal repeats).
Indicates symmetry is within a protomer in an oligomeric complex.
Abbreviations not in main text: PS-II, photosynthetic reaction center II; CDC, cholesterol-dependent cytolysin; CNT, concentrative nucleoside transporter; DEG-eNaC, degenerin epithelial sodium channel family; BR, bacteriorhodopsin; SR-II, sensory rhodopsin II; DASS, divalent anion:Na+ symporters.
Figure 2.

Point symmetry types in membrane protein structures. Structures are shown as cartoon helices, viewed down onto the membrane. Different colors are used to indicate symmetric elements, i.e., independent chains or (*)internal repeats. Non-symmetric elements are in gray. EmrE is an asymmetric homo-dimer (PDB entry: 3B5D), while other transporters are asymmetric as well as pseudo-symmetric (e.g., NCX, 3V5U). Two-fold screw axis (22)-pseudo-symmetry is seen in the Mrp antiporter-like subunits of complex I (see Figure 4). Presenilin is an aspartate protease (4HYG). ModB2 is from the homodimeric molybdate type I ABC importer (2ONK). MalFG is from the heterodimeric MalFGK2 type I ABC importer (2R6G). BsYetJ is a pH-dependent Ca2+ channel from the TM Bax inhibitor motif (TMBIM) family (4PGW). FLAP, or five-lipooxygenase-activating protein, is a member of the family of membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG, 2Q7M). M-PPase is a membrane pyrophosphatase (4AV3). M2 is a H+ channel from influenza A (3LBW, TM domains only). TrkH is a K+ channel from the superfamily of K+ transporters (SKT, 3PJZ). TehA is a SLAC anion channel homolog (3M73 chain A). Connexin-26 is a gap junction (2ZW3); each hemi-channel exhibits C6 symmetry. α-hemolysin is a pore-forming toxin (7AHL). The c8-ring is a F-type ATP synthase membrane rotor (2XND). Symmetry axes were defined using SymD v1.3 (52) and figures were made with Pymol v1.7 (Schrödinger Ltd).
Non-symmetric folds are also found in the protein translocation systems YidC (56) and TatC (93), as well as in a cellulose synthesis and translocation system, the BcsA/BcsB complex (78). In the latter case, although the eight TM helices of BcsA are organized into pairs, they are not related by symmetry.
Finally, the P-type ATPases, one of the largest families of primary transporters (101) are clearly non-symmetric (Table 1), which is unusual among transport proteins (see below).
Given that a third of membrane proteins are monomeric (Figure 1), one might expect this list of non-symmetric proteins (Table 1) to be longer. Could it be that most monomeric proteins contain internal symmetry? It has been estimated that as many as ~25–50% of domains contain no internal pseudo-symmetric (16, 80). However, it is possible that such automated approaches underestimate the occurrence of internal repeats, because their divergence makes them difficult to detect (1, 80). As a telling example, most GPCRs have been classified as non-symmetric (80), even though rhodopsin (also a GPCR) contains a clear structural duplication (16). Thus, it seems that the repeats in GPCRs have diverged significantly. An effective strategy might therefore be to assign pseudo-symmetry to a given structural class based on analysis of all known structures in that structural class, rather than using representative folds.
4.2 Cyclic Symmetry
When surveying the most common symmetry groups in oligomeric structures, Levy et al found that 80% contained dihedral symmetry, while only 20% were cyclic (59). In contrast, internally-duplicated segments are >90% rotationally symmetric (80). A survey of available symmetries in membrane proteins (both internal and oligomeric; Tables 1–3, Figure 2) corroborates previous observations that membrane proteins differ from water-soluble proteins in that the vast majority of membrane protein symmetries are cyclic (16, 80), even for oligomers. Indeed, only two cases with dihedral symmetry were found (see section 5.3). In contrast, almost all imaginable cyclic symmetry groups are found in the available membrane protein structures (Figure 2).
Table 3.
Membrane proteins of known structure with five-fold or higher symmetrical folds
| Order | 5-fold | 6-fold | 7-fold | 8+-fold | ||
|---|---|---|---|---|---|---|
| Group | C5 | pC5 | C6 | D6 | C7 | C8+ or pC8+ |
| Receptor, Enzyme | PmLH-II (C8), RaLH-II (C9), TtLH-I (C16) | |||||
| Primary | F/V/A-ATPase c-rings (C8, C10-C15 & pC11) | |||||
| Secondary | ||||||
| Channel | cys-loop, CorA, SLAC, MscL, FNT | cys-loop | MARVEL, CRAC, UreI | Cx | MscS, CDC | PFT (C12), OMA (C8), OMP (pC8-pC24) |
See legend to Table 1 for details. Higher-order symmetry groups are given in parentheses. Abbreviations not used elsewhere in text: cys-loop, cys-loop receptor; CorA, Mg2+ channel family; SLAC, slow anion channel; FNT, formate/nitrite transporter; MARVEL, myelin and lymphocyte (MAL) and related proteins for vesicle trafficking and membrane link family; CRAC, Ca2+-release activated Ca2+ channel; Cx, connexin; CDC, cholesterol-dependent cytolysin; PmLH2, Phaeospirillum molischianum light-harvesting complex (LHC)-II; RaLH2, Rhodoblastus acidophilus LHC-II; TtLH-I, Thermochromatum tepidum LH-I/PRC complex; OMA, outer membrane auxiliary protein; PFT, pore-forming toxin.
Table 2.
Membrane proteins of known structure with three- or four-fold symmetrical folds
| Order | 3-fold | 4-fold | |||
|---|---|---|---|---|---|
| Group | C3 | pC3 | C4 | pC4 | D4 |
| Receptor, Enzyme | DgkA, MAPEG, plant LHC-II, pMMO, BR | Nitric oxide reductase, cyt c oxidase, pMMO1 | |||
| Primary | M-PPase | ||||
| Secondary | BCCT, RND, EAAT, MFS | MCP | |||
| Channel | P2X-R, DEG-eNaC, RND, dermicin, UT, SLAC1, Amt, OMP | UreI1 | KcsA, NavAb, AMPA-R, M2, MIP | SKT, NMDA-R | AQP-0 |
See legend to Table 1 for details. Abbreviations not elsewhere in text: DgkA, diacylgycerol kinase A; MAPEG, membrane associated proteins in eicosanoid and glutathione mechanism; pMMO, particulate methane mono-oxygenase; P2X-R, P2X receptor; UT, urea transporter; OMP, outer membrane protein (β-barrel); cyt c oxidase, cytochrome c oxidase; cASIC, chicken acid sensing ion channel; AMPA-R, α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; MIP, major intrinsic proteins; NMDA-R, N-methyl-D-aspartate receptor.
4.2.1 Symmetry with the axis perpendicular to the membrane plane
The majority of symmetric membrane proteins contain a rotational symmetry whose axis runs perpendicular to the membrane plane (Figure 2). This type of symmetry axis implies that the N-terminal and C-terminal ends of all involved chains are located on the same side of the membrane, which presumably simplifies the insertion process.
4.2.1.1 Take your partner by the hand: cyclic two-fold (C2) symmetry and pseudo-symmetry with a perpendicular axis
The simplest and among the most common symmetric arrangement in membrane proteins involves a 180° rotation around an axis perpendicular to the membrane (C2, Table 1). As described below, perfect C2 symmetry is found in homo-oligomeric complexes while C2 pseudo-symmetry is observed both in hetero-oligomeric complexes and in internal repeats (Figure 2). In cases where this association is required for function, the symmetric elements almost always create a binding site or pathway at their interface (Table 1).
Signaling receptors such as RTKs, for example, create a ligand binding site at the dimer interface (57, 60). Each protomer contains extracellular and intracellular domains, connected by a single TM helix. Binding of the ligand to the extracellular domains causes dimerization, or triggers a conformational change within a pre-existing dimer (57). Recent structures obtained by nuclear magnetic resonance (NMR) of isolated TM helix homo-dimers for, e.g., ErbB2 (9), and heterodimers of, e.g., ErbB1/ErbB2 (75), are consistent with C2 symmetry or pseudo-symmetry extending into the membrane.
Primary transporters of the ATP-binding cassette (ABC) transporter family assemble using C2 perpendicular symmetry, creating a substrate pathway as well as ATP binding sites at the dimer interface (90). All ABC transporters contain two TM domains (TMDs) and two nucleotide-binding domains (NBDs, or ABCs), assembled from either separate chains or fused domains. Notably, the NBDs create two, off-axis binding sites for ATP using a head-to-tail arrangement, whereas the substrate pathway follows the symmetry axis.
Four different classes of ABC transporter have been identified (67), namely ABC exporters, and three types of ABC importer called type I, type II, and energy coupling factor (ECF) importers. Simple homo-dimers are found in the first three classes, e.g., forming local C2 symmetry in the TM domains of the type I importer ModB2 ((43), Figure 2), and of the type II importer BtuC2 (68). Hetero-dimeric ABC transporters, in contrast, come together in pseudo-C2-symmetric complexes, as exemplified by MalF and MalG, which form the TM segments of the ABC importer MalFGK ((83), Figure 2). In some cases, one NBD has lost the ability to hydrolyze ATP. A structure of an ABC exporter with one of these so-called “degenerate” NBDs contains nucleotide bound only to one site, creating an asymmetry in these soluble domains (42), with intriguing functional implications (34).
Perpendicular pseudo-C2-symmetry is also seen in some secondary transporters (Table 1, Figure 3), notably in the largest class, the major facilitator superfamily (MFS, (85)). The MFS fold was confirmed by structural studies (41) to contain two lobes of six-TM helices each lining a central pathway. Interestingly, the multidrug and toxin extrusion (MATE) transporters, such as NorM (39), and the resistance-nodule-division (RND) transporters exemplified by AcrB (23), also contain two domains of six TM-helices lining a central pathway. Nevertheless, the topological arrangement of the helices differs between the three folds.
Figure 3.

Transmembrane topologies of secondary transporters of known structure. The outside of the cell or organelle is oriented to the top. Protein name, family name, human solute carrier (SLC) nomenclature, and representative PDB identifiers (in parentheses) are given. Helices are represented as cylinders, and strands as arrows. Each inverted-topology repeat is highlighted using a triangle whose base is on the side of the N-terminus. Abbreviations: Vc, Vibrio cholerae; CNT, concentrative nucleoside transporter; APC, amino acid/polyamine/organocation superfamily; AAC, ADP-ATP carrier; PDs, periplasmic domains.
Beyond the aforementioned functional roles, dimerization of membrane proteins also appears to be a common strategy for regulation (Table 1), e.g. by enhancing stability or introducing allostery. Dimerization of class C GPRCs allows them a unique mode of activation (53). In addition, the occurrence and functional relevance of homo- and hetero-oligomerization by class A GPCRs is the focus of intense study (27).
4.2.1.2 Three-fold symmetry is often found in regulatory roles
As discussed below, homo-trimeric assemblies are sometimes found to create channels. These include the P2X ATP-gated ion channels (48) and the outer membrane protein component of the RND efflux systems (20). In addition, membrane proteins with diverse functions are assembled from triplicated internal repeats with pseudo-C3 symmetry (Table 2). These include ion-pumping pyrophosphatases (Figure 2) (49, 63), and mitochondrial carrier proteins (MCP, Figure 3) (86).
Strikingly, no perfectly three-fold-symmetric structures have been reported for transporters, at least not for their core transport units (Table 2). However, homo-trimeric assemblies of secondary transporters and channels are common, apparently for regulatory reasons. For example, trimerization of the Na+-coupled aspartate transporter, a homolog of the excitatory amino acid transporters (EAAT), may help stabilize the protein during its large elevator-like conformational change (17, 92).
Interestingly, asymmetry between protomers is also seen in trimeric transporter assemblies, such as AcrB (Figure 3). In each protomer, two repeats of five TM-helices cycle through three distinct conformational states, resulting in H+ uptake (23). These changes in the membrane domains are mechanically transduced to a periplasmic domain, which also cycles through three distinct states resulting in drug extrusion. The asymmetry in the trimer results from coupling of the transport cycles of the three protomers, owing to an extensive interface between the periplasmic domains. This asymmetric coupling mechanism may minimize the extent of drug backflow (23).
Interactions between the cytoplasmic tails of neighboring protomers (91) also create an asymmetry in the trimeric Na+-coupled betaine transporter, BetP (102), which may contribute to increasing its transport rate in response to osmotic stress (95).
4.2.1.3 Higher cyclic symmetries in channels & other systems
The creation of a central pathway is a common feature of parallel oligomers of membrane proteins (Figure 2), resulting in hollow rings with between three and twelve-fold symmetry (see above; Tables 2–3). Tetramers include the H+ channel M2 from influenza virus (44) and the ionotrophic glutamate receptors such as the Glu2A receptor (98). Many channels are pentameric, e.g., the anion channel TehA from the plant SLAC family (14), although hexamers, like synaptophysin, a MARVEL-domain channel (5), heptamers, e.g., the small mechanosensitive channel, MscS (7), and octamers (19) have been observed. The largest pore-forming oligomers are constructed from toxins such as the cytolysins (88) (Table 3). Indeed, a toxin called perfringolysin O may form a pore-forming complex with 40–50 protomers, which would undoubtedly represent the highest symmetry order in a membrane protein (88, 97).
Rings of membrane proteins perform roles other than pore formation (Table 3). During photosynthesis, for example, LHCs, form ovals or rings in order to transfer the energy harnessed to PRCs located in the center (Table 3)(74, 81). Another example is the rotor ring of the F-type ATP synthases. These rings are very likely plugged by lipids (87) and the ring facilitates H+ or Na+ permeation as it rotates against an adjacent, static subunit (46, 87). Because three ATP molecules are synthesized for every revolution of the ring, the number of subunits (which in most cases equals the number of transported ions) determines the thermodynamic capacity of the enzyme to synthesize ATP (73).
Pseudo-symmetry is less common in α-helical complexes with higher order number (Tables 2–3), although a number of channels and receptors are known to form heteromers (e.g. (47)), as do some of the membrane rings of rotary ATPases (73). The β–strands of outer-membrane β-barrel proteins (OMP) are typically related by an 8- to 24-fold rotational pseudo-symmetry (25) (Table 3).
4.2.2 The membrane-bisecting axis of rotation
A surprising discovery of recent years is of rotational symmetries whose axes run along a plane that bisects the membrane (Figure 2). This symmetry raises energetic conundrums because the protein must either insert non-helical segments deep into the bilayer and risk exposing polar groups to the hydrophobic core, or insert the entire protein with dual TM topologies, seemingly a challenge for the insertion machinery (10).
Most of these pseudo-C2 symmetries relate internal repeats with an odd number of TM helices (Figure 3). As a result, the topologies of these repeats are inverted with respect to one another, placing their N-termini on opposite sides of the membrane (Figure 2, Table 1). The small multidrug resistance (SMR) transporters (Figure 2), and the FluC fluoride channel (99) represent the only well-characterized homo-or hetero-dimers with a symmetry axis oriented in this way.
When comparing inverted-topology folds, an underappreciated distinction emerges, namely the position of the symmetry axis relative to the repeats. The symmetry axis may lie in between the two repeats, in which case each repeat has an independent fold (see EmrE, Figure 2). Alternatively, the symmetry axis may pass through the center of both repeats, and then the helices of the two repeats must interdigitate (see NCX, Figure 2).
4.2.2.1 Adjacent inverted-topology repeats
Interestingly, inverted-repeat folds in which the repeats are adjacent to the axis are mostly found in channels (Table 1). Many of these channels are specific for small polar molecules such as water (Aqp1 (76)), urea (dvUT (58)), or ammonia (AmtB (50)). In addition, adjacent inverted-topology repeats are found in channels and transporters for small anions (e.g., CLCs (21)) and cations, as in the Bax inhibitor homolog, BsYetJ (Figure 2) (13). Unusually, the two three-TM-helix repeats in BsYetJ surround the seventh TM helix, through which the pseudo-symmetry axis also passes (Figure 2).
With the exception of BsYetJ, the interface between adjacent repeats defines two symmetry-equivalent pathways that lead from either side of the membrane (31). This seems an evolutionarily parsimonious strategy for channel formation because a single duplication leads to a narrow pathway that may be especially suitable for small molecule conductance (99).
4.2.2.2 Interdigitating inverted-topology repeats
Proteins containing interdigitating inverted-topology structural repeats have arguably the most complex of the known membrane protein folds (Figure 2), and are mainly involved in secondary transport (Table 1). However, interlocking repeat elements also contribute to subdomains in larger transporters, e.g., in type II ABC importers, four TM helices within each lobe contribute to a pseudo-symmetric inverted repeat (16, 68). In addition, in the MFS fold, each two-fold symmetric lobe is itself composed of interdigitating three-TM-helix inverted-topology repeats (41) (Figure 3). As a result, the MFS fold can be considered to contain interlocking inverted-topology repeats of 6-TM helices (89). As described below, a functional advantage of such interlocking inverted-repeats may be an ability to adopt asymmetric states that fulfill the requirements for alternating-access.
Interdigitation leads to wide spacing between contiguous helices (in sequence), implying that the isolated repeats are unlikely to be well-folded. The resultant interlocked interfaces are indistinguishable from the interior of proteins (37), with minimal hydration and extensive hydrophobic cores, in stark contrast to interfaces of typical oligomers. The folding of these proteins must therefore be a complex process. It would be interesting to examine whether individual inverted-topology repeats can exist as independently-folded units.
The possible evolutionary origins of such a complex fold are also enigmatic. However, their interfaces are reminiscent of domain-swapping water-soluble proteins (64), which may provide a useful starting point for further inquiry.
4.2.2.3 Asymmetry in membrane proteins
Although symmetric and pseudo-symmetric arrangements predominate, a number of membrane proteins exhibit a structural asymmetry that is essential for function (blue in Table 1). Asymmetry in a homo-oligomer has the evolutionary handicap that the asymmetric interface must evolve to optimize interactions with two different environments simultaneously (37); this disadvantage explains their rarity, but appears in some cases to be compensated by the functional advantages.
The first example of functional asymmetry in membrane proteins was provided by the homo-dimeric SMR transporter EmrE (15, 28, 104). In structures of EmrE, the two identical protein chains adopt an antiparallel orientation with different conformations, and as a result create a pathway to one side of the membrane only (Figure 2). This fold is an example of classical asymmetry. Around the same time, the structure of an amino-acid transporter, LeuT in an outward-facing conformation, i.e., also containing one access pathway (111), was found to contain a distinctive asymmetry underlying the formation of the outward-facing state (31, 32). The asymmetry in LeuT was obscured by the pseudo-symmetry of the repeats (111) which contain <10% identical residues, and therefore also exhibit some level of inherent structural divergence. Once detected, however, the asymmetry could be taken advantage of, in a strategy to model an alternate state. Specifically, by threading the sequence of the first repeat onto the structure of the second repeat, and vice versa, the two halves of the protein ‘swap’ their conformations. For LeuT this resulted in a model of an inward-facing state (32), whose overall features are remarkably consistent with structures determined subsequently (54).
The implication of these findings for EmrE and LeuT is that these proteins can adopt two states, consistent with alternating-access, i.e., with pathways leading to one or other side of the membrane, by creating an asymmetry in the two repeats. During the cycle, therefore, the first repeat (or protomer) must adopt the conformation of the second repeat (or protomer), and vice versa (28, 32). Given that physiologically, substrate accumulation is driven only by the balance of substrate concentrations and the associated membrane potentials, all conformations of the transporter need to be accessible from an energetic standpoint, without any additional input (i.e., in contrast to primary transporters). The degeneracy of structural states implied by the asymmetry-exchange mechanism is an elegant solution to this requirement.
The ‘repeat-swapping’ modeling strategy has since been applied to proteins with diverse structural folds (Figure 3) including GltPh (17), NCX from the Ca2+:cation exchanger family (CaCA (62)), lactose permease from the MFS (89), and NhaA from the Na+-H+ antiporter (NPA) family (96). Notably, all these folds comprise inverted-repeats with interdigitating helices (Table 1, Figure 2). In each case, a model of an alternate state was generated, and the predicted global conformational changes have since been validated by structures (29, 30, 92, 106) or other evidence (89, 96). Importantly, NMR spectroscopy data supports the proposal that antiparallel EmrE functions by exchanging between degenerate states (79). Thus, the asymmetry-exchange mechanism underlies alternating-access by both homo-dimers and pseudo-symmetric interdigitating inverted-repeats.
How do asymmetric transporters optimize their pathways to interact equally favorably with two different environments (37), i.e., to participate in protein-protein packing as well as interact with substrate and/or aqueous solution? In GltPh these interfaces comprise smooth surfaces with both polar and hydrophobic character (92), thereby preventing the protein from becoming trapped in one state.
A notable conundrum posed by the asymmetry-exchange mechanism is the role of symmetric states. Let us consider two subunits A and B that can both adopt two conformations, i and j. The asymmetric states of the transporter can thus be defined as AiBj or AjBi. What then prevents the formation of AiBi or AjBj? Might one of these arrangements correspond to an occluded state, or possibly, a leaky state (71)?
Intriguingly, asymmetry has recently been detected in the antiparallel homo-dimeric fluoride channel, FluC (C. Miller, personal communication), although the reason why asymmetry is required by a channel is unknown. Still, pure asymmetry as in EmrE and FluC is rare, and the inverted repeats of asymmetry-exchanging transporters are typically divergent in sequence. Pseudo-symmetry may therefore play an important role in adapting secondary transporters to diverse substrates and conditions. In some cases, the breakdown in symmetry may also reduce the free energy of one state over the other, just enough to provide preferential conformations of the transporter, e.g., while awaiting substrate binding. Analysis of possible common ancestors of asymmetry-exchanging transporters may therefore provide useful insights into the minimal requirements for secondary transport.
4.2.2.4 Face-to-back inverted-topology-repeats
Remarkably, a two-fold screw-axis pseudo-symmetry with a membrane-bisecting axis was recently identified within a membrane protein subunit related to Na+-H+ antiporters of the Mrp family (22)(Table 1). Specifically, the multimeric NADH:ubiquinone oxidoreductase, or complex I of the respiratory chain, was found to contain an five-TM helix internal repeat within each of three subunits (Nqo12, Nqo13 and Nqo14) responsible for proton pumping (Figure 4). However, because they are related by a pseudo-twofold screw axis (22) rather than a rotational axis (C2), the repeats are oriented face-to-back, rather than adjacent or interdigitating (22). Moreover, the three subunits are arranged linearly, and capped at one end by an additional domain (Nqo8), which also contains a repeat (6). It is therefore tempting to speculate that the screw-axis is well suited to creating a linear (in effect, helical) array of subunits with repeated interactions. That is, the interfaces between repeats in Nqo14 and Nqo13 are similar to those between repeats in each subunit (Figure 4). The mechanistic relevance of this face-to-back symmetry remains to be established.
Figure 4.

The membrane domain of complex I from T. thermophilus (PDB entry 4HE8). Inverted-topology repeats related by a two-fold (22) screw-axis are colored blue to red for the first repeat or dark gray for the second repeat. The single ‘repeat’ in subunit Nqo8 has same TM topology as the three ‘first repeats’, but is rotated by 180° around an axis perpendicular to the membrane plane (cf. dark blue helices), and is more tilted. Nqo8 is separated from Nqo14 by a number of additional subunits (purple). Other non-symmetric segments and subunits are colored white.
4.3 Dihedral and plane symmetries
Dihedral and planar symmetries contain within them a two-fold symmetry axis, and are therefore potentially compatible with membranes. However, in dihedral symmetry, there must be rotational two-fold axes both perpendicular and parallel to the membrane (105). Therefore, dihedral symmetry within a protein spanning a single membrane potentially exposes polar groups to the hydrophobic membrane core. Consistent with this constraint, the only two known structures with dihedral symmetry are proteins that span two membranes. In gap junctions, two hemichannels, one per membrane, are stacked up to create a pore crossing the two membranes. Each hemichannel is formed by a hexamer of connexin proteins with C6 symmetry, and the entire gap junction exhibits a dihedral (D6) symmetry (Table 3, Figure 2). Aquaporin-0 (AQP-0) tetramers pack head-to-head into octamers that span the membranes of the eye lens fiber cells creating a dihedral symmetry (4, Table 2) (36).
Plane symmetries, created by translations in two directions, although rare, are found in arrays of membrane proteins at specific cellular structures. These include ribbons of claudin proteins at epithelial tight junctions (33, 100), and arrays of AQP-0 octamers (12). Both specific and non-specific properties of the lipids may be critical for the formation of such arrays (18, 35).
4.4 High-order cubic and space-group symmetries
The planar nature of lipid membranes renders the three-dimensional arrangements with cubic or space group symmetries unavailable to membrane proteins. However, attachments to scaffolding or anchoring proteins may result in assemblies of membrane proteins forming spatially ordered complexes.
SUMMARY POINTS.
Membrane protein structures to date exhibit a wide range of symmetries and pseudo-symmetries from two-fold to planar arrays, but the vast majority conforms to cyclic point group symmetry.
Available membrane protein structures apparently constitute a similar fraction of oligomers as water-soluble proteins, but contain a higher proportion of internal repeats. Overall, membrane proteins may exhibit symmetry more frequently than water-soluble proteins.
Inverted-topology (pseudo-)symmetries create channels and pathways through the membrane; the MFS family is a notable exception where 3-TM helix inverted-topology repeats create the two halves that themselves line the substrate pathway.
Asymmetry has been observed in secondary transporters, and in one channel, whose folds contain inverted topologies.
Both identical (homo-oligomeric) and divergent (pseudo-symmetric internal-repeat) protein sequences use asymmetry-exchange to create degenerate alternate states consistent with alternating-access transport mechanisms.
FUTURE ISSUES.
Systematic analysis of the growing number of membrane protein structures will be needed to further classify symmetries, pseudo-symmetries, oligomerization, and internal-repeats in membrane proteins. Continued efforts in structural biology will be needed to fill in gaps in fold space.
Which factors govern asymmetry in secondary transporters, and how is the balance of asymmetric states affected by substrate binding? How do those factors change for different transporter architectures, and how are they affected by single point mutations?
Do symmetric (or pseudo-symmetric) states play a role in secondary transport by asymmetry-exchange mechanisms, e.g. as occluded/closed or leak states?
What factors define the boundary between channel and transporter functions (with symmetric and asymmetric functional states, respectively), especially in folds such as the CLCs?
What was the evolutionary pathway of inverted-topology interlocking repeats? Are the individual repeats stable as separate entities? Bioinformatic and folding studies of the simplest cases, e.g. NCX, may help identify contributions from, e.g., circular permutation or domain swapping.
Did all inverted-topology folds arise from divergent evolution of a single ancestor, or from convergent evolution from many dual topology proteins ?
Acknowledgments
Thanks to José Faraldo-Gómez for helpful discussions, suggestions and comments on the manuscript. This work was supported by the Intramural Research Program of the NIH, NINDS.
Acronym list
- TM
transmembrane
- PDB
protein data bank
GLOSSARY
- Non-symmetric proteins
Those that show no apparent symmetry-related internal duplications, nor any symmetry relationship between chains in a multi-subunit complex.
- Symmetric proteins
Those composed of identical sequences replicated around the symmetry axis, therefore invariably homo-oligomeric complexes.
- Pseudo-symmetric proteins
Two or more protein segments whose sequences differ, but share similar topological arrangements (folds) of their backbones.
- Inverted-topology repeats
Membrane protein segments inserted in opposite orientations, and related by a two-fold axis running parallel to the membrane.
- Internal repeats
Duplications of a structural element within a single polypeptide chain.
- Asymmetric proteins
Protein segments with similar folds adopting distinct conformations of their backbones within the context of that same fold.
- Topology
The direction of threading of protein segments back and forth across the membrane.
- Oligomers
Assemblies of two or more protein chains, with either the same (homo-oligomers) or different (hetero-oligomers) protein sequences.
- Domain
A functional and structural unit of protein, typically between 100 and 250 amino-acids in length.
- Domain swapping
The positional exchange of one or more secondary structure elements from neighboring protomers in an oligomeric complex.
- Protomer
A single subunit of a homo-oligomeric subunit, as distinct from a monomer, which is a non-oligomeric entity.
- Dihedral symmetry with order N
contains 2xN units related by N 2-fold rotational axes and one N-fold rotational axis.
- Dual-topology
The ability for a protein to insert into the membrane in both orientations with equal probability. Also known as undecided.
Footnotes
- Vinothkumar et al: A systematic and thorough review of structures and functions of membrane proteins up to 2009.
- Myers-Turnbull et al: Recent assessment of internal pseudo-symmetry indicating that membrane proteins are enriched in symmetric structural repeats.
- Goodsell & Olson: An excellent and comprehensive discussion of symmetry in all classes of proteins.
- Fleishmann et al: Structural modeling using electron microscopy data for antiparallel EmrE. The authors propose an asymmetry-exchange mechanism.
- Forrest et al. 2008: Authors propose asymmetry exchange for pseudo-symmetric repeats in LeuT and demonstrate accessibility of proposed pathway.
- Dutzler et al 2002: The first structure of a membrane protein containing inverted-topology repeats.
- Baradaran et al 2013: The first structure of a face-to-back inverted-topology repeat.
- Bowie, 2013: Insightful overview of membrane protein insertion and folding, particularly important for dual-topology proteins.
LITERATURE CITED
- 1.Abraham A-L, Pothier J, Rocha EPC. Alternative to Homo-oligomerisation: The Creation of Local Symmetry in Proteins by Internal Amplification. J Mol Biol. 2009;394(3):522–34. doi: 10.1016/j.jmb.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 2.Almén M, Nordström KJ, Fredriksson R, Schiöth HB. Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009;7(1):50. doi: 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.André I, Strauss CEM, Kaplan DB, Bradley P, Baker D. Emergence of symmetry in homooligomeric biological assemblies. Proc Natl Acad Sci USA. 2008;105(42):16148–52. doi: 10.1073/pnas.0807576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408(6814):796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 5.Arthur CP, Stowell MHB. Structure of Synaptophysin: A Hexameric MARVEL-Domain Channel Protein. Structure. 2007;15(6):707–14. doi: 10.1016/j.str.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–48. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bass RB, Strop P, Barclay M, Rees DC. Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science. 2002;298(5598):1582–87. doi: 10.1126/science.1077945. [DOI] [PubMed] [Google Scholar]
- 8.Benjamini A, Smit B. Robust Driving Forces for Transmembrane Helix Packing. Biophys J. 2012;103(6):1227–35. doi: 10.1016/j.bpj.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, et al. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem. 2008;283(11):6950–56. doi: 10.1074/jbc.M709202200. [DOI] [PubMed] [Google Scholar]
- 10.Bowie JU. Structural biology. Membrane protein twists and turns. Science. 2013;339(6118):398–99. doi: 10.1126/science.1228655. [DOI] [PubMed] [Google Scholar]
- 11.Bracey MH, Cravatt BF, Stevens RC. Structural commonalities among integral membrane enzymes. FEBS Letters. 2004;567(2–3):159–65. doi: 10.1016/j.febslet.2004.04.084. [DOI] [PubMed] [Google Scholar]
- 12.Buzhynskyy N, Sens P, Behar-Cohen F, Scheuring S. Eye lens membrane junctional microdomains: a comparison between healthy and pathological cases. New J Phys. 2011;13(8):085016. [Google Scholar]
- 13.Chang Y, Bruni R, Kloss B, Assur Z, Kloppmann E, et al. Structural basis for a pH-sensitive calcium leak across membranes. Science. 2014;344(6188):1131–35. doi: 10.1126/science.1252043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y-H, Hu L, Punta M, Bruni R, Hillerich B, et al. Homologue structure of the SLAC1 anion channel for closing stomata in leaves. Nature. 2010;467(7319):1074–80. doi: 10.1038/nature09487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y-J, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci USA. 2007;104(48):18999–9004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi S, Jeon J, Yang J-S, Kim S. Common occurrence of internal repeat symmetry in membrane proteins. Proteins. 2008;71(1):68–80. doi: 10.1002/prot.21656. [DOI] [PubMed] [Google Scholar]
- 17.Crisman TJ, Qu S, Kanner BI, Forrest LR. Inward-facing conformation of glutamate transporters as revealed by their inverted-topology structural repeats. Proc Natl Acad Sci USA. 2009;106(49):20752–57. doi: 10.1073/pnas.0908570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies KM, Anselmi C, Wittig I, Faraldo-Gómez JD, Kühlbrandt W. Structure of the yeast F1FO-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1204593109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong C, Beis K, Nesper J, Brunkan-Lamontagne AL, Clarke BR, et al. Wza the translocon for E. coli Capsular polysaccharides defines a new class of membrane protein. Nature. 2006;444(7116):226–29. doi: 10.1038/nature05267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du D, Wang Z, James NR, Voss JE, Klimont E, et al. Structure of the AcrAB-TolC multidrug efflux pump. Nature. 2014;509(7501):512–15. doi: 10.1038/nature13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415(6869):287–94. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 22.Efremov RG, Sazanov LA. Structure of the membrane domain of respiratory complex I. Nature. 2011;476(7361):414–20. doi: 10.1038/nature10330. [DOI] [PubMed] [Google Scholar]
- 23.Eicher T, Seeger MA, Anselmi C, Zhou W, Brandstätter L, et al. Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. eLife. 2014 doi: 10.7554/eLife.03145. inpress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erez E, Fass D, Bibi E. How intramembrane proteases bury hydrolytic reactions in the membrane. Nature. 2009;459(7245):371–78. doi: 10.1038/nature08146. [DOI] [PubMed] [Google Scholar]
- 25.Fairman JW, Noinaj N, Buchanan SK. The structural biology of beta-barrel membrane proteins: a summary of recent reports. Curr Opin Struct Biol. 2011;21(4):523–31. doi: 10.1016/j.sbi.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng L, Yan H, Wu Z, Yan N, Wang Z, et al. Structure of a Site-2 Protease Family Intramembrane Metalloprotease. Science. 2007;318(5856):1608–12. doi: 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- 27.Ferre S, Casado V, Devi LA, Filizola M, Jockers R, et al. G Protein-Coupled Receptor Oligomerization Revisited: Functional and Pharmacological Perspectives. Pharmacol Rev. 2014;66(2):413–34. doi: 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fleishman SJ, Harrington SE, Enosh A, Halperin D, Tate CG, Ben-Tal N. Quasi-symmetry in the Cryo-EM Structure of EmrE Provides the Key to Modeling its Transmembrane Domain. J Mol Biol. 2006;364(1):54–67. doi: 10.1016/j.jmb.2006.08.072. [DOI] [PubMed] [Google Scholar]
- 29.Forrest LR. Structural biology. (Pseudo-)symmetrical transport. Science. 2013;339(6118):399–401. doi: 10.1126/science.1228465. [DOI] [PubMed] [Google Scholar]
- 30.Forrest LR, Krämer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta. 2011;1807(2):167–88. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 31.Forrest LR, Rudnick G. The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology (Bethesda) 2009;24:377–86. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forrest LR, Zhang Y-W, Jacobs MT, Gesmonde J, Xie L, et al. Mechanism for alternating access in neurotransmitter transporters. Proc Natl Acad Sci USA. 2008;105(30):10338–43. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. 1998;143(2):391–401. doi: 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.George AM, Jones PM. Perspectives on the structure-function of ABC transporters: the Switch and Constant Contact models. Prog Biophys Mol Biol. 2012;109(3):95–107. doi: 10.1016/j.pbiomolbio.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, et al. Lipid–protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438(7068):633–38. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonen T, Sliz P, Kistler J, Cheng Y, Walz T. Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature. 2004;429(6988):193–97. doi: 10.1038/nature02503. [DOI] [PubMed] [Google Scholar]
- 37.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–53. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 38.Gross DJ. The role of symmetry in fundamental physics. Proc Natl Acad Sci USA. 1996;93(25):14256–59. doi: 10.1073/pnas.93.25.14256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He X, Szewczyk P, Karyakin A, Evin M, Hong W-X, et al. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature. 2010;467(7318):991–94. doi: 10.1038/nature09408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hennerdal A, Falk J, Lindahl E, Elofsson A. Internal duplications in α-helical membrane protein topologies are common but the nonduplicated forms are rare. Protein Sci. 2010;19(12):2305–18. doi: 10.1002/pro.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirai T, Heymann JAW, Shi D, Sarker R, Maloney PC, Subramaniam S. Three-dimensional structure of a bacterial oxalate transporter. Nat Struct Biol. 2002;9(8):597–600. doi: 10.1038/nsb821. [DOI] [PubMed] [Google Scholar]
- 42.Hohl M, Briand C, Grütter MG, Seeger MA. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat Struct Mol Biol. 2012;19(4):395–402. doi: 10.1038/nsmb.2267. [DOI] [PubMed] [Google Scholar]
- 43.Hollenstein K, Frei DC, Locher KP. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446(7132):213–16. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 44.Hong M, DeGrado WF. Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 2012;21(11):1620–33. doi: 10.1002/pro.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211(5052):969–70. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 46.Junge W, Lill H, Engelbrecht S. ATP synthase: an electrochemical transducer with rotatory mechanics. Trends Biochem Sci. 1997;22(11):420–23. doi: 10.1016/s0968-0004(97)01129-8. [DOI] [PubMed] [Google Scholar]
- 47.Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344(6187):992–97. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawate T, Michel JC, Birdsong WT, Gouaux E. Crystal structure of the ATP-gated P2X(4) ion channel in the closed state. Nature. 2009;460(7255):592–98. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kellosalo J, Kajander T, Kogan K, Pokharel K, Goldman A. The Structure and Catalytic Cycle of a Sodium-Pumping Pyrophosphatase. Science. 2012;337(6093):473–76. doi: 10.1126/science.1222505. [DOI] [PubMed] [Google Scholar]
- 50.Khademi S, O’Connell J, Remis J, Robles-Colmenares Y, Miercke LJW, Stroud RM. Mechanism of Ammonia Transport by Amt/Mep/Rh: Structure of AmtB at 1.35 Å. Science. 2004;305(5690):1587–94. doi: 10.1126/science.1101952. [DOI] [PubMed] [Google Scholar]
- 51.Khafizov K, Staritzbichler R, Stamm M, Forrest LR. A study of the evolution of inverted-topology repeats from LeuT-fold transporters using AlignMe. Biochemistry. 2010;49(50):10702–13. doi: 10.1021/bi101256x. [DOI] [PubMed] [Google Scholar]
- 52.Kim C, Basner J, Lee B. Detecting internally symmetric protein structures. BMC Bioinformatics. 2010;11(1):303. doi: 10.1186/1471-2105-11-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kniazeff J, Prézeau L, Rondard P, Pin J-P, Goudet C. Dimers and beyond: The functional puzzles of class C GPCRs. Pharmacol Therapeutics. 2011;130(1):9–25. doi: 10.1016/j.pharmthera.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 54.Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481(7382):469–74. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krogh A, Larsson B, Heijne von G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 56.Kumazaki K, Chiba S, Takemoto M, Furukawa A, Nishiyama K-I, et al. Structural basis of Sec-independent membrane protein insertion by YidC. Nature. 2014;509(7501):516–20. doi: 10.1038/nature13167. [DOI] [PubMed] [Google Scholar]
- 57.Lemmon MA, Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2010;141(7):1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levin EJ, Quick M, Zhou M. Crystal structure of a bacterial homologue of the kidney urea transporter. Nature. 2009;462(7274):757–61. doi: 10.1038/nature08558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D complex: a structural classification of protein complexes. PLoS Comp Biol. 2006;2(11):e155. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li E, Hristova K. Receptor tyrosine kinase transmembrane domains: Function, dimer structure and dimerization energetics. Cell Adh Migr. 2010;4(2):249–54. doi: 10.4161/cam.4.2.10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2014;493(7430):56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- 62.Liao J, Li H, Zeng W, Sauer DB, Belmares R, Jiang Y. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science. 2012;335(6069):686–90. doi: 10.1126/science.1215759. [DOI] [PubMed] [Google Scholar]
- 63.Lin S-M, Tsai J-Y, Hsiao C-D, Huang Y-T, Chiu C-L, et al. Crystal structure of a membrane-embedded H+-translocating pyrophosphatase. Nature. 2012;484(7394):399–403. doi: 10.1038/nature10963. [DOI] [PubMed] [Google Scholar]
- 64.Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11(6):1285–99. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu Y, Gerstein M, Engelman DM. Transmembrane protein domains rarely use covalent domain recombination as an evolutionary mechanism. Proc Natl Acad Sci USA. 2004;101(10):3495–97. doi: 10.1073/pnas.0307330101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lizak C, Gerber S, Numao S, Aebi M, Locher KP. X-ray structure of a bacterial oligosaccharyltransferase. Nature. 2011;474(7351):350–55. doi: 10.1038/nature10151. [DOI] [PubMed] [Google Scholar]
- 67.Locher KP. Structure and mechanism of ATP-binding cassette transporters. Phil Trans Royal Soc B. 2009;364(1514):239–45. doi: 10.1098/rstb.2008.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296(5570):1091–98. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 69.Lukatsky D, Zeldovich K, Shakhnovich E. Statistically Enhanced Self-Attraction of Random Patterns. Phys Rev Lett. 2006;97(17):178101. doi: 10.1103/PhysRevLett.97.178101. [DOI] [PubMed] [Google Scholar]
- 70.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–55. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 71.Mager S, Min C, Henry DJ, Chavkin C, Hoffman BJ, et al. Conducting states of a mammalian serotonin transporter. Neuron. 1994;12(4):845–59. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 72.Marsden RL, Lee D, Maibaum M, Yeats C, Orengo CA. Comprehensive genome analysis of 203 genomes provides structural genomics with new insights into protein family space. Nucl Acids Res. 2006;34(3):1066–80. doi: 10.1093/nar/gkj494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matthies D, Zhou W, Klyszejko AL, Anselmi C, Yildiz Ö, et al. High-Resolution Structure and Mechanism of an F/V-Hybrid Rotor Ring in a Na+-coupled ATP Synthase. Nature Commun. doi: 10.1038/ncomms6286. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McLuskey K, Roszak AW, Zhu Y, Isaacs NW. Crystal structures of all-alpha type membrane proteins. Eur Biophys J. 2009;39(5):723–55. doi: 10.1007/s00249-009-0546-6. [DOI] [PubMed] [Google Scholar]
- 75.Mineev KS, Bocharov EV, Pustovalova YE, Bocharova OV, Chupin VV, Arseniev AS. Spatial structure of the transmembrane domain heterodimer of ErbB1 and ErbB2 receptor tyrosine kinases. J Mol Biol. 2010;400(2):231–43. doi: 10.1016/j.jmb.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 76.Mitsuoka K, Murata K, Walz T, Hirai T, Agre P, et al. The structure of aquaporin-1 at 4.5-Å resolution reveals short alpha-helices in the center of the monomer. J Struct Biol. 1999;128(1):34–43. doi: 10.1006/jsbi.1999.4177. [DOI] [PubMed] [Google Scholar]
- 77.Mondal S, Johnston JM, Wang H, Khelashvili G, Filizola M, Weinstein H. Membrane Driven Spatial Organization of GPCRs. Sci Rep. 2013;3:2909. doi: 10.1038/srep02909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morgan JLW, Strumillo J, Zimmer J. Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature. 2013;493(7431):181–86. doi: 10.1038/nature11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morrison EA, DeKoster GT, Dutta S, Vafabakhsh R, Clarkson MW, et al. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature. 2012;481(7379):45–50. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Myers-Turnbull D, Bliven SE, Rose PW, Aziz ZK, Youkharibache P, et al. Systematic detection of internal symmetry in proteins using CE-Symm. J Mol Biol. 2014;426(11):2255–68. doi: 10.1016/j.jmb.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Niwa S, Yu L-J, Takeda K, Hirano Y, Kawakami T, et al. Structure of the LH1–RC complex from Thermochromatium tepidum at 3.0 A. Nature. 2014;508(7495):228–32. doi: 10.1038/nature13197. [DOI] [PubMed] [Google Scholar]
- 82.Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics. 2009;10(1):159. doi: 10.1186/1471-2105-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450(7169):515–21. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 84.Pao GM, Wu LF, Johnson KD, Höfte H, Chrispeels MJ, et al. Evolution of the MIP family of integral membrane transport proteins. Mol Microbiol. 1991;5(1):33–37. doi: 10.1111/j.1365-2958.1991.tb01823.x. [DOI] [PubMed] [Google Scholar]
- 85.Pao SS, Paulsen IT, Saier MH. Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62(1):1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ-M, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426(6962):39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 87.Pogoryelov D, Krah A, Langer JD, Yildiz Ö, Faraldo-Gómez JD, Meier T. Microscopic rotary mechanism of ion translocation in the F(o) complex of ATP synthases. Nat Chem Biol. 2010;6(12):891–99. doi: 10.1038/nchembio.457. [DOI] [PubMed] [Google Scholar]
- 88.Popoff MR. Clostridial pore-forming toxins: Powerful virulence factors. Anaerobe. 2014:1–19. doi: 10.1016/j.anaerobe.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 89.Radestock S, Forrest LR. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J Mol Biol. 2011;407(5):698–715. doi: 10.1016/j.jmb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 90.Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10(3):218–27. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ressl S, van Scheltinga ACT, Vonrhein C, Ott V, Ziegler C. Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature. 2009;457(7234):47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 92.Reyes N, Ginter C, Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462(7275):880–85. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rollauer SE, Tarry MJ, Graham JE, Jääskeläinen M, Jäger F, et al. Structure of the TatC core of the twin-arginine protein transport system. Nature. 2012;492(7428):210–14. doi: 10.1038/nature11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saraste M, Walker JE. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Letters. 1982;144(2):250–54. doi: 10.1016/0014-5793(82)80648-0. [DOI] [PubMed] [Google Scholar]
- 95.Schiller D, Rübenhagen R, Krämer R, Morbach S. The C-Terminal Domain of the Betaine Carrier BetP of Corynebacterium glutamicum Is Directly Involved in Sensing K+ as an Osmotic Stimulus. Biochemistry. 2004;43(19):5583–91. doi: 10.1021/bi0359628. [DOI] [PubMed] [Google Scholar]
- 96.Schushan M, Rimon A, Haliloglu T, Forrest LR, Padan E, Ben-Tal N. A model-structure of a periplasm-facing state of the NhaA antiporter suggests the molecular underpinnings of pH-induced conformational changes. J Biol Chem. 2012;287(22):18249–61. doi: 10.1074/jbc.M111.336446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shepard LA, Shatursky O, Johnson AE, Tweten RK. The Mechanism of Pore Assembly for a Cholesterol-Dependent Cytolysin: Formation of a Large Prepore Complex Precedes the Insertion of the Transmembrane β-Hairpins. Biochemistry. 2000;39(33):10284–93. doi: 10.1021/bi000436r. [DOI] [PubMed] [Google Scholar]
- 98.Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462(7274):745–56. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stockbridge RB, Robertson JL, Kolmakova-Partensky L, Miller C. A family of fluoride-specific ion channels with dual-topology architecture. eLife. 2013;2(0):e01084–84. doi: 10.7554/eLife.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, et al. Crystal Structure of a Claudin Provides Insight into the Architecture of Tight Junctions. Science. 2014;344(6181):304–7. doi: 10.1126/science.1248571. [DOI] [PubMed] [Google Scholar]
- 101.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature. 2000;405(6787):647–55. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 102.Tsai C-J, Khafizov K, Hakulinen J, Forrest LR, Krämer R, et al. Structural asymmetry in a trimeric Na+/betaine symporter, BetP, from Corynebacterium glutamicum. J Mol Biol. 2011;407(3):368–81. doi: 10.1016/j.jmb.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 103.Tusnády GE, Dosztányi Z, Simon I. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucl Acids Res. 2005;33(Database issue):D275–78. doi: 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ubarretxena-Belandia I, Baldwin JM, Schuldiner S, Tate CG. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 2003;22(23):6175–81. doi: 10.1093/emboj/cdg611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Venkatakrishnan AJ, Levy ED, Teichmann SA. Homomeric protein complexes: evolution and assembly. Biochem Soc Trans. 2010;38(4):879–82. doi: 10.1042/BST0380879. [DOI] [PubMed] [Google Scholar]
- 106.Waight AB, Pedersen BP, Schlessinger A, Bonomi M, Chau BH, et al. Structural basis for alternating access of a eukaryotic calcium/proton exchanger. Nature. 2013;499(7456):107–10. doi: 10.1038/nature12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444(7116):179–80. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 108.White SH. Rhomboid intramembrane protease structures galore! Nat Struct Mol Biol. 2006;13(12):1049–51. doi: 10.1038/nsmb1206-1049. [DOI] [PubMed] [Google Scholar]
- 109.White SH, editor. Membrane proteins of known 3D structure. http://blanco.biomol.uci.edu/mpstruc/
- 110.Wu Z, Yan N, Feng L, Oberstein A, Yan H, et al. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13(12):1084–91. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- 111.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437(7056):215–23. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]