

Summary
Topoisomerase II is a major component of mitotic chromosomes and its unique decatenating activity has been implicated in many aspects of chromosome dynamics, of which chromosome segregation is the most seriously affected by loss of topo II activity in living cells. There is considerable evidence that topo II plays a role at the centromere including: the centromere-specific accumulation of topo II protein; cytogenetic/ molecular mapping of topo II catalytic activity to active centromeres; the influence of sumoylated topo II on sister centromere cohesion; and its involvement in the activation of a Mad2-dependent spindle checkpoint. Using a conditional-lethal DNA topoisomerase IIα mutant human cell line we find that topo IIα depletion, while leading to a disorganised metaphase plate, does not have any overt effect on general kinetochore assembly. Fluorescence in situ hybridisation suggested that centromeres segregate normally, most segregation errors being chromatin bridges involving longer chromosome arms. Strikingly, a linear human X centromere-based minichromosome also displayed a significantly increased rate of missegregation. This sensitivity to topo IIα depletion may be linked to structural alterations within the centromere domain, as indicated by a significant shortening of the distance across metaphase sister centromeres, and the abnormal persistence of PICH-coated connections between segregating chromatids.
Keywords: topoisomerase, PICH, chromosome, centromere, condensation, segregation
Introduction
DNA topoisomerases have essential roles through altering the structure of double-stranded DNA. They are classified into two categories, topo I and II, based on the mode of the enzymatic reaction. Only topo II can both remove positive or negative supercoils and catenate or decatenate DNA duplexes. Due to its unique, ATP-dependent, DNA strand passing activity, topo II plays a role in virtually all processes involving double-stranded DNA and functions at multiple steps in the assembly of mitotic chromosomes from interphase nuclei (Wang, 2002; Porter and Farr, 2004)
While yeast and Drosophila have a single topo II gene, there are two distinct isoforms in higher eukaryotes, designated topo IIα (170 kDa) and topo IIβ (180 kDa) that are encoded by different genes. The two isoforms have identical catalytic activities in vitro and both can complement yeast topo II function. However, the two isoforms show differences in cell cycle expression and have different distributions in vivo. Topo IIα is associated with mitotic chromosomes and is preferentially expressed in proliferating cells, with levels varying throughout the cell cycle, increasing in S, peaking in G2-M and diminishing in G1 (Heck et al., 1988) The IIβ isoform is expessed in both proliferating and differentiated cells and its amount and stability show no significant fluctuations through the cell cycle. Localisation of IIβ, however, is controversial; while the bulk is diffusely nucleoplasmic in interphase and mitosis (Chaly et al., 1996; Meyer et al., 1997) a minor fraction of IIβ appears to be associated with mitotic chromatin (Christensen et al., 2002; Null et al., 2002) . Nevertheless, while topo IIα is essential for mitosis and cell division, topo IIβ is dispensable in cultured cells (Woessner et al., 1991; Chaly et al., 1996; Grue et al., 1998; Yang et al., 2000; Akimitsu et al., 2003).
A distinctive feature of topo II (especially IIα) is that it accumulates at mitotic centromeres in prometaphase and remains there until early anaphase (Taagepera et al., 1993; Gorbsky, 1994; Rattner et al., 1996; Sumner, 1996; Christensen et al., 2002; Null et al., 2002). A number of studies have suggested that topo II may have a role in influencing centromere organisation (Rattner et al., 1996; Toyoda and Yanagida, 2006), centromeric cohesion (Bachant et al., 2002; Takahashi et al., 2006) and a Mad2-dependent spindle checkpoint (Mikhailov et al., 2004; Skoufias et al., 2004; Toyoda and Yanagida, 2006). This view has been strengthened by the identification, through cytogenetic and molecular studies, of topo II cleavage activity at active centromeres in vertebrates and in parasitic protozoa (Floridia et al., 2000; Andersen et al., 2002; Spence et al., 2002; Agostinho et al., 2004; Spence et al., 2005; Kelly et al., 2006; Obado et al., 2007). However, no overt effect of topo II depletion on general kinetochore assembly, or on anaphase centromere separation, has been reported (Chang et al., 2003; Sakaguchi and Kikuchi, 2004; Toyoda and Yanagida, 2006). Here we investigate the role of topo II in mitotic chromosome dynamics using a human cell line (designated HTETOP) conditionally null mutant for topo IIα (Carpenter and Porter, 2004). Our focus has been on the influence of topo II within the centromere domain and on anaphase chromosome segregation. The latter is of particular interest given the recent description of ultrafine DNA connections, extending between the centromeres of otherwise well separated chromatids in normal anaphases, which stain for the PICH (Plk-1-interacting checkpoint) and BLM (Bloom's syndrome) helicases (Baumann et al., 2007; Chan et al., 2007).
While no gross effects on mitotic chromosome structure, or general kinetochore assembly, were detected, the segregation behaviour of a “sensitised” minichromosome in the topo IIα-depleted background suggested a centromere effect. Further analysis revealed that depletion of topo IIα, as well as promoting bridging through a failure to efficiently decatenate chromosome arms, also leads to an altered centromere topology. This is reflected in a shortening of the distance across sister centromeres under tension, together with the abnormal persistence of PICH-coated connections between segregating chromosomes in topo IIα-depleted human cells.
Materials & Methods
Cell culture, fusion and transfection
HTETOP cells were cultured as previously described (Carpenter and Porter, 2004) The 2.7 Mb minichromosome was transferred into the HTETOP background by whole cell fusion between it and a DT40 hybrid donor cell line (1aA1, (Spence et al., 2006) HTETOP:minichromosome hybrids were selected using 2.5 μg/ml blasticidin S (ICN). The transfer of the minichromosome into two independently derived hybrids was confirmed by fluorescence in situ hybridisation (FISH) using a DXZ1 DNA probe and by pulsed field gel electrophoresis and Southern blotting of uncut HMW DNA.
Etoposide (Sigma) was dissolved in 100% DMSO at 10 mM and stored in the dark at −20°C. The inhibitor was added to exponentially growing cells to the specified final concentration and incubated at 37°C for the time indicated. An equivalent volume of 100% DMSO was added as the no drug control. Doxycycline (dox) (Sigma) was dissolved in water and stored in the dark at −20°C for up to 2 weeks. It was used at a final concentration of 1 μg/ml. MG132 (Calbiochem) was dissolved in DMSO at 10 mM and stored at −20°C for up to 4 weeks. HTETOP cells were arrested using a final concentration of 10 μM for 2 hours 30 minutes. Colcemid (KaryoMax, Gibco/Invitrogen) was used to arrest HTETOP cells at a final concentration of 100 ng/ml for 1 hour for immunofluorescence (where used), 2 hours for inter-kinetochore measurements and overnight for mitotic chromosome structural integrity assays.
In siRNA experiments human topo IIβ was depleted using 5′-GGCCCAGAUUUUAAUUAUAtt-3′. Two other target sequences were also selected and gave similar results (5″-GGCAUCGCAUCUUGUUUAGAtt-3′ and 5′-GGUUUAUACAAGAUCUUUGtt-3′) (Ambion). Transfection was done by nucleofection according to the manufacturer's instructions (Amaxa).
Pulsed field gel electrophoresis (PFGE) and Southern blot hybridisation
High molecular weight (HMW) DNA was resolved in 0.7% chromosomal grade agarose (BioRad 162-0136), 0.25x TBE, over 72 hour at 11°C using the following parameters: a pulse time of 350 - 50 seconds (changing logarithmically), a rotor angle of 110° - 100° (decreasing linearly), a voltage of 120 V - 50 V (decreasing linearly) (Rotaphor TypeV, Biometra). Southern transfers were performed onto nylon membranes (Hybond N+, GE BioSiences) and DNA probes were radioactively labelled by random-priming. Southern blot hybridisations were carried out in a Hybaid oven and membranes were washed at high stringency (0.2x SSC/0.1% SDS at 65°C). The DNA probe used in hybridisations was DXZ1, a 2.0 kb BamHI α-satellite DNA fragment from pSV2X5.
Fluorescence in situ hybridisation (FISH)
FISH was carried out as previously described (Spence et al., 2002) Probe DNA was labelled with DIG-11-dUTP (Roche). Specimens mounted in Vectashield (Vector Labs) with DAPI counterstain. Fluorochromes used were anti-DIG FITC (Roche) and avidin Texas Red (Vector Labs). Telomeric DNA was detected using a fluorescently-conjugated telomere PNA probe (DakoCytomation). Other DNA probes used were as follows: a pan-centromere chromosome paint (Cambio); a mouse rDNA probe from plasmid pA; the human centromere probes D11Z1, D6Z1 and DXZ1; and paints for human chromosomes 6, X, 19 and 20 (Star-FISH™ Cambio). For FISH with rDNA slides were pre-treated as follows: RNase (100 μg/ml) in 2x SSC at 37°C for 1 hour), 3 washes in 2x SSC, 1 wash in PBS at 37°C, pepsin treated (100 μg/ml in 10 mM HCl, 10 minutes at 37°C), washed in PBS, fixed in 1% formaldehyde/PBS for 10 minutes followed by a final rinse in PBS. Slides were dried in an ethanol series before FISH.
For measurements across bioriented sister centromeres, MG132-arrested cells growing on slides in QuadriPerm slide chambers (VivaScience Ltd) were fixed using absolute methanol for 10 minutes, followed by 2% formaldehyde/PBS and a final fix in 3:1 methanol: acetic acid. FISH signals were detected for the 11 centromere and Z-stacks of multi-channel images captured at 0.1 μm intervals using Leica FW4000 software. The distance across sister centromere pairs, to the outer edges of the D11Z1 signal, was measured using Leica Deblur software.
Indirect immunofluorescence (IF)
To localise topo IIα, tubulin, Aurora B/AIM-1, CENP-A, CENP-C, CENP-E, CENP-F and acetyl histone H3 cells were swollen in 75 mM KCl, spun onto poly-L-lysine coated slides (BDH) in a Cytospin cytocentrifuge (Shandon) and fixed in methanol. Antibodies were diluted in KCM (120 mM KCl, 20 mM NaCl, 10 mM Tris-HCl pH 8.0, 0.5 mM EDTA, 0.01% Triton X-100) plus 10% foetal bovine serum. Washes were in KCTo localise PICH cells growing on slides were fixed and permeabilised in PTEMF (20 mM PIPES pH 6.8, 4% fomaldehyde, 0.2% Triton X-100, 10 mM EGTA, 1 mM MgCl2). Antibodies were diluted in 3% BSA in PBS + 0.05% Tween 20 (PBST) and washes were in PBST. SMC2 IF was carried out using a version of the solvent spreading method [Earnshaw, 1989 #114]. Colcemid-arrested cells were swollen with 75 mM KCl for 10 minutes and then fixed in three changes of 3:1 methanol: acetic acid before being dropped onto slides and air dried. Spreads were rehydrated in PBS for 15 minutes before being swollen in TEEN (1 mM triethanolamine-HCl pH 8.5, 0.2 mM NaEDTA, 25 mM NaCl) for 7 minutes. The primary and secondary antibodies were diluted (1:100, 1:200) in TEEN + 0.5% foetal bovine serum, washes were in TEEN. Final washing was in KB (10 mM Tris-HCl pH 7.7, 0.15 M NaCl, 0.5% foetal bovine serum).
For measurements of spindle length dox-treated cells (0 or 72 hours) growing on slides were arrested using MG132, fixed in methanol and stained using mouse anti-tubulin (gamma or alpha). Pole-to-pole measurements for bipolar cells where the spindle was perpendicular to the light path were made using the line tool in the Leica Deblur software on maximum intensity projections of z-stacks. CENP-A quantification was carried out as previously described (Spence et al., 2006)
Antibodies
Primary antibodies for IF were as follows: rabbit anti-human CENP-A (1:400) (Valdivia et al., 1998); mouse anti-human CENP-A (1:500) (Abcam); Aurora B/AIM-1 (1:200) (BD); Acetyl histone H3 (1:100)(Upstate); CENP-C (554, 1:100) (Saitoh et al., 1992); CENP-B (mACA-1, 1:1000) (Cooke et al., 1990); SMC2 (ScII A, 1:200) (Saitoh et al., 1994); Topo IIα (1:100)(Topogen); β-tubulin (1:200) (Sigma); γ-tubulin (1:500)(Sigma); α-tubulin (1:500) (Abcam); CENP-F (D10, 1:750) (Liao et al., 1995); CENP-E (mAb177, 1:250) (Yen et al., 1991); PICH (raised in rabbit, 1:200) (Baumann et al., 2007). Secondary antibodies for IF (1:200) were as follows: goat anti-mouse Texas Red and goat anti-rabbit Texas Red (Molecular Probes); rabbit anti-mouse FITC, swine anti-rabbit FITC, or swine anti-rabbit conjugated to biotin (Dako); sheep anti-mouse digoxigenin (Chemicon). Biotin-labelled secondary antibodies were visualized using fluorescein-avidin DN (Vector Labs) or avidin Cy5 (Amersham Bioscience) and digoxigenin with sheep anti-digoxigenin FITC (Roche).
Mitotic chromosome structural integrity assay
For assessment of mitotic chromosome structural integrity by treatment with chromosome compacting/unfolding buffers, cells were grown on slides in Quadriperm slide chambers and assayed as described in (Hudson et al., 2003). Chromosomes are first unfolded by exposure to a low ionic strength solution lacking divalent cations and containing EDTA (0.5x TEEN: 0.5 mM triethanolamine-HCl, pH 8.5, 0.2 mM NaEDTA, 12.5 mM NaCl). They are then refolded by the addition of a low ionic strength buffer containing Mg2+ (RSB buffer: 10 mM Tris:HCl pH 7.4, 10 mM NaCl, 5 mM MgCl2). The TEEN-RSB cycle was then repeated. For microscopy slides were fixed in 2% formaldehyde and stained with DAPI.
Imaging
Images were captured using a Leica DM6000B microscope driven by FW4000 software, or a Zeiss Axioskop 2 using the Cytovision system (Applied Imaging).
Statistics
Comparison of means was done by unpaired Student's t tests, assuming equal variance, unless otherwise indicated. Results were handled in Excel.
Results
Chromosomes depleted of topo IIα retain a structural memory
Topo II and the condensin complex are major non-histone components of the mitotic chromosome scaffold. Recently it was shown that depletion of condensin results in mitotic chromosomes that are structurally disrupted and in which other scaffold proteins, including topo IIα, are mislocalised (Hudson et al., 2003). We therefore examined the effect of topo IIα depletion (72 hours dox) on condensin localisation. Immunofluorescence (IF) using antibodies against the condensin I and II subunit SMC2 revealed a characteristic axial distribution in mitotic chromosomes even when depleted of topo IIα (Fig. 1). Some variation in the intensity of SMC2 staining was seen between mitotic cells, but this appeared to be independent of doxycycline exposure. To investigate whether a condensin phenotype would be exposed if both topo IIα and β were depleted simultaneously, siRNA was used to transiently deplete IIβ from HTETOP cells. (Depletion of topo IIβ was confirmed by IF, western blotting and realtime PCR and levels estimated to be ~20% of normal at 72 hour post siRNA transfection, Fig. S1). Condensin localisation was unaffected by the depletion of both topo II isoforms (data not shown).
Fig. 1.

(A) Immunofluorescence for topo IIα on HTETOP metaphase cells grown in the absence (topo IIαON) and presence (topo IIαOFF) of doxycycline for 48 hours. (B) Immunofluorescence for the SMC2 subunit of condensin on HTETOP metaphase spreads isolated from cells grown in the presence and absence of doxycycline for 72 hours. The normal axial distribution of SMC2 is detected following topo IIα depletion. Scale bar, 10 μm.
We then examined the effect of topo IIα depletion on chromosome integrity. The assay used relies on the ability of chromosomes to recover their morphology following complete unfolding of the chromatin (Cole, 1967; Hudson et al., 2003). Chromosomes, in cells grown on coverslips and arrested overnight with colcemid, are first unfolded by exposure to a low ionic strength solution lacking divalent cations and containing EDTA. They are then refolded by the addition of a low ionic strength buffer containing 5 mM Mg2+. Under this assay the term structural integrity is defined as being “the ability of chromosomes to remain morphologically indistinguishable at the level of the light microscope during repeated cycles of swelling and shrinking” (Vagnarelli et al., 2004). HTETOP cells in which topo IIα had been depleted (72-120 hours dox) were subjected to successive cycles with chromatin-unfolding and chromatin-compacting buffers. No effect on the ability of mitotic chromosomes to expand and to subsequently recover their gross structural integrity was detected following depletion of topo IIα (Fig.2). Consistent with previous observations, the extent of contraction achieved in this assay by chromosomes depleted of topo IIα was slightly less than for normal chromatin, with chromosomes generally being thinner with less well resolved sister chromatids (Fig. 2). However, ≥98% of chromosomes were seen to be contracted following two rounds of incubation in the expansion and contraction buffers, irrespective of whether or not the cells had been exposed to doxycycline. This suggests that depletion of topo IIα does not affect the ability of chromosomes to recover a compact structure. Depletion of topo IIβ, either in the presence or absence of topo IIα, had no detectable effect on the ability of HTETOP chromosomes to recover their native morphology under these assay conditions (Fig. 2 and data not shown).
Fig. 2.

Topo II is not required for the structural integrity of mitotic chromosomes. Shown are representative images following the expansion and refolding assay. Stages (1)–(3) represent the first cycle of expansion and refolding, and (4) is the refolded status after cycle 2. Stage (1) colcemid-arrested metaphase before treatment; stage (2) first round expansion in the low salt + EDTA TEEN buffer; stage (3) first round contraction in the low salt + Mg2+ RSB buffer; stage (4) appearance after a second round of expansion and contraction. Topo II-depleted chromosomes (both α-only depleted/120 hours dox, and α- plus β-depleted/120 hours dox + 72 hours siRNA) recover their normal starting morphology, which is a slightly thinner and more ribbon-like appearance than that displayed by the control chromosomes (Carpenter and Porter, 2004). All images are shown at the same magnification, with some regions enlarged for clarity. Scale bar, 10 μm.
Many topo IIα depleted cells have a disorganised metaphase plate
In many metaphase cells following depletion of topo IIα one or more chromosome arms extended away from the compact mass of chromatin, often stretching towards the pole (Fig. 3B-E). This behaviour, which has previously been described in Drosophila cells transiently depleted of topo II (Chang et al., 2003), was particularly clear in doxycycline-treated cells arrested in metaphase under tension using the proteasome inhibitor MG132. After 48-72 hours doxycycline 20-30% of MG132-arrested metaphases had at least one arm protruding polewards, compared with a background frequency of 0-4%. Similar observations were made following the depletion of both topo IIα and IIβ (72 hours dox + siRNA) (Fig. 3 D,E). Staining for the inner kinetochore protein CENP-C suggested that, in most cases, these protrusions were not associated with the pulling activity of the centromere.
Fig. 3.
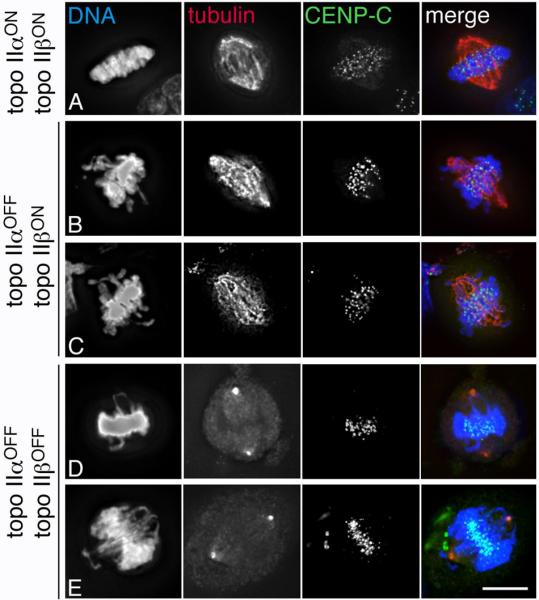
Many metaphases in topo II-depleted cells have one or more elongated chromosome arms stretched towards the spindle pole. (A) A control (topo IIα and IIβ-expressing) HTETOP metaphase cell; (B)-(C) HTETOP topo IIα-depleted metaphase cells (48 hours dox); (D)-(E) topo IIα (72 hours dox) and topo IIβ (72 hours siRNA)-depleted cells arrested in metaphase with MG132 (DNA/DAPI – blue; tubulin, α or γ as indicated – red; CENP-C – green). Scale bar, 10 μm.
Missegregation following topo IIα depletion arises from effects both on chromosome arms and at centromeres
After 48-72 hours of topo IIα repression most HTETOP anaphase cells are aberrant, with frequent lagging and bridging of chromatin (Carpenter and Porter, 2004). Nevertheless, despite the lethal nature of topo IIα-depletion, these cells do manage to separate the bulk of their chromosomes. This is in contrast to the situation when both topo II isoforms are depleted simultaneously, where the bulk of the chromatin remains unsegregated (Sakaguchi and Kikuchi, 2004; Toyoda and Yanagida, 2006) (data not shown). This suggests that topo IIβ contributes substantially to sister chromatid decatenation, as has been reported previously (Sakaguchi and Kikuchi, 2004).
To address the nature of the segregation errors arising from depletion of topo IIα and the origin of the DNA in the chromatin bridges, FISH was undertaken using: a pancentromere DNA probe; centromere-specific DNA probes; paints for human chromosomes 6, 19, 20 and the X; a PNA:telomeric (TTAGGG)n repeat, and a rDNA probe. This work has been undertaken on HTETOP and a hybrid derivative carrying an extra human X centromere-based minichromosome.
Analysis of bridged anaphases using a pan-centromeric DNA suggested that centric regions segregate normally following the depletion of topo IIα (Fig. 4A). We then examined the segregation behaviour of the ribosomal RNA gene arrays. In S.cerevisiae TOP2-dependent decatenation of the rDNA appears to occur later than for the rest of the genome (Sullivan et al., 2004) and in top2 mutants hyper-recombination within the rDNA cluster has been described (Christman et al., 1988). However, in HTETOP cells there was no evidence for the rDNA loci being disproportionately involved in chromatin bridges (Fig. 4B). For example, although 96% of anaphases in topo IIα-depleted (72 hours dox) cells exhibit chromatin bridging (48/50 anaphase cells), rDNA was detected in the chromatin bridge in only 6% of cases (3/50). In all remaining cells the nucleolar organiser regions segregated correctly. Telomeric DNA was frequently observed within the chromatin bridges arising from topo IIα depletion, consistent with a failure to decatenate the distal regions of some chromosome arms. The occasional detection of chromatin bridges that lack (TTAGGG)n hybridisation signals could be due either to a hybridisation failure, or could reflect bridging events arising from chromosomal rearrangements, such as breakage-fusion-bridge cycles, rather than from a failure of decatenation (Fig. 4C)
Fig. 4.
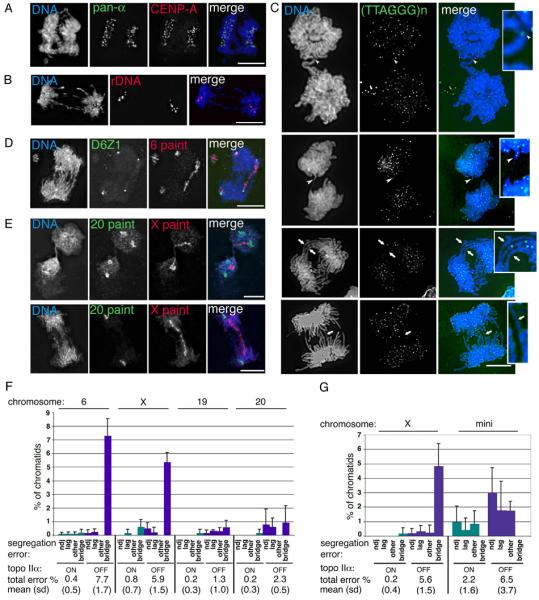
The effect of topo IIα depletion on human chromosome segregation. (A)-(E) HTETOP topo IIα-depleted (48 hours dox) segregating cells. (A) immuno-FISH showing co-localisation of segregating signals for the DNA pan-alphoid probe (green) and anti-CENP-A (red); (B) typical FISH image showing segregation of the rDNA loci (red); (C) four post-metaphase cells showing the segregation behaviour of telomeric regions, detected using a PNA anti-telomere probe (green). White arrowheads indicate telomeric DNA in chromatin bridges. White arrows point to chromatin bridges that lack detectable (TTAGGG)n signals. These regions have been enlarged for clarity. (D) in this anaphase the centromeres (green) of both copies of chromosome 6 have segregated, while the 6 paint (red) reveals that for one of these chromosomes the sister chromatids are bridged. This cell also appears to contain an acentric fragment of chromosome 6. (E) Two examples of segregating cells in which chromosome 20 (green) has separated correctly (although the second cell appears to have only one copy of this chromosome), while the sister chromatids of the single X chromosome (red) form a bridge. DNA is counterstained with DAPI – blue. Scale bar, 10 μm. (F) Chromosome paints and centromere-specific DNA probes were used to analyse the segregation of four of the endogenous HTETOP chromosomes: 6, the X, 19 and 20. For each chromosome the numbers of sister chromatids analysed from either untreated (topoIIαON) or 48 hours doxycycline-treated (topo IIαOFF) cells ranged from 141 to 447 per experiment. Shown are the means from 3 independent experiments (with s.d.). The total number of chromatids assessed for each chromosome under topo IIα ON/OFF conditions were: Chr. 6 - 1230 and 1183; Chr. X – 657 and 592; Chr. 19 – 1146 and 1058; Chr. 20 – 1298 and 1006. Segregation errors were classified as non-disjunction (ndj), lagging, bridging, or others (uncharacterised, apparent 1:0, segregation events, which might reflect absence of replication, chromosome loss or hybridisation failure). The combined segregation errors for each chromosome are summarised below the graph (mean percentage, s.d.). (G) The segregation behaviour of a 2.7 Mb human X centromere-based minichromosome was analysed following its transfer (by cell fusion) into HTETOP cells. Segregation data for the endogenous X chromosome in this hybrid background is also presented (mean and s.d.). For both the mini and X chromosomes the total number of sister chromatids examined from either untreated or 48 hours doxycycline-treated cells was ~1300 (based on data collected from 3 experiments for each of two independently-derived hybrid cell lines). The total levels of chromatid missegregation following topo IIα depletion are significantly higher, both for the whole X and for the minichromosome (Paired Student's t-Test, p ≤0.005).
Chromosome-specific paints, used in conjunction with centromere-specific α-satellite probes, revealed that following topo IIα depletion, segregation errors increased for all chromosomes studied, but were especially marked for the larger chromosomes (Fig. 4 D-F). For example, for chromosome 6 (overall size ~176 Mb with arms of ~65 and ~111 Mb) and the X (~160 Mb with arms of ~60 and ~100 Mb)] 7.7% and 5.9 % of chromatids were missegregated in cells depleted of topo IIα, while for chromosomes 19 and 20 (overall size ~70 Mb with arms of ~30 and ~40 Mb) topo IIα depletion induced missegregation rates of 1.3% and 2.3% of chromatids respectively. For chromosome 6 and the X chromatin bridging made up the bulk of the missegregation events (95% and 89% respectively), while for chromosomes 19 and 20 bridging events contributed only 44% and 39% of the total errors (the rest being nondisjunction, chromosome lagging, or apparent 1:0 segregation events).
To test further the notion that the bridging caused by topo IIα depletion is a function of chromosome arm length, we examined the segregation of an “armless” 2.7 Mb human minichromosome following its transfer into HTETOP cells. This linear minichromosome is a truncated version of the human X chromosome: the arms have been deleted and it's nontelomeric DNA is composed totally of centric and pericentromeric sequences (Spence et al., 2006). Two independently derived hybrids were analysed and gave similar results. Consistent with a correlation between arm length and chromosome bridging, the minichromosome displayed no bridging, before or after topo IIα depletion. Strikingly, however, other forms of missegregation, to which the minichromosome is particularly prone, were exacerbated by topo IIα depletion. Thus when topo IIα was depleted (48 hours dox) the missegregation rate of the minichromosome increased from 2.2 % to 6.5%, compared with <0.2% to 5.6% for the X (Fig. 4G). While for the X chromosome most errors involved chromatin bridging (86%), for the minichromosome the errors detected were a mixture of nondisjunction, lagging and apparent 1:0 segregation events. To rule out the possibility that doxycycline exposure might by itself affect chromosome segregation, the behaviour of the minichromosome was assessed in the parental HT1080 background following 72 hours doxycycline; there was no detectable effect on the rate of minichromosome missegregation (<1% both in the presence and absence of doxycycline). These data therefore suggest that, in addition to a crucial role in preventing the accumulation of catenations in chromosome arms, topo IIα is required for normal centromere function.
Prominent PICH connections in topo IIα-depleted anaphase cells
To investigate general kinetochore assembly, HTETOP cells in which topo IIα has been depleted (72-96 hours doxycycline) were examined by IF using antibodies against CENP-A, -C, -E, –F, Aurora B and acetylated histone H3. The depletion of topo IIα does not have any obvious effect on the localisation of these proteins, or on the underacetylated nature of the centromere region (Fig. 5). Quantification of the centromere-specific histone H3 variant CENP-A at randomly selected centromeres, by indirect IF, revealed no significant change in cells depleted of topo IIα (n 459) relative to cells expressing topo IIα (n 906): topo IIαOFF median = 3608 Fluorescence Units (FU) (average absolute deviation 912) compared to topo IIαON median = 3560 FU (average absolute deviation 799). CENP-A localisation was also examined in HTETOP cells depleted of IIβ and in cells depleted of both isoforms simultaneously. CENP-A was detected at centromeres in all cases (data not shown) Thus, there appears to be no obvious effect of topo II depletion on the localisation of CENP-A, or of the other kinetochore components examined.
Fig. 5.

Immunofluorescence of topo IIα-expressing and topo IIα-depleted (72 hours dox) HTETOP cells. (A) Metaphases (cytospun, no colcemid) stained using an antibody against CENP-A (green); (B) partial chromosome spreads (colcemid) stained for CENP-F (green); (C) partial metaphase spreads (colcemid) stained for CENP-E (green)); (D) typical metaphase cells (grown in situ, no colcemid) stained for CENP-C (green) and Aurora B (red); (E) immuno-FISH of partial chromosome spreads (colcemid) showing the typical banding pattern generated using an antibody raised against acetylated histone H3 (Lysines 1-21) (green). The centromeric regions have been identified using a pan-alphoid DNA probe (red). The white arrowheads highlight examples of hypoacetylated centromeric and pericentromeric domains. DNA/DAPI - blue. Scale bars, 10 μm.
We then examined the effect of topo IIα depletion on the localisation of PICH (Plk1-interacting checkpoint helicase) (Baumann et al., 2007). PICH was found at the centromere in metaphase and PICH-positive threads were detected in anaphase irrespective of the presence/absence of topo IIα (Fig. 6). Strikingly the number of PICH-positive threads detected post-metaphase increased substantially in cells depleted of topo IIα (48 hours dox) (Fig. 6B,C). Thus, following depletion of topo IIα the mean number of threads per anaphase cell increased 5-fold (from 0.9 to 4.8 PICH threads/cell), with the proportion of PICH thread-negative anaphases decreasing from 74% to 10%. In some cases these PICH-positive threads could be distinguished from the DAPI-stained chromatin bridges (examples indicated in Fig. 6B).
Fig. 6.

The effect of topo IIα depletion on anaphase PICH-coated threads. HTETOP cells expressing topo IIα (A), or depleted of topo IIα (48 hours dox) (B) were fixed and permeabilised before being co-stained for PICH (green) and CENP-B (red). Shown are cells in anaphase, or undergoing aberrant cytokinesis. Arrowheads indicate examples of PICH threads that are not associated with DAPI-stained chromatin bridges. Scale bar, 10 μm. (C) Quantification of the number of PICH threads for anaphase cells expressing topo IIα (topo IIαON) and for cells treated for 48 hours with doxycycline (topo IIαOFF). Fifty anaphase cells were analysed for each and the stage was determined by the distance between segregating sister kinetochores.
Both isoforms contribute to centromeric topo II cleavage activity
Topo IIα protein has been shown to be concentrated at the centromere of mitotic vertebrate chromosomes and to have major cleavage sites within centromeric DNA. To examine the effect of depleting topo IIα on centromeric DNA cleavage, this activity was assayed within the centromere of the 2.7 Mb human X centromere-based minichromosome. This allowed us to examine a molecularly-defined haploid centromere, resolvable using PFGE. Two independently-derived HTETOP:minichromosome hybrids were analysed and gave similar results.
There was a noticeable difference in the ethidium bromide-staining pattern of HMW DNA depending on whether the cells had been treated with doxycycline. In the absence of etoposide (irrespective of doxycycline exposure) the bulk of the DNA was restricted to the slots and region of the gel above the compression zone, consistent with it being intact HMW DNA. Following etoposide exposure the DNA extracted from cells expressing topo IIα showed a substantial accumulation of fragments in the 200 kb–2 Mb size range. In cells from which most of the topo II has been depleted the bulk of the DNA from etoposide-exposed cells was >2 Mb. This is consistent with all, or most, of the double strand breaks trapped by etoposide being created by the action of topo II (Fig. 7A).
Fig. 7.

The effect of topo II depletion on centromeric topo II cleavage activity. The HTETOP: minichromosome hybrid cells expressing, or depleted (72 hours dox) for, topo IIα and/or topo IIβ (72 hours siRNA) were exposed in culture to etoposide (0 (DMSO only), 100 or 500 μM) for 60 minutes at 37° C and embedded in agarose. (A) Undigested HMW DNA was resolved by PFGE and stained using ethidium bromide. (B) After transfer the Southern blot was probed using the X α-satellite DNA DXZ1 to detect the 2.7 Mb X centromere-based minichromosome ( ). An etoposide-specific DXZ1-hybridising fragment of ~1.85 Mb (<) could be detected, in addition to a more general smear of hybridisation (this was more noticeable in DNA from cells expressing topo IIα). At the lower etoposide concentration (100 μM) a signal in the 1.85 Mb range could only be detected in cells expressing topo IIα (either alone, or together with the β isoform); at the higher etoposide concentration (500 μM) the 1.85 Mb signal was barely detectable after depletion of both isoforms, but could still be detected following depletion of topo IIα alone (although the signal was reduced relative to that of the intact minichromosome in the same sample). This suggests that both isoforms contribute to this cleavage site within the centromeric DNA.
). An etoposide-specific DXZ1-hybridising fragment of ~1.85 Mb (<) could be detected, in addition to a more general smear of hybridisation (this was more noticeable in DNA from cells expressing topo IIα). At the lower etoposide concentration (100 μM) a signal in the 1.85 Mb range could only be detected in cells expressing topo IIα (either alone, or together with the β isoform); at the higher etoposide concentration (500 μM) the 1.85 Mb signal was barely detectable after depletion of both isoforms, but could still be detected following depletion of topo IIα alone (although the signal was reduced relative to that of the intact minichromosome in the same sample). This suggests that both isoforms contribute to this cleavage site within the centromeric DNA.
Southern blotting allowed us to focus on etoposide-specific cleavage fragments originating from within the centromeric α-satellite (DXZ1) DNA array on the minichromosome. Previous work has shown that exposure of growing cells to etoposide (0, 100 or 500 μM, 60 minutes at 37°C) results in partial cleavage of the minichromosome, generating 1.85 and 0.85 Mb DXZ1-hybridising fragments, together with a smear of more randomly cleaved products (this smear often masks the weaker 0.85 Mb hybridisation band) (Spence et al., 2002; Spence et al., 2005). In the HTETOP background following 72 hours doxycycline the amount of DXZ1 cleavage product was reduced substantially (and was barely detectable using 100 μM etoposide), although cleavage could still be detected using 500 μM etoposide (but the 1.85 Mb signal was reduced relative to that of the intact 2.7 Mb minichromosome). In order to determine the origin of the residual cleavage we investigated the effect of depleting IIβ, which has been estimated by others to be present at ~13% of IIα protein levels (Sakaguchi and Kikuchi, 2004). Depletion of topo IIβ alone had no detectable effect in this assay, but following depletion of both isoforms simultaneously very little, if any, DXZ1 cleavage was detectable, even at the higher etoposide concentration (Fig. 7B). This is consistent with etoposide trapping double strand breaks generated by both isoforms of topo II, and suggests that both isoforms can contribute to cleavage within centromeric DNA.
A shortening in the distance across topo IIα depleted-bioriented sister centromeres is consistent with impeded decatenation
To determine whether topo IIα depletion has any effect on the structure of sister kinetochores under tension, HTETOP cells were arrested using MG132, fixed and subjected to FISH using a chromosome 11-specific centromere probe (D11Z1). The α-satellite DNA of biorented sister chromosomes appeared either as separate dots or stretched continuously across the centromere domain. The distance across sister centromere pairs, to the outer edges of the D11Z1 signal, was measured after 72 hours doxycycline, in parallel with untreated controls (Fig. 8A,B).
Fig. 8.

The effect of topo IIα depletion on metaphase inter-kinetochore distance. (A) Distances between sister centromeres of human chromosome 11 detected by FISH using D11Z1 DNA as a probe were assessed using Leica Deblur software. Scale bar, 10 μm. (B) Measurements made of treated cells in parallel with untreated controls (treated/ untreated pairs indicated by brackets) are presented as a boxplot showing the third and first quartiles, with the median indicated by a cross in each box. The maximum and minimum values are indicated by the ends of the vertical lines. Each plot is based on ≥40 measurements. In HTETOP topo IIα was depleted by 72 hours doxycycline exposure. Cells were arrested using MG132 (10 μM for 2 hours 30 minutes) allowing distances under tension to be measured. For the doxycycline-treated/ topo IIα-depleted HTETOP cells data from 3 independent experiments is presented. In each case the decrease in the distance across the centromere domain under tension following topo IIα-depletion is significant (p = 0.001, 0.0001, 0.01 respectively). To deplete topo IIβ HTETOP cells were transiently transfected with siRNA (72 hours). Colcemid-treated HTETOP cells served as a control where no spindle or tension existed. The lack of any effect from doxycycline-treatment itself was confirmed by analysis of MG132-arrested parental HT1080 cells. Clone J is a derivative of HTETOP that constitutively expresses topo IIα as a fusion with the C terminus of eGFP (Carpenter and Porter, 2004). (C) Quantification of spindle lengths in HTETOP cells arrested using MG132 (10 μM for 2 hours 30 minutes) following 0 (topo IIαON) or 72 hours (topo IIαOFF) doxycycline exposure. Measurements of pole-to-pole distance, based in γ-tubulin and DAPI staining, were collected from 3 independent experiments (topo IIαONn154, topo IIαOFF n151).
The distances across paired centromeres under tension in HTETOP cells decreased significantly (~10-25%) in topo IIα-depleted cells during metaphase (Fig 8B). In topo IIαON HTETOP cells grown in the absence of doxycycline and arrested using MG132 the median distance between sister centromeres was 1.76 μm (average absolute deviation 0.39, n163), while in topo IIαOFF HTETOP cells the distance was significantly less (1.46 μm, average absolute deviation 0.35, n188, p <0.0001] (based on the 3 independent experiments presented in Fig. 8B). No significant difference in the distance across sister centromeres was detected in the absence of tension (data collected from colcemid-arrested topoIIαON and topoαOFF HETETOP cells), or following transient depletion of topo IIβ (72 hours siRNA + MG132).
To rule out the possibility that this decrease was due to an effect of doxycycline exposure, rather than due to depletion of topo IIα, distances were measured in the same way in the parental HT1080 cell line grown with and without doxycycline addition and arrested using MG132. There was no significant difference in inter-kinetochore distance indicating that the decrease is specific to depletion of topo IIα (Fig. 8B). Further support for this is provided by the observation that in HTETOP cells rescued from doxycycline-sensitivity by constitutive expression of the topo IIα protein (as a fusion with the C terminus of eGFP in cell line HTETOP/GFP-topoIIα Clone J, (Carpenter and Porter, 2004) the distance across sister centromeres is restored (i.e. there is no significant difference in sister centromere distances in clone J cells grown in the presence or absence of doxycycline, arrested using MG132) (Fig. 8B).
Since influences that perturb centromere organisation sometimes have an impact on the metaphase spindle, spindle length was examined in HTETOP cells stained with γ-tubulin. Data collected from three independent experiments revealed a small, but significant, increase in the pole-to-pole spindle distance in cells depleted of topo IIα: topo IIαON cells (0 hours dox) mean spindle length = 8.29 μm (sd 2.25, n154); topo IIαOFF cells (72 hours dox) mean distance = 9.91 μm (sd 3.38, n150) p = 0.0001 (Fig. 8C). A similar significant increase in spindle length was detected where distances between focused spindle poles were estimated based on α-tubulin staining (data not shown). Although, following 72 hours of topo IIα-depletion, many nuclei had an abnormal appearance (Carpenter and Porter, 2004) estimates of nuclear size/polyploidy in the two cell populations (based on the DAPI-stained area in interphase cells) revealed no significant change (topo IIαON: mean area 271 μm2, sd 89.1, n329 compared with topo IIαOFF mean 275 μm2, sd 111, n343).
Discussion
Topo IIα is not essential for the maintenance of gross structural integrity
Several reports have now shown that in living cells extensive chromosome condensation occurs in the absence of topo II, although in general the process seems to take longer, with the final extent of compaction being less (Chang et al., 2003; Carpenter and Porter, 2004; Sakaguchi and Kikuchi, 2004; Toyoda and Yanagida, 2006) Nevertheless, the chromosomes look remarkably normal. Moreover, unlike depletion of condensin, which leads to altered topo II localisation (Hudson et al., 2003; Vagnarelli et al., 2006) topo II depletion (in either human or Drosophila cells) does not affect the axial distribution of condensin (this report and (Chang et al., 2003). Therefore, in order to probe topo IIα-depleted chromosomes for possible effects on other aspects of structure they were exposed to buffer conditions that, when applied to normal chromosomes, result in the alternate disruption and restoration of the chromosome architecture. Following depletion of condensin from vertebrate cells, chromosomes are unable to recover their condensed morphology after unfolding under these conditions (Hudson et al., 2003). However, upon removal of topo IIα (alone, or with IIβ) mitotic chromosomes, although generally longer and thinner than normal, nervertheless retained the ability to recondense after extensive unfolding. Although we do rule out the possibility that these compacted structures may be, to some extent, abnormal our observations suggest that in human cells topo II does not have an essential scaffolding role. This is consistent with earlier observations that topo II (i) can be extracted from condensed chromosomes assembled in vitro without any observed change in structure (Hirano and Mitchison, 1993) and (ii) interacts very dynamically with mitotic chromatin (Christensen et al., 2002; Tavormina et al., 2002).
Topo IIα is required for a compact metaphase plate
When we examined the organisation of the metaphase plate in the absence of topo IIα we observed that in many cells one or more chromosome arms extended away from the compact mass of chromatin, often stretching towards the pole, apparently independent of centromere activity. A similarly disorganised metaphase plate has been reported in topo II-depleted S2 cells (Chang et al., 2003; Savvidou et al., 2005). This suggests that the chromosome arms may be trapped in the vicinity of the pole and are unable to retract, or be pushed back, towards the metaphase plate. Whether this chromosome arm congression defect reveals a conserved role for the centrosomal topo II protein described by others (Barthelmes et al., 2000) remains to be explored.
Topo IIα ensures that long chromosome arms are resolved in anaphase
The major topo IIα-depletion phenotype is a defect in chromosome segregation, with virtually all cells displaying one or more anaphase chromatin bridges. We have shown that chromosomes with longer arms are involved in bridging more frequently (5-10-fold) than smaller chromosomes. We found no evidence of either centromeric DNA or the rDNA loci being disproportionately involved in these bridges. One explanation for the positive correlation between bridging and chromosome arm length would be that, as sister chromatids are drawn apart by the poleward spindle forces, residual catenations are either ruptured, or slide along the arms, allowing separation to proceed from the centromere outwards. The longer the arm the more likely it is that residual intertwinings, accumulating in the distal regions, may fail to be resolved during anaphase, resulting in a bridge. It is also possible that reduced chromosome condensation in topo IIα-depleted cells contributes to the bridging phenotype. Similar observations about long arms preventing intertwinings from passively resolving off the end during segregation have been made in S.cerevisiae topo II mutants (Spell and Holm, 1994).
A linear minichromosome reveals a topo IIα-depletion effect at the centromere
The concentration of topo IIα at the mitotic centromere raises the question as to whether this isoform has a specific role in some aspect of centromere biology. In our system topo IIα is gradually and asynchronously depleted from cells and as a result it is not possible to study the impact of topo IIα depletion on the centromere independently from its effects on arm decatenation. However, we were able to study the segregation behaviour of a linear minichromosome derived from the centromeric and pericentromeric regions of the human X chromosome. Since FISH had revealed a positive correlation between bridging and arm length and suggested normal centromere separation we were surprised at the relatively high level of missegregation displayed by the minichromosome. Given that this structure would have no/ or few arm catenations to resolve, this suggests a topo IIα-depletion defect at the centromere, to which our minichromosome is sensitised compared with normal chromosomes. This observation, together with earlier reports of drug-inhibition of topo II affecting kinetochore structure (Rattner et al., 1996) and the more recent detection of major sites of topo II cleavage activity within human centromeric DNA (Floridia et al., 2000; Spence et al., 2002; Spence et al., 2005) led us to examine centromere behaviour in the absence of topo IIα in more detail.
Topo IIα depletion is associated with a shortening in the distance across bioriented sister centromeres
We detected a significant shortening (~10-25%) in the distance across sister centromeres following the depletion of topo IIα. Transient depletion of IIβ alone had no detectable effect. Depleting both isoforms simultaneously resulted in a highly disordered metaphase plate, making the identification of sister centromere pairs difficult. However, where bioriented sister centromere pairs could be identified, the distance between them was found to decrease to a similar extent to that observed following IIα depletion (data not shown). This is consistent with the 20-30% shortening reported previously following either the depletion, or the ICRF193-induced inhibition, of both topo II isoforms simultaneously (Toyoda and Yanagida, 2006).
This decreased distance across bioriented centromeres argues against a role for topo II in centromere cohesion. Instead it would be consistent with depletion of topo II impeding decatenation and resulting in the persistence of highly intertwined duplexes with reduced flexibility (Skibbens et al., 1993; Shelby et al., 1996). Intriguingly, a small but significant increase in the length of the spindle was also detected following topo IIα depletion. While many nuclei had an abnormal appearance, the increased spindle length did not appear to be accounted for by any significant increase in nuclear size. Thus, depletion of topo IIα results in decreased stretching of bioriented chromosomes and elongation of the mitotic spindle. In this regard it is intriguing that recent affinity purification experiments have revealed a potential interaction between Drosophila topo II and Ndc80 (Przewloka et al., 2007). This raises the possibility that topo IIα may exert an effect on kinetochore-MT interactions through the Ndc80 complex. Further work is required to elucidate the function of topoIIα within the centromere domain and to understand the mechanism underlying the decreased distance across bioriented sister centromeres.
TopoIIα has role in the generation and/ or resolution of anaphase PICH connections
Consistent with observations on the depletion of the single topo II isoform in S2 cells (Chang et al., 2003), we found that depletion of topo IIα (alone, or with IIβ) had no detectable effect on generalised kinetochore assembly in human cells. There was also no obvious effect, at the level of the condensed metaphase chromosome, on the hypoacetylated status of the centromere/pericentromeric domain. We then examined the distribution of the recently identified inner centromere protein PICH (Baumann et al., 2007). PICH could be detected at the centromere domain of metaphase chromosomes and was seen extending, in thread-like structures, between segregating chromosomes in anaphase cells. Strikingly, when topo IIα levels are depleted the number of PICH threads increased substantially in segregating cells. This is consistent with the observation that if topo II is inhibited using ICRF193 or ICRF159, PICH threads becoming very prominent and supports the idea that PICH, a putative chromatin-remodelling enzyme, associates with catenated DNA, that is stretched under tension, until it is resolved during anaphase (Baumann et al., 2007; Chan et al., 2007) . At least some of the PICH threads detected in topo IIα-depleted anaphases were clearly distinct from chromatin bridges, suggesting that this protein is not simply associated with residual arm catenations. Others have shown that the PICH-coated ultrafine anaphase connections contain DNA and colocalise with the Bloom's syndrome helicase BLM and its cellular partners topo IIIα and hRMI1, suggesting that topo IIIα may have a key role in their resolution (Chan et al., 2007). Moreover, these extensions are often capped by immunofluorescence signals for the outer kinetochore protein Hec1, suggesting that they may involve DNA originating from the centromere (Baumann et al., 2007; Chan et al., 2007). Intriguingly, evidence has been presented, from vertebrates and parasitic protozoa, for a concentration of topo II cleavage activity within centromeric DNA (Floridia et al., 2000; Spence et al., 2002; Spence et al., 2005; Kelly et al., 2006; Obado et al., 2007). We have shown here that in human cells both topo II isoforms contribute to this cleavage activity. Establishing what relationship, if any, exists between this topo II-susceptible centric subdomain and the PICH/BLM-coated ultrafine anaphase connections awaits future investigation.
Concluding remarks
It is well known that perturbation of topo IIα activity has serious consequences for chromosome segregation. This study has shown that the DNA caught up in anaphase chromatin bridges arises predominantly from a failure in chromosome arm decatenation, with longer chromosome arms being particularly susceptible. In addition to this, however, we detected a surprisingly high rate of missegregation by a <3 Mb “arm-less” minichromosome. Closer examination of the impact topo IIα depletion on chromosome segregation revealed a number of more subtle effects including a defect in chromosome arm congression, a shortening of the distance across bioriented sister centromeres, an increase in the length of the mitotic spindle and an increase in the incidence of PICH-coated connections in anaphase cells. Currently, we do not understand which of these phenotypes impacts on the mitotic stability of the minichromosome, how they are related, or the underlying mechanism(s). It is possible that the shortening of interkinetochore distance and lengthening of the spindle reflect effects of topo IIα depletion on the stability of kinetochore-microtubule attachments. Previous work has indicated that the minichromosome has a compromised kinetochore (based on reduced levels of CENP-A and Aurora B) (Spence et al., 2006), which may make it especially susceptible to destabilising effects.
The concentration of topo IIα at mitotic centromeres should serve to ensure that there is efficient decatenation of residual intertwinings at anaphase onset. This would appear to be critical for maintaining genome integrity, since the powerful forces exerted by the kinetochore microtubules on this part of the chromosome during poleward movement could lead to rupture of the duplex, especially if the capacity for removing centromeric catenations through displacement along the arms is restricted by the presence of the kinetochore. However, identification of the PICH protein revealed that in mitosis it is normal for connections to persist between segregating sisters well into anaphase. Why this occurs is perplexing. The suggestion that PICH may regulate access of the decatenating activity and/ or protect catenated centromeric DNA from non-specific rupture (Baumann et al., 2007) presents challenges for future experiments to test the link between the various topoisomerases (topo II and topo III), helicases (PICH and BLM) and cohesin in chromosome alignment and separation.
Acknowledgements
We are grateful to the following for their generous gifts of antibodies: W.C. Earnshaw (CENP-C, CENP-B, SMC2); C. Baumann and E.A. Nigg (PICH); M. Valdivia (CENP-A) and T.Yen (CENP-E, CENP-F). We thank P. Vagnarelli and M. Johnson for reagents, discussions and advice. These experiments were supported by projects grants from Cancer Research-UK (C9609/A3527) and the Biotechnology and Biological Sciences Research Council (BBS/B/04994).
References
- Agostinho M, Rino J, Braga J, Ferreira F, Steffensen S, Ferreira J. Human topoisomerase IIα: targeting to subchromosomal sites of activity during interphase and mitosis. Mol. Biol. Cell. 2004;15:2388–2400. doi: 10.1091/mbc.E03-08-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimitsu N, Adachi N, Hirai H, Hossain MS, Hamamoto H, Kobayashi M, Aratani Y, Koyama H, Sekimizu K. Enforced cytokinesis without complete nuclear division in embryonic cells depleting the activity of DNA topoisomerase IIα. Genes Cells. 2003;8:393–402. doi: 10.1046/j.1365-2443.2003.00643.x. [DOI] [PubMed] [Google Scholar]
- Andersen CL, Wandall A, Kjeldsen E, Mielke C, Koch J. Active, but not inactive, human centromeres display topoisomerase II activity in vivo. Chromosome Res. 2002;10:305–312. doi: 10.1023/a:1016571825025. [DOI] [PubMed] [Google Scholar]
- Bachant J, Alcasabas A, Blat Y, Kleckner N, Elledge SJ. The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol. Cell. 2002;9:1169–1182. doi: 10.1016/s1097-2765(02)00543-9. [DOI] [PubMed] [Google Scholar]
- Barthelmes HU, Grue P, Feineis S, Straub T, Boege F. Active DNA topoisomerase IIα is a component of the salt-stable centrosome core. J. Biol. Chem. 2000;275:38823–38830. doi: 10.1074/jbc.M007044200. [DOI] [PubMed] [Google Scholar]
- Baumann C, Korner R, Hofmann K, Nigg EA. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell. 2007;128:101–114. doi: 10.1016/j.cell.2006.11.041. [DOI] [PubMed] [Google Scholar]
- Carpenter AJ, Porter AC. Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIα mutant human cell line. Mol. Biol. Cell. 2004;15:5700–5711. doi: 10.1091/mbc.E04-08-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaly N, Chen X, Dentry J, Brown DL. Organization of DNA topoisomerase II isotypes during the cell cycle of human lymphocytes and HeLa cells. Chromosome Res. 1996;4:457–466. doi: 10.1007/BF02265053. [DOI] [PubMed] [Google Scholar]
- Chan KL, North PS, Hickson ID. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007;26:3397–3409. doi: 10.1038/sj.emboj.7601777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CJ, Goulding S, Earnshaw WC, Carmena M. RNAi analysis reveals an unexpected role for topoisomerase II in chromosome arm congression to a metaphase plate. J. Cell Sci. 2003;116:4715–4726. doi: 10.1242/jcs.00797. [DOI] [PubMed] [Google Scholar]
- Christensen MO, Larsen MK, Barthelmes HU, Hock R, Andersen CL, Kjeldsen E, Knudsen BR, Westergaard O, Boege F, Mielke C. Dynamics of human DNA topoisomerases IIα and IIβ in living cells. J. Cell Biol. 2002;157:31–44. doi: 10.1083/jcb.200112023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman MF, Dietrich FS, Fink GR. Mitotic recombination in the rDNA of S. cerevisiae is suppressed by the combined action of DNA topoisomerases I and II. Cell. 1988;55:413–425. doi: 10.1016/0092-8674(88)90027-x. [DOI] [PubMed] [Google Scholar]
- Cole A. Chromosome structure. Theor. Biophys. 1967;1:305–375. [Google Scholar]
- Cooke CA, Bernat RL, Earnshaw WC. CENP-B: a major human centromere protein located beneath the kinetochore. J. Cell Biol. 1990;110:1475–1488. doi: 10.1083/jcb.110.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floridia G, Zatterale A, Zuffardi O, Tyler-Smith C. Mapping of a human centromere onto the DNA by topoisomerase II cleavage. EMBO Rep. 2000;1:489–493. doi: 10.1093/embo-reports/kvd110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbsky GJ. Cell cycle progression and chromosome segregation in mammalian cells cultured in the presence of the topoisomerase II inhibitors ICRF-187 [(+)-1,2-bis(3,5-dioxopiperazinyl-1-yl)propane; ADR-529] and ICRF-159 (Razoxane) Cancer Res. 1994;54:1042–1048. [PubMed] [Google Scholar]
- Grue P, Grasser A, Sehested M, Jensen PB, Uhse A, Straub T, Ness W, Boege F. Essential mitotic functions of DNA topoisomerase IIα are not adopted by topoisomerase IIβ in human H69 cells. J. Biol. Chem. 1998;273:33660–33666. doi: 10.1074/jbc.273.50.33660. [DOI] [PubMed] [Google Scholar]
- Heck MM, Hittelman WN, Earnshaw WC. Differential expression of DNA topoisomerases I and II during the eukaryotic cell cycle. Proc. Natl. Acad. Sci. USA. 1988;85:1086–1090. doi: 10.1073/pnas.85.4.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Mitchison TJ. Topoisomerase II does not play a scaffolding role in the organization of mitotic chromosomes assembled in Xenopus egg extracts. J. Cell Biol. 1993;120:601–612. doi: 10.1083/jcb.120.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson DF, Vagnarelli P, Gassmann R, Earnshaw WC. Condensin is required for nonhistone protein assembly and structural integrity of vertebrate mitotic chromosomes. Dev. Cell. 2003;5:323–336. doi: 10.1016/s1534-5807(03)00199-0. [DOI] [PubMed] [Google Scholar]
- Kelly JM, McRobert L, Baker DA. Evidence on the chromosomal location of centromeric DNA in Plasmodium falciparum from etoposide-mediated topoisomerase-II cleavage. Proc. Natl. Acad. Sci. USA. 2006;103:6706–6711. doi: 10.1073/pnas.0510363103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ. CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J. Cell Biol. 1995;130:507–518. doi: 10.1083/jcb.130.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KN, Kjeldsen E, Straub T, Knudsen BR, Hickson ID, Kikuchi A, Kreipe H, Boege F. Cell cycle-coupled relocation of types I and II topoisomerases and modulation of catalytic enzyme activities. J. Cell Biol. 1997;136:775–788. doi: 10.1083/jcb.136.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov A, Shinohara M, Rieder CL. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J. Cell Biol. 2004;166:517–526. doi: 10.1083/jcb.200405167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Null AP, Hudson J, Gorbsky GJ. Both alpha and beta isoforms of mammalian DNA topoisomerase II associate with chromosomes in mitosis. Cell Growth Differ. 2002;13:325–333. [PubMed] [Google Scholar]
- Obado SO, Bot C, Nilsson D, Andersson B, Kelly JM. Repetitive DNA is associated with centromeric domains in Trypanosoma brucei but not Trypanosoma cruzi. Genome Biol. 2007;8:R37. doi: 10.1186/gb-2007-8-3-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter AC, Farr CJ. Topoisomerase II: untangling its contribution at the centromere. Chromosome Res. 2004;12:569–583. doi: 10.1023/B:CHRO.0000036608.91085.d1. [DOI] [PubMed] [Google Scholar]
- Przewloka MR, Zhang W, Costa P, Archambault V, D'Avino PP, Lilley KS, Laue ED, McAinsh AD, Glover DM. Molecular analysis of core kinetochore composition and assembly in Drosophila melanogaster. PLoS ONE. 2007;2:e478. doi: 10.1371/journal.pone.0000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner JB, Hendzel MJ, Furbee CS, Muller MT, Bazett-Jones DP. Topoisomerase II alpha is associated with the mammalian centromere in a cell cycle-and species-specific manner and is required for proper centromere/kinetochore structure. J. Cell Biol. 1996;134:1097–1107. doi: 10.1083/jcb.134.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh H, Tomkiel J, Cooke CA, Ratrie H, 3rd, Maurer M, Rothfield NF, Earnshaw WC. CENP-C, an autoantigen in scleroderma, is a component of the human inner kinetochore plate. Cell. 1992;70:115–125. doi: 10.1016/0092-8674(92)90538-n. [DOI] [PubMed] [Google Scholar]
- Saitoh N, Goldberg IG, Wood ER, Earnshaw WC. ScII: an abundant chromosome scaffold protein is a member of a family of putative ATPases with an unusual predicted tertiary structure. J. Cell Biol. 1994;127:303–318. doi: 10.1083/jcb.127.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi A, Kikuchi A. Functional compatibility between isoform alpha and beta of type II DNA topoisomerase. J. Cell Sci. 2004;117:1047–1054. doi: 10.1242/jcs.00977. [DOI] [PubMed] [Google Scholar]
- Savvidou E, Cobbe N, Steffensen S, Cotterill S, Heck MM. Drosophila CAP-D2 is required for condensin complex stability and resolution of sister chromatids. J. Cell Sci. 2005;118:2529–2543. doi: 10.1242/jcs.02392. [DOI] [PubMed] [Google Scholar]
- Shelby RD, Hahn KM, Sullivan KF. Dynamic elastic behavior of alpha-satellite DNA domains visualized in situ in living human cells. J. Cell Biol. 1996;135:545–557. doi: 10.1083/jcb.135.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibbens RV, Skeen VP, Salmon ED. Directional instability of kinetochore motility during chromosome congression and segregation in mitotic newt lung cells: a push-pull mechanism. J. Cell Biol. 1993;122:859–875. doi: 10.1083/jcb.122.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoufias DA, Lacroix FB, Andreassen PR, Wilson L, Margolis RL. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol. Cell. 2004;15:977–990. doi: 10.1016/j.molcel.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Spell RM, Holm C. Nature and distribution of chromosomal intertwinings in Saccharomyces cerevisiae. Mol. Cell. Biol. 1994;14:1465–1476. doi: 10.1128/mcb.14.2.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence JM, Fournier RE, Oshimura M, Regnier V, Farr CJ. Topoisomerase II cleavage activity within the human D11Z1 and DXZ1 alpha-satellite arrays. Chromosome Res. 2005;13:637–648. doi: 10.1007/s10577-005-1003-8. [DOI] [PubMed] [Google Scholar]
- Spence JM, Mills W, Mann K, Huxley C, Farr CJ. Increased missegregation and chromosome loss with decreasing chromosome size in vertebrate cells. Chromosoma. 2006;115:60–74. doi: 10.1007/s00412-005-0032-6. [DOI] [PubMed] [Google Scholar]
- Spence JM, Critcher R, Ebersole TA, Valdivia MM, Earnshaw WC, Fukagawa T, Farr CJ. Co-localization of centromere activity, proteins and topoisomerase II within a subdomain of the major human X alpha-satellite array. EMBO J. 2002;21:5269–5280. doi: 10.1093/emboj/cdf511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M, Higuchi T, Katis VL, Uhlmann F. Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell. 2004;117:471–482. doi: 10.1016/s0092-8674(04)00415-5. [DOI] [PubMed] [Google Scholar]
- Sumner AT. The distribution of topoisomerase II on mammalian chromosomes. Chromosome Res. 1996;4:5–14. doi: 10.1007/BF02254938. [DOI] [PubMed] [Google Scholar]
- Taagepera S, Rao PN, Drake FH, Gorbsky GJ. DNA topoisomerase II alpha is the major chromosome protein recognized by the mitotic phosphoprotein antibody MPM-2. Proc. Natl. Acad. Sci. USA. 1993;90:8407–8411. doi: 10.1073/pnas.90.18.8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Yong-Gonzalez V, Kikuchi Y, Strunnikov A. SIZ1/SIZ2 control of chromosome transmission fidelity is mediated by the sumoylation of topoisomerase II. Genetics. 2006;172:783–794. doi: 10.1534/genetics.105.047167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavormina PA, Come MG, Hudson JR, Mo YY, Beck WT, Gorbsky GJ. Rapid exchange of mammalian topoisomerase IIα at kinetochores and chromosome arms in mitosis. J. Cell Biol. 2002;158:23–29. doi: 10.1083/jcb.200202053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda Y, Yanagida M. Coordinated requirements of human topo II and cohesin for metaphase centromere alignment under Mad2-dependent spindle checkpoint surveillance. Mol. Biol. Cell. 2006;17:2287–2302. doi: 10.1091/mbc.E05-11-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnarelli P, Morrison C, Dodson H, Sonoda E, Takeda S, Earnshaw WC. Analysis of Scc1-deficient cells defines a key metaphase role of vertebrate cohesin in linking sister kinetochores. EMBO Rep. 2004;5:167–171. doi: 10.1038/sj.embor.7400077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnarelli P, Hudson DF, Ribeiro SA, Trinkle-Mulcahy L, Spence JM, Lai F, Farr CJ, Lamond AI, Earnshaw WC. Condensin and Repo-Man-PP1 co-operate in the regulation of chromosome architecture during mitosis. Nat. Cell Biol. 2006;8:1133–1142. doi: 10.1038/ncb1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia MM, Figueroa J, Iglesias C, Ortiz M. A novel centromere monospecific serum to a human autoepitope on the histone H3-like protein CENP-A. FEBS Lett. 1998;422:5–9. doi: 10.1016/s0014-5793(97)01583-4. [DOI] [PubMed] [Google Scholar]
- Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- Woessner RD, Mattern MR, Mirabelli CK, Johnson RK, Drake FH. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991;2:209–214. [PubMed] [Google Scholar]
- Yang X, Li W, Prescott ED, Burden SJ, Wang JC. DNA topoisomerase IIβ and neural development. Science. 2000;287:131–134. doi: 10.1126/science.287.5450.131. [DOI] [PubMed] [Google Scholar]
- Yen TJ, Compton DA, Wise D, Zinkowski RP, Brinkley BR, Earnshaw WC, Cleveland DW. CENP-E, a novel human centromere-associated protein required for progression from metaphase to anaphase. EMBO J. 1991;10:1245–1254. doi: 10.1002/j.1460-2075.1991.tb08066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]