

Abstract
Ligand-induced activation of G protein-coupled receptors (GPCRs) is a key mechanism permitting communication between cells and organs. Enormous progress has recently elucidated the structural and dynamic features of GPCR transmembrane signaling. Nanobodies, the recombinant antigen-binding fragments of camelid heavy-chain-only antibodies, have emerged as important research tools to lock GPCRs in particular conformational states. Active-state stabilizing nanobodies have elucidated several agonist-bound structures of hormone-activated GPCRs and have provided insight into the dynamic character of receptors. Nanobodies have also been used to stabilize transient GPCR transmembrane signaling complexes, yielding the first structural insights into GPCR signal transduction across the cellular membrane. Beyond their in vitro uses, nanobodies have served as conformational biosensors in living systems and have provided novel ways to modulate GPCR function. Here, we highlight several examples of how nanobodies have enabled the study of GPCR function and give insights into potential future uses of these important tools.
Keywords: nanobody, G protein-coupled receptor, conformational plasticity, receptor activation, crystallographic chaperone, intrabody
INTRODUCTION
The G protein-coupled receptor (GPCR) superfamily comprises more than 800 distinct human cell surface receptors that share a common seven-transmembrane α-helical fold (1–3). As highly versatile membrane sensors, GPCRs respond to a variety of extracellular signals, including photons, ions, sensory stimuli, lipids, neurotransmitters, hormones, and large proteins (4). These receptors convert various signaling cues into cellular responses by engaging with intracellular G proteins, β-arrestins, and other downstream effectors (5, 6). About 450 human sensory GPCRs mediate olfaction, taste, light perception, or pheromone signaling (7). The remaining approximately 350 nonsensory GPCRs mediate cell-to-cell signaling and are the targets for a large fraction of clinically used drugs (8, 9), although only a minority of these receptors are exploited therapeutically. About 120 human receptors are still orphan receptors with unknown endogenous ligands and/or function (10).
GPCRs relay diverse extracellular signals into cells by activating associated heterotrimeric G proteins. Agonist binding causes the formation of an active transmembrane signaling complex and results in the exchange of bound GDP for GTP by the Gα subunit of the G protein and the concomitant dissociation and release of the Gα and Gβγ subunits (11, 12). The separate Gα and Gβγ subunits then promote the production of and consequent signaling by second-messenger systems, such as those involving cyclic AMP (13), diacylglycerol (14), or calcium (15). Signaling is terminated by phosphorylation of the cytoplasmic loops and the C-terminal tail of the receptor by GPCR kinases (GRKs). Receptor phosphorylation triggers a cascade of events that include the recruitment of arrestins, uncoupling of GPCRs from heterotrimeric G proteins, receptor internalization, and activation of G protein-independent signal transduction pathways (16, 17). In recent years, evidence has accumulated that GPCRs can form homo-and heterodimers as well as higher-order oligomeric assemblies under physiological conditions (18, 19). It is now well accepted that family C GPCRs form constitutive homo-or heteromers (20).
Researchers have made remarkable progress in the field of GPCR structural biology during the past decade, with more than 100 GPCR structures reported for more than 30 unique receptor subtypes (21). Several obstacles to generating diffraction-quality crystals of GPCRs have been overcome by innovative methods such as recombinant expression systems (22), protein engineering (23–27), lipid-based crystallography (28), and novel methods for collecting X-ray diffraction data (29, 30). These methodological innovations have been combined with novel reagents, including compounds tailored for crystallography (31) and detergents that improve receptor stability (32). Most GPCR structures solved to date represent the most energetically stable, inactive, antagonist-bound conformations. These structures have increased our understanding of ligand binding and selectivity (33). However, a mechanistic understanding of GPCR signaling requires structural insights into the active state as well as other ligand-specific states of the receptor responsible for the broad array of GPCR functions. This review focuses on the instrumental role of nanobodies as tools to study the structural and dynamic features of GPCR transmembrane signaling.
GPCR SIGNALING COMPLEXITY AND CONFORMATIONAL PLASTICITY
Views on activation of GPCRs have evolved considerably over the past 20 years (34). The earliest models consisted of two receptor conformations: an inactive state in equilibrium with an active state (35). In these binary models, receptor signaling correlates directly with this equilibrium. Agonists and partial agonists shift the equilibrium toward the active state and thereby induce activation of heterotrimeric G proteins. Inverse agonists shift the equilibrium to the inactive state and suppress basal activation of G proteins. Although many aspects of GPCR function can be explained by a simple two-state model, accumulating evidence supports more complex multistate behavior (see the sidebar, Conformational Plasticity of Common Drug Targets) (36). Many GPCRs are capable of signaling through multiple intracellular pathways. These include canonical second-messenger signaling pathways mediated by G protein activation as well as G protein-independent pathways. Ligands acting on a given GPCR can induce varying levels of response (37). Some ligands, termed biased agonists, are capable of preferentially activating one signaling pathway among many (38). In addition to ligand-directed activation of a broad range of signaling responses, many other elements can influence GPCR function, including changes in cell membrane lipid composition, tension, and fluidity (39–41) as well as membrane voltage (42). The function of GPCRs is also regulated by pH (43) and common ions such as sodium (44). This broad complexity in function has suggested that GPCRs sample a continuum of conformational ensembles rather than discrete on and off states. A wide variety of biophysical studies—spanning X-ray crystallography, NMR spectroscopy (45–48), double electron-electron resonance (DEER) spectroscopy (49), single-molecule fluorescence (50, 51), and electron microscopy (52–55)—have confirmed and further examined the role of this conformational complexity in GPCR function.
Ligand Efficacy and Biased Signaling is Dependent on GPCR Conformational Complexity
Although highly complex, the conformational dynamics of GPCR signaling can be conceptualized by free-energy landscapes that provide a schematic relation between structure, dynamics, function, and ligand efficacy (56). The relative populations of different states are indicated by the depth of each energy well, and the rate of transition between different states is illustrated by the height of the energy barriers. Recent single-molecule fluorescence (50, 51), NMR (48), DEER spectroscopy (49), and molecular dynamics experiments (57) on the β2-adrenergic receptor (β2AR), a prototypical class A GPCR, indicate that the ligand-free transmembrane core of the receptor is highly flexible and exists in separate inactive conformations that exchange within hundreds of microseconds. Basal activity most likely results from a small fraction of activated receptors that exist within the conformational ensemble of the ligand-free receptor (Figure 1). Surprisingly, agonist binding does not reduce this conformational heterogeneity but rather increases conformational dynamics with states that exchange on a millisecond timescale (58). It thus appears that ligands bind particular conformations of the conformational ensemble preferentially, at the expense of conformational states from the pool of possible conformations for which the ligand has a lower affinity, creating a bias in the reference ensemble by mass action (59). Proteomics-based studies have also demonstrated that different ligands stabilize qualitatively different receptor conformational ensembles (60).
Figure 1.
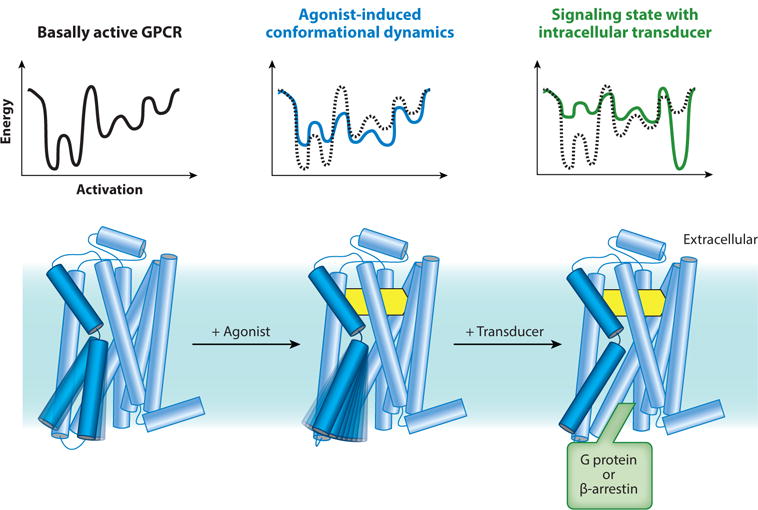
Conformational complexity in G protein-coupled receptor (GPCR) function. Unliganded and antagonist-bound GPCRs are conformationally dynamic. Agonists further induce receptor dynamics to varying degrees, and the crystallographically observed active state is stabilized only in the presence of a G protein or an active state-stabilizing chaperone (49). Energy diagrams illustrate the conformational complexity of GPCRs and highlight the inherent difficulty in capturing agonist-bound receptors in conformationally homogeneous crystallographic lattices.
NANOBODY-BASED STABILIZATION OF GPCR CONFORMATIONS
The intrinsic conformational heterogeneity of GPCRs presented a major hurdle in obtaining the first crystal structures of hormone-activated receptors (61). Most GPCR structures solved to date have relied on high-affinity, slowly dissociating antagonists or the introduction of mutations to limit this inherent conformational flexibility. As most agonists are predicted to induce even further GPCR conformational heterogeneity (58), it is not surprising that the majority of structures solved to date represent inactive conformations (33). In an effort to stabilize the active conformation of agonist-bound GPCRs, we and others have used recombinant camelid single-domain antibody fragments called nanobodies.
Nanobodies: Single-Domain Antibodies that Bind Conformational Epitopes
In addition to conventional heterotetrameric antibodies, camelids (e.g., llamas) also produce functional heavy-chain-only antibodies devoid of light chains (62). The identical heavy chains of these homodimeric antibodies consist of one variable domain and two constant domains (Figure 2). Nanobodies, the recombinant variable domains of the heavy-chain antibodies, are monomeric single-domain proteins encoded by single-gene fragments and harbor the full antigen-binding capacity of the parental antibody (63). Nanobodies are produced easily in microorganisms (64–67), mammalian cell lines (68), and plants (69, 70) and exhibit superior stability compared to recombinant derivatives of conventional antibodies such as single-chain variable fragments (71).
Figure 2.
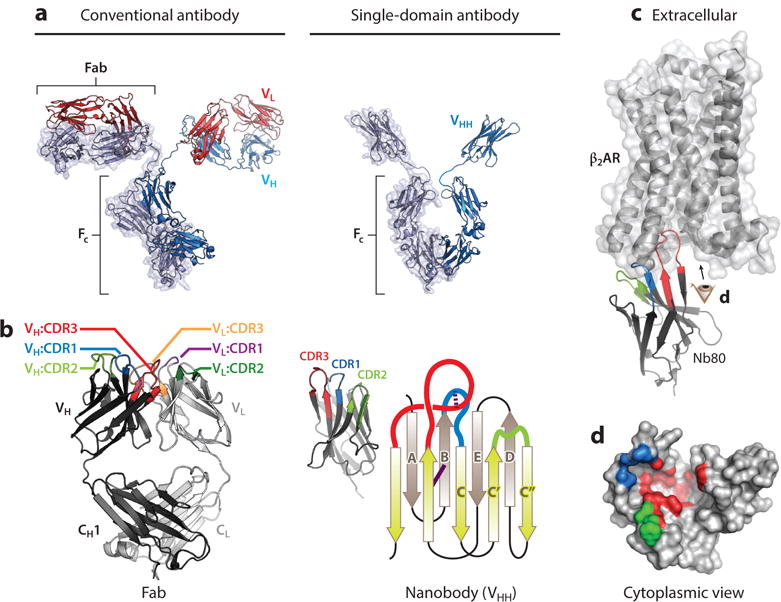
Nanobody structure and function and comparison to conventional antibodies. (a) Comparison of conventional antibodies to camelid single-domain antibodies. Conventional antibodies are heterotetrameric molecules consisting of two heavy chains (VH) and two light chains (VL) with a conserved domain called the crystallizable fragment (FC). Variable loops responsible for antigen binding are within the distal tips of the Fab domain. Camelid single-chain antibodies contain a single immunoglobulin domain (VHH) that binds antigens individually. (b) Comparison of the minimal binding domain of conventional antibodies (Fab) and single-domain antibodies (VHH or nanobody). The antigen-binding region of a Fab is composed of six complementarity-determining regions (CDRs), with three in each VH and VL. Correct VH/VL pairing is required for antigen binding. In contrast, nanobodies contain three CDRs, and the single immunoglobulin fold is sufficient for antigen binding. The nanobody immunoglobulin fold is built from a pair of antiparallel β sheets with a conserved disulfide bond (solid purple line). The CDRs originate from loops between individual strands. Many nanobodies contain an extra interloop disulfide bond that restricts the flexibility of CDR1 and CDR3 (dotted purple line). (c) The prolate structure of the nanobody forms a convex paratope surface, which allows it to access antigenic cavities. In the β2-adrenergic receptor·Nanobody80 (β2AR·Nb80) complex shown here [Protein Data Bank (PDB) ID: 3P0G], CDR3 of Nb80 inserts into the cytoplasmic surface of active β2AR, with additional interactions made by CDR1 and CDR2. The resulting β2AR epitope recognized by Nb80, viewed from the cytoplasmic surface (eye symbol), is displayed in panel d. Note that each CDR binds different regions of the complex three-dimensional epitope that is discontinuous in β2AR sequence. CDRs and framework residues in this figure have been defined according to the International ImMunoGeneTics Information System (IMGT) (138).
Antigen-specific nanobodies are obtained by immunization of a camelid followed by cloning the variable VHH gene repertoire from peripheral blood lymphocytes upon induction of a sufficient immune response. Nanobodies of desired function are then obtained by selection through one of many combinatorial biology methods, which include phage display (72), yeast display (73), and ribosome display (74, 75). Because the full immune repertoire of antigen-specific heavy-chain antibodies in a given llama is approximately 1 × 106, libraries covering the complete nanobody repertoire of an immunized animal can be derived from the total number of B cells contained in a 50-mL sample of blood.
Nanobodies have gained significant traction as alternatives to conventional antibodies. Because of their compact prolate shape, nanobodies expose a convex paratope and have access to cavities or clefts on the surfaces of proteins that are inaccessible to conventional antibodies (76). These cryptic epitopes can often be recognized readily by the long complementarity-determining region 3 (CDR3) loop of the nanobody (77). Perhaps most importantly, camelids immunized with natively folded proteins produce nanobodies that can bind conformational epitopes, which are often composed of discontinuous amino acid segments and occur only within the fully native protein (Figure 2). Nanobodies have been developed to trap unstable structural intermediates along the fibrillation pathway of amyloidogenic proteins (78, 79). Multidomain proteins are more rigid in complex with a nanobody than the multidomain protein is by itself (80). In complex with a nanobody, the total amount of structured polypeptide increases, thus providing a much better starting point for the crystallization of intrinsically unfolded proteins (79, 81). Nanobodies can also be used to stabilize the protomers of larger protein assemblies (82).
Building on their unique properties, several laboratories have demonstrated that nanobodies are highly useful reagents for examining dynamic biological systems. Nanobodies have been used to crystallize flexible membrane proteins (83–87), transient multiprotein assemblies (88–90), and individual substates of conformationally complex proteins (91–95). Although nanobodies may select high-energy, low-population conformations of a dynamic protein, the probability of an in vivo matured nanobody inducing a non-native conformation of the antigen is low. Immature B cells require engagement of displayed antibodies with antigen to proliferate and differentiate during clonal selection (96). Antibodies that induce non-native conformations of the antigen pay a substantial energetic penalty in this process, and B cell clones displaying such antibodies will have a significantly lower probability of proliferation and differentiation into mature antibody-secreting B lymphocytes. This notion is supported by recent experiments that identified several nanobodies raised by immunization against the prokaryotic lactose permease transporter LacY. Although each nanobody stabilizes a unique conformation of LacY, the stabilized states are within the broad conformational ensemble of the transporter. This observation gives credence to the notion that nanobodies bind antigens primarily by conformational selection and not induced fit (97).
Active State-Stabilizing Nanobodies
The first structural insights into GPCR activation came from the light-activated GPCR rhodopsin. Rhodopsin is activated by absorption of a photon, which causes the isomerization of 11-cis-retinal to all-trans-retinal. Upon activation, rhodopsin converts rapidly to metarhodopsin II, which is the conformational state capable of activating the G protein transducin (98). Hydrolysis of the Schiff base between rhodopsin and retinal occurs spontaneously, and subsequent dissociation of retinal leaves the apoprotein opsin. To stabilize the active conformation of opsin for crystallography, previous studies utilized low pH (99, 100) and the C-terminal peptide of transducin (101). Subsequent efforts elucidated the structure of retinal-bound metarhodopsin II, both alone and in complex with the C-terminal peptide of transducin (102, 103).
Although these structures revealed considerable insight into receptor activation, the strategies used to capture the active conformation of rhodopsin were not easily transferable to other GPCRs. Most hormone-activated GPCRs are too dynamic to crystallize in the presence of agonist alone. Spectroscopic studies highlighted above demonstrated that a large fraction of agonist-bound β2AR exists in an inactive conformation (104); indeed, our efforts to crystallize the β2AR covalently bound to an engineered agonist resulted in an inactive conformation of the receptor (105). Similarly, other groups have pursued strategies to conformationally stabilize active state-receptors by introducing mutations at key positions (106, 107). These strategies have succeeded in determining the structures of agonist-occupied receptor, but the receptor conformation remains in the inactive states (108).
Building on the unique features of nanobodies outlined above, our labs initially explored the use of these single-domain antibodies as chaperones for stabilizing the active, agonist-bound conformation of the β2AR. We posited that these nanobodies may function as structural surrogates of native signaling partners (109), most notably for the heterotrimeric G protein. In our efforts to generate such nanobodies, we immunized llamas with agonist-bound β2AR reconstituted into lipid vesicles (72). The entire repertoire of in vivo matured nanobodies was cloned and subjected to phage display to identify conformational antibodies that recognized agonist-bound β2AR selectively. Several such nanobodies were identified and one clone, Nanobody80 (Nb80), was characterized extensively by spectroscopic and pharmacological assays. Binding of Nb80 to fluorophore-labeled β2AR induced a change in signal consistent with receptor activation, as was observed previously with the heterotrimeric G protein Gs (104). In pharmacological assays, Nb80 recapitulated a key feature of GPCR allostery. Researchers have previously shown that G proteins allosterically increase the affinity of agonists at a receptor (11, 110). In the presence of Nb80, purified and reconstituted β2AR demonstrated an increase in the affinity of the agonist isoproterenol, similar to what occurs in the presence of Gs (Figure 3). These lines of evidence suggested strongly that Nb80 stabilizes an active-state conformation of β2AR comparable to that in the Gs complex. Subsequent identification of nanobodies with similar properties at the M2 muscarinic receptor (M2R) (92) and the μ-opioid receptor (μOR) (94) have demonstrated that this approach is generalizable and can likely be extended to the broader GPCR family (Figure 3).
Figure 3.
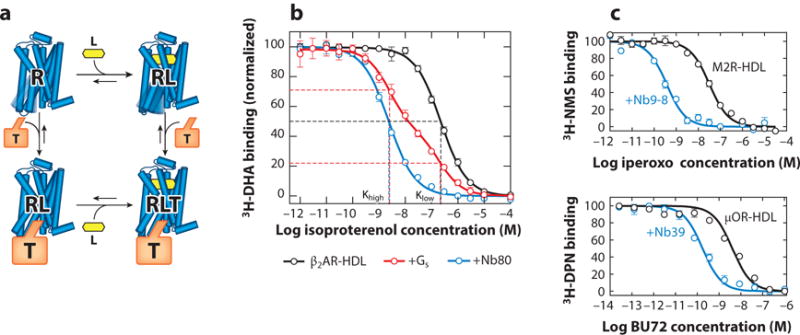
Active state-stabilizing nanobodies recapitulate the G protein-coupled receptor ternary complex model. (a) In the ternary complex model, agonist (L) binding results in increased affinity for the intracellular transducer, most commonly one of the heterotrimeric G proteins. Conversely, binding of a transducer (T) to the receptor (R) induces a reciprocal increase in agonist affinity. (b) The β2-adrenergic receptor (β2AR) reconstituted into high-density lipoprotein (HDL) particles shows a monophasic competition curve resulting from its affinity for the agonist isoproterenol. Addition of Gs results in a biphasic curve, with a fraction of receptor displaying an increase in agonist affinity induced by the G protein. Nanobody80 (Nb80), an active state-stabilizing nanobody for the β2AR, induces a similar high-affinity agonist state. (c) Similar nanobodies were identified subsequently for the M2 muscarinic receptor (M2R) (Nb9-8) and the μ-opioid receptor (μOR) (Nb39). In each case, active state-stabilizing nanobodies increase the affinity of agonists. Other abbreviations: 3H-DHA, 3H-dihydroalprenolol; 3H-DPN, 3H-diprenorphine; 3H-NMS, 3H-N-methylscopolamine.
Nanobodies Facilitate Crystallization of Active-State GPCRs
The first structure of active β2AR was determined using Nb80 and a high-affinity, slowly dissociating agonist BI167107 (91). This structure revealed several important features of GPCR activation. Since this initial use of Nb80 to stabilize the β2AR active state, several nanobodies have been derived to obtain active-state structures of several GPCRs. Although Nb80 successfully enabled crystallization of the β2AR bound to BI167107, crystallization of the same receptor bound to its low-affinity endogenous agonist adrenaline remained challenging. To facilitate crystallization with low-affinity agonists, Nb80 was affinity matured using yeast surface display and novel conformational selection strategies. The resulting affinity-matured nanobody, Nb6B9, displayed slower dissociation kinetics at the β2AR and was used successfully to capture the structure of β2AR bound to adrenaline (93). Nanobodies raised against M2R (92), μOR (94), and the viral chemokine receptor US28 (95) demonstrated subsequently that active state-stabilizing nanobodies are a robustly general tool for crystallizing agonist-bound receptors. This holds true even for constitutively active GPCRs such as US28, in which the presence of a nanobody (Nb7) improved the diffraction quality significantly compared to the US28·CX3CL1 complex alone (95).
In each case, active state-stabilizing nanobodies bind to the intracellular domain of the receptor in a cavity similar to that occupied by the G protein α subunit and arrestin (54) (Figure 4). The nanobody CDRs engage with the intracellular loops of the receptor, and the net binding energy stabilizes the receptor in an active conformation. These structures have revealed several conserved features of activated GPCRs, the most notable being a 9–11-Å outward displacement of transmembrane helix 6 (TM6) accompanied by slight inward movement of TM5 and a rearrangement of the NPxxY motif in TM7 (Figure 4). In each active-state structure, the nanobody stabilizes a conformation of the receptor where two key tyrosine residues, Y5.58 and Y7.53, interact via a conserved hydrogen-bonding network mediated by a water molecule (see Reference 111 for a recent update of the generic Ballesteros-Weinstein numbering of GPCR residues). These structures also reveal a recurring rearrangement of a cluster of conserved hydrophobic and aromatic residues including I/L/V3.40, P5.50, and F6.44, called the transmission switch (112), that connects the intracellular nanobody-binding domain to the orthosteric ligand-binding site (Figure 4). Although the overall structural changes are conserved in the intracellular domain, each receptor displays unique modes of agonist recognition in the orthosteric-binding pocket. Activation of the β2AR and μOR is driven by subtle changes in the orthosteric pocket induced by agonists (91, 92). In comparison, activation of the M2R by the agonist iperoxo is driven by a dramatic closure of the orthosteric ligand-binding pocket and a pivoting motion of TM6 (94).
Figure 4.
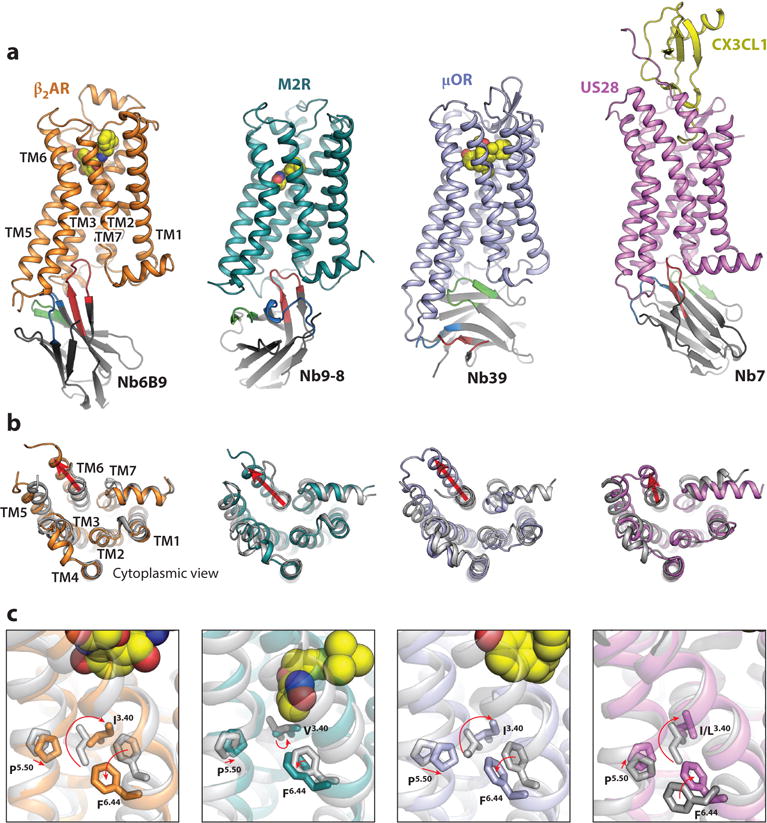
Structural basis of G protein-coupled receptor (GPCR) activation revealed by nanobody-assisted crystallography. (a) Active-state structures of agonist-bound, nanobody-stabilized GPCRs. Each nanobody binds the intracellular surface of the receptor in a unique orientation. Whereas complementarity-determining region 3 (CDR3) (red) most commonly extends into the core of the receptor, Nanobody39 (Nb39) uses framework residues, CDR1, and CDR2 to engage with the μ-opioid receptor (μOR). (b) Cytoplasmic view comparing inactive [grey; Protein Data Bank (PDB) IDs: 2RH1, 3UON, 4DKL, 4XT1] and active receptors (colored; PDB IDs: 4LDE, 4MQS, 5C1M, 4MBS). Key conserved features include an outward displacement of transmembrane helix 6 (TM6), an inward movement of TM5, and a rearrangement of TM7. (c) Active-state GPCR structures reveal a conserved mode of allostery between the ligand-binding pocket and the cytoplasmic domain. The agonists (yellow spheres) induce conformational changes in the ligand-binding pocket, which are relayed via a conserved set of transmission switch residues (F6.44, P5.50, and I/L/V3.40) that undergo similar rearrangements upon activation.
Based on the above successes, we anticipate that nanobody-assisted receptor crystallization will continue to offer new insights into GPCR function. Most previous inactive-state GPCR crystal structures have utilized receptor fusions to a highly stable soluble protein that facilitates crystal lattice contacts (23, 24). In the case of the M2R, μOR, and US28, the active state-stabilizing nanobody provides sufficient hydrophilic contact surface area to facilitate receptor crystallization, thereby obviating the need for extensive receptor engineering. Unlike stabilization by G proteins, nanobody-based stabilization of GPCRs is not dependent on low-affinity interactions or complicated by nucleotides required for G protein function. As a result, nanobodies provide an efficient means to add hydrophilic lattice contacts simultaneously and to conformationally stabilize biologically interesting GPCR conformations.
GPCR Dynamics Revealed by Nanobodies
Multiple lines of pharmacological, biochemical, and spectroscopic evidence suggest that GPCRs adopt a multitude of conformations that interchange rapidly on timescales ranging from microseconds to hundreds of milliseconds. Determining how ligands alter this conformational landscape will be critical to a full understanding of GPCR allostery. Whereas crystallographic studies provide a high-resolution view of distinct conformations within this landscape, they provide static descriptions of highly dynamic behavior. Several recent spectroscopic studies have started to examine the complex conformational landscape of GPCR function. Early fluorescence studies established that different ligands induce unique conformations of the β2AR (113, 114). Ligand-specific conformations of β2AR have been examined subsequently by more sophisticated spectroscopic techniques, including single-molecule fluorescence spectroscopy (50, 51), 13C-dimethyllysine spectroscopy (115), 19F-NMR spectroscopy (47, 116), and 13C-ε-methionine NMR spectroscopy (117). These studies have established clearly that different agonists can induce unique spectroscopic signatures in the β2AR, further supporting the hypothesis that a given GPCR can adopt unique conformations in the presence of different agonists.
As GPCRs are likely highly dynamic in both the presence and absence of agonists, spectroscopic studies of the receptor alone do not allow precise correlation of spectroscopic signals to specific structural states. Agonists may increase the population of the active state but rarely allow sufficient conformational stabilization to uniquely assign a given signal to the conformation observed by crystallographic study. Our groups and others have therefore utilized active state-stabilizing nanobodies to assign spectroscopic signals to specific receptor conformations. Using Nb80, we demonstrated previously that the β2AR achieves the crystallographically observed conformation only in the presence of both a high-affinity agonist and the nanobody. These experiments were performed using several complementary techniques, including 13C-ε-methionine NMR (48), 19F-NMR (49, 118, 119), and DEER spectroscopy (49), with additional evidence provided by molecular dynamics simulations (105). In each case, addition of Nb80 provided a useful control to determine signals originating from a fully active β2AR. As anticipated, both NMR and DEER experiments revealed that binding of Nb80 stabilizes a single active conformation with minimal residual β2AR dynamics. A similar approach has even been applied successfully to assess the fully active conformation of a turkey β1AR mutant thermostabilized in the inactive conformation (45). In this case, addition of Nb80 provided sufficient binding energy to overcome inactive-state conformational stabilization, and the authors were able to use backbone signals obtained in the presence of Nb80 to examine the active conformation of the turkey β1AR. In a more extreme example, we observed that 13C-dimethyllysine spectroscopy of the μOR revealed minimal changes in the cytoplasmic domain with agonists alone (46). Conformational changes associated with receptor activation were observed only in the presence of a μOR active state-stabilizing nanobody, Nb33. The last example highlights the necessity of assigning fully active states with either signaling proteins or active state-stabilizing nanobodies during spectroscopic studies (Figure 5). In the absence of such controls, it remains challenging, if not impossible, to fully assess the dynamic range of the receptor in a spectroscopic experiment.
Figure 5.
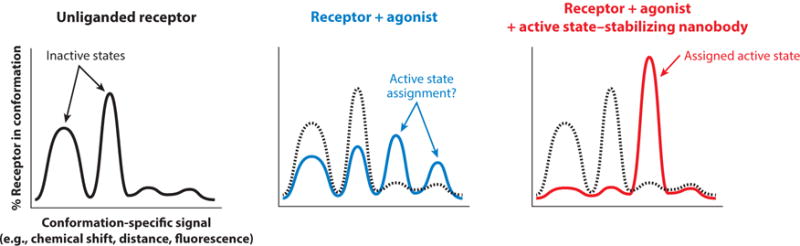
Nanobody-based assignment of G protein-coupled receptor conformational states. In many spectroscopic experiments, distinct signals arise from unique conformations of a given receptor. In the absence of ligand, multiple signals reflect conformational heterogeneity. Addition of agonists is associated with increased conformational heterogeneity and the presence of new signals arising from a greater number of conformations. Assignment of a specific signal to a crystallographically observed conformation or a pharmacologically characterized state is challenging (middle panel). Active state-stabilizing nanobodies can conformationally stabilize the receptor, thereby allowing clear assignment of signals and further interpretation of spectroscopic data.
STABILIZATION OF GPCR SIGNALING COMPLEXES
Active state-stabilizing nanobodies have revealed several conserved features of GPCR activation. However, the most extensive insight into receptor-mediated activation of G protein signaling required a structure of a GPCR bound to its cognate G protein. The structure of the agonist-bound β2AR·Gs complex was instrumental in understanding the cooperative interactions across the membrane between agonist, receptor, and G protein that cause agonist binding at the extracellular side to activate the cytosolic Gα subunit (90). Because of the difficulty in stabilizing multiple conformationally dynamic proteins, each requiring disparate biochemical conditions for stability, the crystallization of this complex necessitated a highly multidisciplinary approach (61). Several inventive steps were needed, including biochemical optimization for purification of a nucleotide-free complex, identification of an ultrahigh-affinity agonist, and development of new detergents (30) and mesophase lipids (120), as well as new GPCR protein engineering (121). Despite these innovations, single-particle electron microscopy data of the purified complex suggested that the α-helical domain of the Gαs subunit is conformationally heterogeneous in the complex (53).
To compensate for this heterogeneity, we attempted to identify nanobodies that would rigid-ify the β2AR·Gs complex. Llamas were immunized with the agonist-bound β2AR·Gs complex cross-linked with bis(sulfosuccinimidyl)glutarate (72). Nanobodies were selected for binding to the nucleotide-free complex reconstituted into high-density lipoprotein particles (also known as Nanodiscs). Panning identified several binders, and two nanobodies called Nb35 and Nb37 were selected for their ability to selectively bind the β2AR·Gs complex but not the receptor alone. Most importantly, Nb35 and Nb37 prevented dissociation of the nucleotide-free complex by the nonhydrolyzable GTP analog GTPγS (90). With the use of Nb37, electron microscopy of the β2AR·Gs complex revealed the conformational flexibility of the Gas α-helical domain more convincingly (53). Addition of Nb35 to the T4L-β2AR·Gs complex resulted in a marked improvement in crystallogenesis. The resulting crystals diffracted to 2.9 Å and yielded the structure of the first GPCR-G protein complex (90). In the final refined structure, Nb35 bound at the interface of the Gα and the Gβ subunits, thereby stabilizing the two domains. The nanobody also provided key lattice contacts to adjacent repeating units.
NANOBODIES ILLUMINATE GPCR FUNCTION IN LIVING CELLS
As highlighted in the sections above, structural and biophysical studies over the past 15 years have yielded significant insight into the molecular details of GPCR function. Indeed, researchers have determined over 100 crystal structures of 30 unique GPCRs in complex with small molecule ligands, peptides, or antibodies (21). Recent efforts have elucidated the first structures of GPCRs in complex with important intracellular signaling proteins (52, 54, 90). Accompanying these structures are numerous biophysical, biochemical, and pharmacological studies that have examined the complexity of GPCR signaling. More in-depth understanding of GPCR signaling, however, requires correlation of these molecular and structural insights to the function of receptors in native living systems. Because nanobodies can be functionally expressed in the cytoplasm of eukaryotic cells (122, 123), recent studies have utilized active state-stabilizing nanobodies to probe conformational changes in receptors within living cells to correlate structure with signaling more fully. In the first example of such an approach, Irannejad and coworkers (124) fused Nb80 to green fluorescent protein (GFP) to follow the activation of the β2AR by the agonist isoproterenol. The same study also utilized a Nb37-GFP fusion, which recognizes nucleotide-free Gs in the agonist-bound β2AR·Gs signaling complex (53), to probe the activation of the β2AR and its cognate G protein directly (124). By fusing Nb80 or Nb37 to GFP and expressing it as a genetically encoded intrabody at a suitably low concentration in the cytoplasm, the authors could show that isoproterenol promotes receptor and G protein activation in the plasma membrane. Intriguingly, the same reporters also demonstrated that β2AR can activate G proteins in the early endosome membrane. Thus, internalized receptors can apparently also contribute to the overall cellular cyclic AMP response within several minutes after agonist application.
In a similar set of studies, nanobodies that bind selectively to active (agonist-bound) or inactive (antagonist-bound) β2AR were generated and characterized (125). When expressed as intrabodies, these nanobodies inhibit G protein activation, GRK-mediated receptor phosphorylation, β-arrestin recruitment, and receptor internalization to varying extents. These functional effects were likely due to either steric blockade of downstream effector interactions or stabilization of specific receptor conformations that do not support effector coupling. In addition to imaging applications, intrabodies may provide novel ways of modulating GPCR signaling with exquisite selectivity for a given receptor and signaling pathway. As more conformation-selective nanobodies become available that are specific for other GPCRs, G proteins, or arrestin, they will likely find broad utility in such applications examining GPCR signaling. Ultimately, these nanobodies may be useful in imaging and modulating GPCR signaling within transgenic animals, thereby allowing correlation of structure and function at the whole-organism level.
THERAPEUTIC TARGETING OF GPCRs USING NANOBODIES
As discussed above, nanobodies are highly useful tools to understand GPCR signaling. Several studies have also demonstrated that nanobodies directed against GPCRs may have therapeutic potential. For example, picomolar-affinity nanobodies have been identified that inhibit CXCR4-mediated signaling competitively and antagonize the chemoattractant effect of the CXCR4 agonist CXCL12 (126). These nanobodies bind extracellular loops of human CXCR4 and display strong antiretroviral activity against T cell-tropic and dual-tropic HIV-1 strains. Additionally, these antibodies induce stem cell mobilization in vivo, perhaps providing utility as alternatives for currently approved small-molecule drugs such as plerixafor. Antagonist nanobodies that bind other chemokine receptors, including CXCR2 or CXCR7, have also been described (127, 128). As nanobodies are only approximately 15 kDa, they may provide better penetration into tumors, the central nervous system, or other bodily compartments difficult to access by conventional antibodies. Depending on the desired indication, half-life extension methods such as PEGylation (129) or fusion to serum albumin (130) could be used for tailoring the half-life of nanobodies and increase their therapeutic window depending on the clinical indication. Also, their small size (gene and domain) and monomeric and soluble behavior render single-domain antibodies ideal building blocks to generate multivalent or multispecific constructs with subsequent fusion to an Fc, enzyme, or toxin (131). Finally, nanobodies that constrain GPCRs in specific, biologically relevant conformations may enable the binding of small-molecule fragments that would otherwise not be able to gain a foothold on the flexible target (132).
CONFORMATIONAL PLASTICITY OF COMMON DRUG TARGETS.
Protein structures elucidated through X-ray crystallography give the impression that proteins are rigid entities. Although static structures are known for many proteins, the functions of proteins are governed ultimately by their dynamic character (133). Allostery, the coupling between ligand binding and protein conformational change, is at the heart of biological networks, and conformational flexibility is key to the function and pharmacology of the majority of current and future drug targets including GPCRs, ion channels, nuclear receptors, and kinases. Agonists, inverse agonists, and biased ligands bind different structural conformations of the same GPCR (37). Allosteric transitions also occur in many therapeutic ion channels (134). A typical ion channel shuttles between open and closed states, and the conformational transition between open and closed states is referred to as gating. Channel opening is triggered by specific stimuli, such as the voltage or pH difference across the cell membrane, and modulated by the binding of ligands (135). In the kinase family, allosteric druggable pockets other than the ATP binding site are formed when these enzymes adopt specific conformations (136). Allosteric effects also govern the biological activity of nuclear receptors (137).
SUMMARY POINTS.
Camelid heavy-chain-only antibody fragments (nanobodies) show promise as tools for stabilizing discrete GPCR conformations and as chaperones for crystallogenesis.
Several structures of agonist-bound GPCRs in the active state have been obtained by nanobody-assisted X-ray crystallography. These structures provide insight into ligand-induced receptor activation.
Nanobodies were instrumental in solving the structure of the agonist-bound β2AR·Gs transmembrane signaling complex.
Most nanobodies can be functionally expressed as genetically encoded intrabodies within a eukaryotic cell. This makes nanobodies ideal tools to correlate structural or dynamic features observed in vitro by biophysical measurements with functional observations from living cells.
Characterizing GPCR conformational states may facilitate the development of more selective drugs capable of modulating a specific signaling pathway, thereby improving therapeutic activity and minimizing undesirable side effects.
FUTURE ISSUES.
GPCRs signal through multiple intracellular pathways, and some ligands can induce selective signaling down one pathway. It remains to be seen whether nanobodies that stabilize ligand-specific receptor conformations can be identified.
Nanobodies stabilizing the β2AR·Gs complex have provided highly useful tools for structural studies as well as imaging GPCR signaling. Nanobody-based stabilization of GPCR complexes with other G proteins, GRKs, or β-arrestin would be valuable to study the structural and functional features of GPCR transmembrane signaling.
Evidence has accumulated that GPCRs can form dimers or multimers. Nanobodies that bind monomers or multimers selectively would allow investigation of oligomerization and its effects on ligand recognition, receptor signaling, and intracellular trafficking.
Allostery, the coupling between ligand binding and protein conformational change, is key to the function and pharmacology of many drug targets. Nanobodies that constrain target proteins in specific, biologically relevant conformations may ultimately prove to be of great value to the pharmaceutical industry as next-generation biologicals or as tools that enable the discovery of conformer-selective drugs.
Acknowledgments
A.M. acknowledges support from the Stanford Medical Scientist Training Program. B.K.K. was funded by National Institutes of Health grants R01GMO83118 and R01NS028471. J.S. received support from INSTRUCT, the Hercules Foundation Flanders, Fonds Wetenschappelijk Onder-zoek-Vlaanderen (FWO)-Flanders (G011110N and G049512N), the Belgian Federal Science Policy Office (IAP7-40), Innoviris Brussels (BRGEOZ132), and Instituut voor Innovatie door Wetenschap en Technologie-Vlaanderen (IWT) (SBO program IWT120026).
Glossary
- G protein-coupled receptors (GPCRs)
a large eukaryotic protein family of membrane receptors that sense molecules outside the cell and activate inside signal transduction pathways
- Arrestins
a family of regulatory proteins that arrest GPCR signaling and are responsible for G proteinindependent signaling
- G protein-coupled receptor kinase (GRK)
a specialized set of kinases that recognize and phosphorylate activated GPCRs
- Nanobody (Nb)
recombinant singledomain antibody fragment that contains the unique structural and functional properties of naturally occurring heavy-chain-only antibodies in camelids
- Biased ligand
ligands that engage some signaling pathways selectively while avoiding, or even inactivating, other pathways mediated by the same receptor
- Ligand efficacy
the efficacy of a drug is determined by its ability to induce a quantifiable biological response
- β2AR
β2 -adrenergic receptor
- Camelids
the biological family Camelidae comprises camels (Camelus dromedarius and Camelus bactrianus), llamas (Lama glama and Lama guanicoe), and vicuñas (Vicugna vicugna and Vicugna pacos)
- Single-chain variable fragment (scFv)
the paired VH and VL domains of conventional antibodies tethered via an oligopeptide
- Conformational epitope
the surface making contact with the nanobody that is composed of discontinuous amino acid residues brought together by the protein folding of the antigen
- M2R
M2 muscarinic receptor
- μOR
μ-opioid receptor
- BallesterosWeinstein number
generic GPCR transmembrane residue number in X.YY format; X is the helix number and YY is the residue position relative to the most conserved residue in helix Xdesignated X.50
Footnotes
DISCLOSURE STATEMENT
A.M. is a founder of Ab Initio Biotherapeutics. B.K.K. serves as an advisor for Ab Initio Biotherapeutics and is a founder of ConfometRx. J.S. is a founder of Confo Therapeutics.
LITERATURE CITED
- 1.Lagerström MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–57. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 2.Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25:413–22. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 4.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–56. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–50. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 6.Wisler JW, Xiao K, Thomsen ARB, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mombaerts P. Genes and ligands for odorant, vomeronasal and taste receptors. Nat Rev Neurosci. 2004;5:263–78. doi: 10.1038/nrn1365. [DOI] [PubMed] [Google Scholar]
- 8.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–96. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 9.Rask-Andersen M, Masuram S, Schioth HB. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu Rev Pharmacol Toxicol. 2014;54:9–26. doi: 10.1146/annurev-pharmtox-011613-135943. [DOI] [PubMed] [Google Scholar]
- 10.Int Union Basic Clin Pharmacol./Br. Pharmacol. Soc.(IUPHAR/BPS) The IUPHAR/BPS Guide to PHARMACOLOGY. Edinburgh, UK: IUPHAR/BPS; 2016. http://www.guidetopharmacology.org/ [Google Scholar]
- 11.De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J Biol Chem. 1980;255:7108–17. [PubMed] [Google Scholar]
- 12.Flock T, Ravarani CN, Sun D, Venkatakrishnan AJ, Kayikci M, et al. Universal allosteric mechanism for Gaactivation by GPCRs. Nature. 2015;524:173–79. doi: 10.1038/nature14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα·GTPγS. Science. 1997;278:1907–16. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 14.Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, et al. Kinetic scaffolding mediated by a phospholipase C-β and Gq signaling complex. Science. 2010;330:974–80. doi: 10.1126/science.1193438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berridge MJ, Irvine RF. Inositol phosphates and cell signalling. Nature. 1989;341:197–205. doi: 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- 16.Kang DS, Tian X, Benovic JL. Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr Opin Cell Biol. 2014;27:63–71. doi: 10.1016/j.ceb.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–34. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 18.Lohse MJ. Dimerization in GPCR mobility and signaling. Curr Opin Pharmacol. 2010;10:53–58. doi: 10.1016/j.coph.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, et al. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. 2014;66:413–34. doi: 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prezeau L, Pin JP. Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat Struct Mol Biol. 2004;11:706–13. doi: 10.1038/nsmb794. [DOI] [PubMed] [Google Scholar]
- 21.Isberg V, Mordalski S, Munk C, Rataj K, Harpsoe K, et al. GPCRdb: an information system for G protein-coupled receptors. Nucleic Acids Res. 2016;44:D356–64. doi: 10.1093/nar/gkv1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maeda S, Schertler GF. Production of GPCR and GPCR complexes for structure determination. Curr Opin Struct Biol. 2013;23:381–92. doi: 10.1016/j.sbi.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 2007;318:1266–73. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 24.Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–76. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. PNAS. 2008;105:877–82. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tate CG. A crystal clear solution for determining G-protein-coupled receptor structures. Trends Biochem Sci. 2012;37:343–52. doi: 10.1016/j.tibs.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Vaidehi N, Grisshammer R, Tate CG. How can mutations thermostabilize G-protein-coupled receptors? Trends Pharmacol Sci. 2016;37:37–46. doi: 10.1016/j.tips.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–31. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowler MW, Guijarro M, Petitdemange S, Baker I, Svensson O, et al. Diffraction cartography: Applying microbeams to macromolecular crystallography sample evaluation and data collection. Acta Crystallogr D Biol Crystallogr. 2010;66:855–64. doi: 10.1107/S0907444910019591. [DOI] [PubMed] [Google Scholar]
- 30.Liu W, Wacker D, Gati C, Han GW, James D, et al. Serial femtosecond crystallography of G protein-coupled receptors. Science. 2013;342:1521–24. doi: 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weichert D, Gmeiner P. Covalent molecular probes for class A G protein-coupled receptors: advances and applications. ACS Chem Biol. 2015;10:1376–86. doi: 10.1021/acschembio.5b00070. [DOI] [PubMed] [Google Scholar]
- 32.Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Chandra R, et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods. 2010;7:1003–8. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooke RM, Brown AJ, Marshall FH, Mason JS. Structures of G protein-coupled receptors reveal new opportunities for drug discovery. Drug Discov Today. 2015;20:1355–64. doi: 10.1016/j.drudis.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci. 2011;32:213–18. doi: 10.1016/j.tips.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leff P. The two-state model of receptor activation. Trends Pharmacol Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- 36.Deupi X, Kobilka B. Activation of G protein-coupled receptors. Adv Protein Chem. 2007;74:137–66. doi: 10.1016/S0065-3233(07)74004-4. [DOI] [PubMed] [Google Scholar]
- 37.Galandrin S, Oligny-Longpre G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–30. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2012;12:205–16. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 39.Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. PNAS. 2006;103:15463–68. doi: 10.1073/pnas.0607224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dawaliby R, Trubbia C, Delporte C, Masureel M, Van Antwerpen P, et al. Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat Chem Biol. 2016;12:35–39. doi: 10.1038/nchembio.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oates J, Watts A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr Opin Struct Biol. 2011;21:802–7. doi: 10.1016/j.sbi.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 42.Mahaut-Smith MP, Martinez-Pinna J, Gurung IS. A role for membrane potential in regulating GPCRs? Trends Pharmacol Sci. 2008;29:421–29. doi: 10.1016/j.tips.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Ghanouni P, Schambye H, Seifert R, Lee TW, Rasmussen SGF, et al. The effect of βH on β2 adrenoceptor function: evidence for protonation-dependent activation. J Biol Chem. 2000;275:3121–27. doi: 10.1074/jbc.275.5.3121. [DOI] [PubMed] [Google Scholar]
- 44.Liu W, Chun E, Thompson AA, Chubukov P, Xu F, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–36. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Isogai S, Deupi X, Opitz C, Heydenreich FM, Tsai CJ, et al. Backbone NMR reveals allosteric signal transduction networks in the β1 -adrenergic receptor. Nature. 2016;530:237–41. doi: 10.1038/nature16577. [DOI] [PubMed] [Google Scholar]
- 46.Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, et al. Propagation of conformational changes during μ-opioid receptor activation. Nature. 2015;524:375–78. doi: 10.1038/nature14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. Biased signaling pathways in (β2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–10. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, et al. The dynamic process of β2 -adrenergic receptor activation. Cell. 2013;152:532–42. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, et al. Structural insights into the dynamic process of β2 -adrenergic receptor signaling. Cell. 2015;161:1101–11. doi: 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamichhane R, Liu JJ, Pljevaljcic G, White KL, van der Schans E, et al. Single-molecule view of basal activity and activation mechanisms of the G protein-coupled receptor β2 AR. PNAS. 2015;112:14254–59. doi: 10.1073/pnas.1519626112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bockenhauer S, Furstenberg A, Yao XJ, Kobilka BK, Moerner WE. Conformational dynamics of single G protein-coupled receptors in solution. J Phys Chem B. 2011;115:13328–38. doi: 10.1021/jp204843r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–22. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westfield GH, Rasmussen SGF, Su M, Dutta S, DeVree BT, et al. Structural flexibility of the Gas α-helical domain in the β2-adrenoceptor Gs complex. PNAS. 2011;108:16086–91. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang Y, Zhou XE, Gao X, He Y, Liu W, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–67. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang L, Yang D, de Graaf C, Moeller A, West GM, et al. Conformational states of the full-length glucagon receptor. Nat Commun. 2015;6:7859. doi: 10.1038/ncomms8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deupi X, Kobilka BK. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology. 2010;25:293–303. doi: 10.1152/physiol.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, et al. Activation mechanism of the β2-adrenergic receptor. PNAS. 2011;108:18684–89. doi: 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manglik A, Kobilka BK. The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr Opin Cell Biol. 2014;27C:136–43. doi: 10.1016/j.ceb.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kenakin T. Efficacy at G-protein-coupled receptors. Nat Rev Drug Discov. 2002;1:103–10. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- 60.Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, et al. Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat Chem Biol. 2011;7:692–700. doi: 10.1038/nchembio.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobilka B. The structural basis of G-protein-coupled receptor signaling (Nobel Lecture) Angew Chem Int Ed Engl. 2013;52:6380–88. doi: 10.1002/anie.201302116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446–48. doi: 10.1038/363446a0. First description of functional heavy-chain-only antibodies in camelids. [DOI] [PubMed] [Google Scholar]
- 63.Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–97. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 64.Arbabi-Ghahroudi M, Tanha J, MacKenzie R. Prokaryotic expression of antibodies. CancerMetas-tasis Rev. 2005;24:501–19. doi: 10.1007/s10555-005-6193-1. [DOI] [PubMed] [Google Scholar]
- 65.Frenken LGJ, van der Linden RHJ, Hermans PWJJ, Bos JW, Ruuls RC, et al. Isolation of antigen specific Llama VHH antibody fragments and their high level secretion by Saccharomyces cerevisiae. J Biotechnol. 2000;78:11–21. doi: 10.1016/s0168-1656(99)00228-x. [DOI] [PubMed] [Google Scholar]
- 66.Frenken LGJ, Hessing JGM, Van den Hondel CAMJJ, Verrips CT. Recent advances in the large-scale production of antibody fragments using lower eukaryotic microorganisms. Res Immunol. 1998;149:589–99. doi: 10.1016/s0923-2494(98)80011-4. [DOI] [PubMed] [Google Scholar]
- 67.Joosten V, Gouka RJ, Van den Hondel CAMJJ, Verrips CT, Lokman BC. Expression and production of llama variable heavy-chain antibody fragments (VHHS) by Aspergillus awamori. Appl Microbiol Biotechnol. 2005;66:384–92. doi: 10.1007/s00253-004-1689-0. [DOI] [PubMed] [Google Scholar]
- 68.Agrawal V, Slivac I, Perret S, Bisson L, St-Laurent G, et al. Stable expression of chimeric heavy chain antibodies in CHO cells. In: Saerens D, Muyldermans S, editors. Single Domain Antibodies: Methods and Protocols. Totowa, NJ: Humana Press; 2012. pp. 287–303. [DOI] [PubMed] [Google Scholar]
- 69.Ismaili A, Jalali-Javaran M, Rasaee MJ, Rahbarizadeh F, Forouzandeh-Moghadam M, Memari HR. Production and characterization of anti-(mucin MUC1) single-domain antibody in tobacco (Nicotiana tabacum cultivar Xanthi) Biotechnol Appl Biochem. 2007;47:11–19. doi: 10.1042/BA20060071. [DOI] [PubMed] [Google Scholar]
- 70.De Buck S, Nolf J, De Meyer T, Virdi V, De Wilde K, et al. Fusion of an Fc chain to a VHH boosts the accumulation levels in Arabidopsis seeds. Plant Biotechnol J. 2013;11:1006–16. doi: 10.1111/pbi.12094. [DOI] [PubMed] [Google Scholar]
- 71.Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, et al. Single-domain antibody fragments with high conformational stability. Protein Sci. 2002;11:500–15. doi: 10.1110/ps.34602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pardon E, Laeremans T, Triest S, Rasmussen SGF, Wohlkonig A, et al. A general protocol for the generation of nanobodies for structural biology. Nat Protoc. 2014;9:674–93. doi: 10.1038/nprot.2014.039. Laboratory protocols for the generation of in vivo matured nanobodies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ryckaert S, Pardon E, Steyaert J, Callewaert N. Isolation of antigen-binding camelid heavy chain antibody fragments (nanobodies) from an immune library displayed on the surface of Pichia pastoris. J Biotechnol. 2010;145:93–98. doi: 10.1016/j.jbiotec.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 74.Fleetwood F, Devoogdt N, Pellis M, Wernery U, Muyldermans S, et al. Surface display of a single-domain antibody library on Gram-positive bacteria. Cell Mol Life Sci. 2013;70:1081–93. doi: 10.1007/s00018-012-1179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pellis M, Pardon E, Zolghadr K, Rothbauer U, Vincke C, et al. A bacterial-two-hybrid selection system for one-step isolation of intracellularlyfunctional nanobodies. Arch Biochem Biophys. 2012;526:114–23. doi: 10.1016/j.abb.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 76.Lauwereys M, Arbabi Ghahroudi M, Desmyter A, Kinne J, Holzer W, et al. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998;17:3512–20. doi: 10.1093/emboj/17.13.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Genst E, Silence K, Decanniere K, Conrath K, Loris R, et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. PNAS. 2006;103:4586–91. doi: 10.1073/pnas.0505379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Domanska K, Vanderhaegen S, Srinivasan V, Pardon E, Dupeux F, et al. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic β2-microglobulin variant. PNAS. 2011;108:1314–19. doi: 10.1073/pnas.1008560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abskharon RN, Giachin G, Wohlkonig A, Soror SH, Pardon E, et al. Probing the N-terminal β-sheet conversion in the crystal structure of the human prion protein bound to a nanobody. J Am Chem Soc. 2014;136:937–44. doi: 10.1021/ja407527p. [DOI] [PubMed] [Google Scholar]
- 80.Korotkov KV, Pardon E, Steyaert J, Hol WG. Crystal structure of the N-terminal domain of the secretin GspD from ETEC determined with the assistance of a nanobody. Structure. 2009;17:255–65. doi: 10.1016/j.str.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Loris R, Marianovsky I, Lah J, Laeremans T, Engelberg-Kulka H, et al. Crystal structure of the intrinsically flexible addiction antidote MazE. J Biol Chem. 2003;278:28252–57. doi: 10.1074/jbc.M302336200. [DOI] [PubMed] [Google Scholar]
- 82.Rivera-Calzada A, Fronzes R, Savva CG, Chandran V, Lian PW, et al. Structure of a bacterial type IV secretion core complex at subnanometre resolution. EMBO J. 2013;32:1195–204. doi: 10.1038/emboj.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ehrnstorfer IA, Geertsma ER, Pardon E, Steyaert J, Dutzler R. Crystal structure of a SLC11 (NRAMP) transporter reveals the basis for transition-metal ion transport. Nat Struct Mol Biol. 2014;21:990–96. doi: 10.1038/nsmb.2904. [DOI] [PubMed] [Google Scholar]
- 84.Smirnova I, Kasho V, Jiang X, Pardon E, Steyaert J, Kaback HR. Outward-facing conformers of LacY stabilized by nanobodies. PNAS. 2014;111:18548–53. doi: 10.1073/pnas.1422265112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Geertsma ER, Chang YN, Shaik FR, Neldner Y, Pardon E, et al. Structure of a prokaryotic fumarate transporter reveals the architecture of the SLC26 family. Nat Struct Mol Biol. 2015;22:803–8. doi: 10.1038/nsmb.3091. [DOI] [PubMed] [Google Scholar]
- 86.Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, et al. X-ray structure of the mouse serotonin 5-HT3 receptor. Nature. 2014;512:276–81. doi: 10.1038/nature13552. [DOI] [PubMed] [Google Scholar]
- 87.Li L, Park E, Ling J, Ingram J, Ploegh H, Rapoport TA. Crystal structure of a substrate-engaged SecY protein-translocation channel. Nature. 2016;531:395–99. doi: 10.1038/nature17163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rostislavleva K, Soler N, Ohashi Y, Zhang L, Pardon E, et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science. 2015;350:aac7365. doi: 10.1126/science.aac7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pathare GR, Nagy I, Sledz P, Anderson DJ, Zhou HJ, et al. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. PNAS. 2014;111:2984–89. doi: 10.1073/pnas.1400546111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–55. doi: 10.1038/nature10361. First structure of a GPCR·G protein complex solved by nanobody-assisted crystallography. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rasmussen SGF, Choi HJ, Fung JJ, Pardon E, Casarosa P, et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature. 2011;469:175–80. doi: 10.1038/nature09648. First description of a G protein-mimicking nanobody that stabilizes the agonist-bound active state of β2AR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–6. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, et al. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502:575–79. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, et al. Structural insights into μ-opioid receptor activation. Nature. 2015;524:315–21. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, et al. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science. 2015;347:1113–17. doi: 10.1126/science.aaa5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–58. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 97.Smirnova I, Kasho V, Jiang X, Pardon E, Steyaert J, Kaback HR. Transient conformers of LacY are trapped by nanobodies. PNAS. 2015;112:13839–44. doi: 10.1073/pnas.1519485112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–67. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–87. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 100.Vogel R, Siebert F. Conformations of the active and inactive states of opsin. J Biol Chem. 2001;276:38487–93. doi: 10.1074/jbc.M105423200. [DOI] [PubMed] [Google Scholar]
- 101.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauß N, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 102.Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, et al. Crystal structure of metarhodopsin II. Nature. 2011;471:651–55. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 103.Standfuss J, Edwards PC, D’Antona A, Fransen M, Xie G, et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–60. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yao XJ, Vélez Ruiz G, Whorton MR, Rasmussen SGF, DeVree BT, et al. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. PNAS. 2009;106:9501–6. doi: 10.1073/pnas.0811437106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature. 2011;469:236–40. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–25. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.White JF, Noinaj N, Shibata Y, Love J, Kloss B, et al. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–13. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lebon G, Warne T, Tate CG. Agonist-bound structures of G protein-coupled receptors. Curr Opin Struct Biol. 2012;22:482–90. doi: 10.1016/j.sbi.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 109.Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol. 2011;21:567–72. doi: 10.1016/j.sbi.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Murphree LJ, Marshall MA, Rieger JM, MacDonald TL, Linden J. Human A2A adenosine receptors: high-affinity agonist binding to receptor-G protein complexes containing Gβ4. Mol Pharmacol. 2002;61:455–62. doi: 10.1124/mol.61.2.455. [DOI] [PubMed] [Google Scholar]
- 111.Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, et al. Generic GPCR residue numbers-aligning topology maps while minding the gaps. Trends Pharmacol Sci. 2015;36:22–31. doi: 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Deupi X, Standfuss J. Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr Opin Struct Biol. 2011;21:541–51. doi: 10.1016/j.sbi.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 113.Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, et al. Functionally different agonists induce distinct conformations in the G protein coupling domain of the β2 adrenergic receptor. J Biol Chem. 2001;276:24433–36. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- 114.Yao X, Parnot C, Deupi X, Ratnala VR, Swaminath G, et al. Coupling ligand structure to specific conformational switches in the ß2-adrenoceptor. Nat Chem Biol. 2006;2:417–22. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 115.Bokoch MP, Zou Y, Rasmussen SGF, Liu CW, Nygaard R, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–12. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Horst R, Stanczak P, Stevens RC, Wüthrich K. β2-Adrenergic receptor solutions for structural biology analyzed with microscale NMR diffusion measurements. Angew Chem Int Ed Engl. 2013;52:331–35. doi: 10.1002/anie.201205474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kofuku Y, Ueda T, Okude J, Shiraishi Y, Kondo K, et al. Efficacy of the β2-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat Commun. 2012;3:1045. doi: 10.1038/ncomms2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim TH, Chung KY, Manglik A, Hansen AL, Dror RO, et al. The role of ligands on the equilibria between functional states of a G protein-coupled receptor. J Am Chem Soc. 2013;135:9465–74. doi: 10.1021/ja404305k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chung KY, Day PW, Vélez-Ruiz G, Sunahara RK, Kobilka BK. Identification of GPCR-interacting cytosolic proteins using HDL particles and mass spectrometry-based proteomic approach. PLOS ONE. 2013;8:e54942. doi: 10.1371/journal.pone.0054942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Misquitta LV, Misquitta Y, Cherezov V, Slattery O, Mohan JM, et al. Membrane protein crystallization in lipidic mesophases with tailored bilayers. Structure. 2004;12:2113–24. doi: 10.1016/j.str.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 121.Zou Y, Weis WI, Kobilka BK. N-terminal T4 lysozyme fusion facilitates crystallization of a G protein coupled receptor. PLOS ONE. 2012;7:e46039. doi: 10.1371/journal.pone.0046039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods. 2006;3:887–89. doi: 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- 123.Vercruysse T, Pawar S, De Borggraeve W, Pardon E, Pavlakis GN, et al. Measuring cooperative Rev protein-protein interactions on Rev responsive RNA by fluorescence resonance energy transfer. RNA Biol. 2011;8:316–24. doi: 10.4161/rna.8.2.13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495:534–38. doi: 10.1038/nature12000. First application of nanobodies as biosensors to monitor conformational changes of GPCRs in living cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Staus DP, Wingler LM, Strachan RT, Rasmussen SGF, Pardon E, et al. Regulation of β2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol Pharmacol. 2014;85:472–81. doi: 10.1124/mol.113.089516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jähnichen S, Blanchetot C, Maussang D, Gonzalez-Pajuelo M, Chow KY, et al. CXCR4nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. PNAS. 2010;107:20565–70. doi: 10.1073/pnas.1012865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maussang D, Mujic-Delic A, Descamps FJ, Stortelers C, Vanlandschoot P, et al. Llama-derived single variable domains (nanobodies) directed against chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J Biol Chem. 2013;288:29562–72. doi: 10.1074/jbc.M113.498436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bradley ME, Dombrecht B, Manini J, Willis J, Vlerick D, et al. Potent and efficacious inhibition of CXCR2 signaling by biparatopic nanobodies combining two distinct modes of action. Mol Pharmacol. 2015;87:251–62. doi: 10.1124/mol.114.094821. [DOI] [PubMed] [Google Scholar]
- 129.Chapman AP, Antoniw P, Spitali M, West S, Stephens S, King DJ. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotechnol. 1999;17:780–83. doi: 10.1038/11717. [DOI] [PubMed] [Google Scholar]
- 130.Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003;21:484–90. doi: 10.1016/j.tibtech.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 131.Saerens D, Ghassabeh GH, Muyldermans S. Single-domain antibodies as building blocks for novel therapeutics. Curr Opin Pharmacol. 2008;8:600–8. doi: 10.1016/j.coph.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 132.Lawson AD. Antibody-enabled small-molecule drug discovery. Nat Rev Drug Discov. 2012;11:519–25. doi: 10.1038/nrd3756. [DOI] [PubMed] [Google Scholar]
- 133.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–72. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 134.Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–50. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 135.Zhou HX, McCammon JA. The gates of ion channels and enzymes. Trends Biochem Sci. 2010;35:179–85. doi: 10.1016/j.tibs.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chene P. Challenges in design of biochemical assays for the identification of small molecules to target multiple conformations of protein kinases. Drug Discov Today. 2008;13:522–29. doi: 10.1016/j.drudis.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 137.Gronemeyer H, Bourguet W. Allosteric effects govern nuclear receptor action: DNA appears as a player. Sci Signal. 2009;2:pe34. doi: 10.1126/scisignal.273pe34. [DOI] [PubMed] [Google Scholar]
- 138.Lefranc MP, Pommié C, Ruiz M, Giudicelli V, Foulquier E, et al. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol. 2003;27:55–77. doi: 10.1016/s0145-305x(02)00039-3. [DOI] [PubMed] [Google Scholar]