

Abstract
Detergents serve as useful tools for membrane protein structural and functional studies. Their amphipathic nature allows detergents to associate with the hydrophobic regions of membrane proteins whilst maintaining the proteins in aqueous solution. However, widely used conventional detergents are limited in their ability to maintain the structural integrity of membrane proteins and thus there are major efforts underway to develop novel agents with improved properties. We prepared mesitylene-cored glucoside amphiphiles (MGAs) with three alkyl chains and compared these agents with previously developed xylene-linked maltoside agents (XMAs) with two alkyl chains and a conventional detergent (DDM). When these agents were evaluated for four membrane proteins including a G protein-coupled receptor (GPCR), some agents such as MGA-C13 and MGA-C14 resulted in markedly enhanced stability of membrane proteins compared to both DDM and the XMAs. This favourable behaviour is due likely to the increased hydrophobic density provided by the extra alkyl chain. Thus, this study not only describes new glucoside agents with potential for membrane protein research, but also introduces a new detergent design principle for future development.
Keywords: amphiphile design, detergents, membrane proteins, protein solubilisation, protein stabilization
Introduction
Membrane proteins play a variety of key in cellular function, including molecular transport, signal transduction and cell-to-cell communication. Aberrant function of membrane proteins is implicated in many diseases, reflected by the fact that more than half of all pharmaceuticals target these important molecules.[1] Three-dimensional structures of membrane proteins are of major interest in drug discovery,[2] but only a few hundred membrane protein structures are currently available, while there are more than 120,000 structures of soluble proteins.[3] The slow progress in membrane protein structural studies is strongly associated with their high propensity to aggregate and denature in an aqueous medium. Conventional detergents such as n-octyl-β-D-glucopyranoside (OG), n-dodecyl-β-D-maltopyranoside (DDM) and lauryldimethylamine-N-oxide (LDAO) are widely used to avoid such protein aggregation and denaturation.[4] However, many membrane proteins surrounded even by these popular detergents tend to undergo structural degradation over time.[5] This is particularly true for eukaryotic membrane proteins and membrane protein complexes. Thus, new detergents with enhanced properties are necessary to advance membrane protein science.[6]
A number of non-conventional amphiphiles have been developed over the last two decades. Representatives include polymer-based nano-assemblies (amphiphilic polymers (amphipols),[7a,b] nanodics (NDs)[7c] and nanolipodisq particles[7d]) and peptide-based amphiphiles (e.g., lipopeptide detergents (LPDs)[8a] and β-peptides (BPs)[8b]). Small amphipathic molecules have also been invented as exemplified by tripod amphiphiles (TPAs),[9a–c] facial amphiphiles (FAs),[9d] glyco-diosgenin (GDN),[9e] neopentyl glycol (NG) class detergents (glucose neopentyl glycols (GNGs),[9f,g] maltose neopentyl glycols (MNGs),[9h–j] neopentyl glycol-derived triglucosides (NDTs)[9k]) and pentasaccharide detergents (PSEs).[9l] Of these small agents, NG class agents (GNGs and MNGs) have proved particularly interesting, as class members (e.g., MNG-3 and GNG-3) have contributed to the crystal structure determinations of more than 25 membrane proteins. This clearly illustrates the important role of new amphiphiles in membrane protein research.[10] In a recent study, we described xylene-linked maltoside amphiphiles (XMAs)[11] with a p-xylene linker that had a favourable stabilizing effect on some membrane proteins, but were suboptimal for others, such as the leucine transporter (LeuT) and the β2 adrenergic receptor (β2AR) (Figure S1a, Supporting Information). We hypothesize that the presence of two alkyl chains as the hydrophobic group in the XMAs is not ideal for tight packing of detergent alkyl chains around a target membrane protein, and this limits the protein stability conferred by these detergents in an aqueous environment. One strategy to address this issue is to enhance the hydrophobic density of the detergent molecules by increasing the number of alkyl chains. Thus, we developed three alkyl chain-bearing amphipathic agents, designated mesitylene-cored glucoside amphiphiles (MGAs) that contain the mesitylene group as a linker. These agents bear one additional alkyl chain and one more branched saccharide headgroup around the central benzene ring compared to the previously developed XMAs.[11] In our evaluation with four membrane protein systems, these new agents conferred markedly enhanced stability for all target proteins relative to the previously reported XMAs[11] and DDM, indicating a critical role of alkyl chain density in detergent’s ability to stabilise membrane proteins.
Results and Discussion
Detergent structures and physical characterizations
Initially, we prepared glucoside versions of the previously reported XMAs, designated xylene-linked glucoside amphiphiles (XGAs) (Figure S1b, Supporting Information); however, initial characterization identified several issues with these agents. First, XGA synthesis was much more difficult than that for the XMAs, giving overall yields of less than 10%. The synthetic efficiency was significantly lower than that of the XMAs (≈60%), mainly due to poor levels of glycosylation (≈15%). Second, the XGAs showed unexpectedly low water solubility, with the C7 alkyl chain XGA (XGA-C7) only marginally soluble in water (<1%). Thus, we could use only shorter alkyl chain agents (XGA-C4, XGA-C5 and XGA-C6) for evaluation with membrane proteins. In addition, the preliminary results of these evaluations indicated that the glucoside versions of XMAs (i.e., XGAs) are unfavourable for membrane protein solubilisation and stabilization (see below). These agents tend to form self-aggregates with diameters of 150–200 nm, significantly larger than standard detergent micelles (Table S1 and Figure S2a, Supporting Information), a feature likely to be associated with their limited use for membrane protein manipulation. These unfavourable results prompted us to develop an additional class of glucoside amphiphiles with a mesitylene linker rather than p-xylene linker (MGAs; Scheme 1). Detergent alkyl chain length varied from C10 to C15 and this is incorporated into the detergent designation. Owing to the core structure change from p-xylene to mesitylene, three alkyl chains and three branched diglucosides could be successfully implemented into the periphery of the central benzene ring. Each agent could be prepared without synthetic difficulty using a protocol comprising five synthetic steps, giving overall yields of ≈25%. Interestingly, water solubility was dramatically increased for the MGAs; the di-alkylated glucosides (XGAs) were poorly water-soluble with a C7 alkyl chain (<1%) while the tri-alkylated glucosides (MGAs) were more than 10% water-soluble even with a C15 alkyl chain. A similar trend was observed in a comparison with the di-alkylated maltoside versions (XMAs); in this scaffold, a C12 alkyl chain was the maximum chain length for good water solubility. Note that the XMAs contain a higher number of glucose units than the MGA counterparts (8 vs. 6). Thus, these results indicate that the MGAs possess a more water-soluble architecture than the XMAs/XGAs, a feature likely to contribute to their favourable properties for membrane protein manipulation.
Scheme 1.
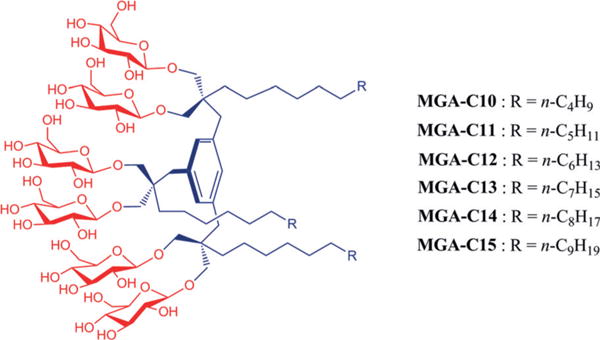
Chemical structures of MGAs (MGA-C10, MGA-C11, MGA-C12, MGA-C13, MGA-C14 and MGA-C15) with alkyl chain length variation from C10 to C15. The number of carbons in the alkyl chain was utilized for detergent designation.
Detergent micelles were characterized in terms of critical micelle concentrations (CMCs) and micelle size, both of which were estimated through fluorophore encapsulation using diphenylhexatriene (DPH)[12] and dynamic light scattering (DLS), respectively. The results are summarized in Table 1. The CMC values of all MGAs (1.5–8.0 μm) were significantly smaller than that of DDM (≈170 μm), indicating a strong tendency to form self-assemblies. Within the MGA series, the detergent CMC values decreased with alkyl chain length, giving MGA-C10 with the shortest alkyl chain the largest values (≈8.0 μm) and MGA-C15 with the longest alkyl chain the smallest value (≈1.5 μm). This is likely due to the increased hydrophobicity of the lipophilic groups with increasing alkyl chain length. Micelles formed by all the MGAs were smaller than or comparable to those formed by DDM (2.9–3.6 vs. 3.4 nm). Detergent micelle size tends to increase with alkyl chain length because of a gradual change in the geometry from conical to cylindrical shape. Yet, all of the MGAs prepared in this study self-assembled into micellar structures with a diameter smaller than or comparable to DDM, mainly due to the presence of the large headgroup (i.e., three branched diglucosides). As can be seen in the DLS profiles, the micelles formed by these MGAs showed a single set of size populations with a narrow distribution, indicative of high homogeneity (Figure S2b, Supporting Information).
Table 1.
Molecular weights (MWs) and critical micelle concentrations (CMCs) of MGAs (MGA-C10, MGA-C11, MGA-C12, MGA-C13, MGA-C14 and MGA-C15) and a conventional detergent (DDM), and hydrodynamic radii (Rh; n=5) of their micelles.
| Detergent | MW[a] | CMC [μM] | CMC [wt%] | Rh [nm][b] |
|---|---|---|---|---|
| MGA-C10 | 1736.1 | ≈ 8 | ≈0.0014 | 2.9 ± 0.01 |
| MGA-C11 | 1778.2 | ≈5 | ≈ 0.009 | 3.0 ± 0.04 |
| MGA-C12 | 1820.3 | ≈3.5 | ≈ 0.0006 | 3.1 ± 0.03 |
| MGA-C13 | 1862.3 | ≈3 | ≈0.0006 | 3.2 ± 0.03 |
| MGA-C14 | 1904.4 | ≈2 | ≈ 0.0004 | 3.4 ± 0.04 |
| MGA-C15 | 1946.5 | ≈ 1.5 | ≈ 0.0003 | 3.6 ± 0.02 |
| DDM | 510.1 | ≈170 | ≈ 0.0087 | 3.4 ± 0.02 |
Molecular weight of detergents.
Hydrodynamic radius of detergents measured at 1.0 wt% by dynamic light scattering.
Detergent evaluation with multiple membrane proteins
In order to characterize the MGAs in terms of detergent efficacy for membrane protein stabilization, the new agents were first evaluated with a protein transporter, uric acid-xanthine/H+ symporter (UapA)[13] from Aspergillus nidulans. The protein expressed in Saccharomyces cerevisiae was extracted by DDM from the membranes and then purified in 0.03% (w/v) of the same detergent. The DDM-purified UapA protein was diluted into buffer solutions containing individual MGAs or DDM to give a final detergent concentration of the individual CMC+ 0.04 wt%. After dilution, the residual DDM concentration was 0.0002 wt%, far smaller than the CMC of DDM (0.0087 wt%). Protein thermostability was assessed over time using the dye, N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM).[14] This non-fluorescent maleimide-containing molecule becomes highly fluorescent following specific conjugation with the sulfhydryl group of cysteine residues. Upon protein unfolding, a sulfhydryl group embedded in the protein interior tends to become exposed on the protein surface, leading to an increase in fluorescence intensity. Thus, the CPM assay provides a convenient means to estimate the change in the amounts of accessible sulfhydryl groups in a reaction medium, directly associated with the change in the relative amounts of unfolded target protein. As shown in Figure 1, all six MGAs were superior to DDM in terms of maintaining the folded structure of the protein at the individual CMC+0.04 wt%. No noticeable difference was observed between the MGAs at this rather low detergent concentration. When detergent concentration was increased to the individual CMC+0.2 wt%, however, efficacy differences between the MGAs became more obvious. Of the MGAs, MGA-C13 and MGA-C14 were most effective followed by MGA-C15. All the other MGAs (MGA-C10, MGA-C11 and MGA-C12) containing a rather short alkyl chain were less favourable although still better than DDM. This result indicates the favourable architecture of the MGAs for preserving the folded state of the transporter.
Figure 1.

Thermal denaturation profile of UapA solubilized in a MGA (MGA-C10, MGA-C11, MGA-C12, MGA-C13, MGA-C14, or MGA-C15) and DDM used at two different concentrations: CMC+0.04 wt% (a) and CMC+0.2 wt% (b). The relative amount of folded protein was monitored as a function of time using the CPM assay carried out at 40°C for 120 min. The data is representative of two independent experiments.
The six MGAs were also evaluated with Salmonella typhimurium melibiose permease (MelBSt).[15] E. coli membranes containing MelBSt were treated with 1.5 wt% individual MGA or DDM and incubated for 90 min at 0°C for direct protein extraction. The amounts of soluble MelBSt after ultracentrifugation were determined by Western blot. The amount of soluble MelBSt in individual detergents was expressed as a percentage of total MelBSt in the untreated membrane sample (Figure 2). At 0°C, DDM showed high efficiency of extraction of the permease while all MGAs were inferior to DDM, indicating suboptimal behaviour of the MGAs. When the incubation temperature was increased to 45°C, however, the amount of soluble MelBSt significantly increased for all the MGAs. It is notable that those MGAs with longer alkyl chains (MGA-C13, MGA-C14, or MGA-C15) extracted soluble transporter as efficiently as DDM. Detergent efficacy between DDM and the MGAs was clearly discerned by a further temperature increase to 55°C. At this higher temperature, DDM yielded only a small amount of soluble MelBSt (<10%), implying that most DDM-extracted transporter aggregated or denatured during the 90 min incubation. In contrast, all the MGAs retained substantial amounts of soluble MelBSt, with the remarkable performance observed for MGA-C13 and MGA-C14. In the case of MGA-C15, the amount of soluble MelBSt was slightly decreased at 55°C. This result suggests that MGA-C13 and MGA-C14 have optimal structures for retaining MelBSt solubility. When the di-alkylated versions of MGAs (i.e., XGAs; XGA-C4, XGA-C5 and XGA-C6) were tested for this protein (Figure S3, Supporting Information), all the XGAs were inferior to DDM with regard to extracting MelBSt from the membranes at 0°C and retaining the permease solubility at the elevated temperatures. These results clearly demonstrate that the tri-alkylated MGA architecture is superior to that of the di-alkylated XGAs at preserving MelBSt solubility/stability.
Figure 2.

Thermostability of MelBSt solubilized in a MGA (MGA-C10, MGA-C11, MGA-C12, MGA-C13, MGA-C14, or MGA-C15) or DDM. The protein was extracted from E. coli membranes by treatment with 1.5 wt% individual detergent at four different temperatures (0, 45, 55, and 65°C) for 90 min. The solubilized MelBSt was separated by ultracentrifugation and visualized by Western blot (a). The amounts of the soluble MelBSt were expressed as percentages of total MelBSt in the untreated membrane (Memb) and presented in histograms (b). Error bars, SEM, n=3.
The favourable results obtained for UapA and MelBSt encouraged us to evaluate the MGAs with the leucine transporter (LeuT)[16] from Aquifex aeolicus. MGA-C10 was inferior to the other MGAs in the evaluation with UapA and MelBSt and thus was not further evaluated. The transporter expressed in E. coli membranes was first extracted from the membranes using 1.0 wt% DDM and purified in the presence of 0.05 wt% of the same detergent. The DDM-purified transporter was mixed with buffer solutions including individual MGAs or DDM to give a final detergent concentration of the individual CMC+ 0.04 wt%. The residual DDM concentration was 0.005 wt%. The protein samples solubilized in the individual detergents were incubated for 12 days at room temperature and substrate binding activity of the transporter was measured at regular intervals by scintillation proximity assay (SPA) using a radio-labelled substrate ([3H]-Leu).[17] As can be seen in Figure 3a, the DDM-solubilized transporter gave a gradual loss in substrate binding activity over time, resulting in ≈40% retention of the initial activity after the 12 day incubation. All the MGAs were better than DDM at maintaining LeuT activity, with the best performance achieved for MGA-C11 followed by MGA-C12 and MGA-C13. The MGA-C11-solubilized transporter gave almost full retention in the substrate binding activity (≈90%) after the 12 day incubation. Of the tested MGAs, MGA-C15 with the longest alkyl chain was the poorest at retaining transporter activity, but still slightly better than DDM. As detergent concentration was increased to the individual CMC+0.2 wt%, the DDM-solubilized transporter more rapidly lost substrate binding activity (Figure 3b). A similar trend was observed for the MGAs with a long alkyl chain (MGA-C14 and MGA-C15); these agents resulted in a significant decrease in the transporter activity (40–50% after 12 day incubation). In contrast, the MGAs with a short alkyl chain (MGA-C11, MGA-C12 and MGA-C13) were still very effective at preserving the activity of the transporter (≈90% after the 12 day incubation). Thus, the outcome of the MGAs was dependent on detergent alkyl chain length. When the di-alkylated versions (XGAs; XGA-C4, XGA-C5 and XGA-C6) were evaluated with LeuT, none of these agents were better than DDM (Figure S4, Supporting Information), consistent with the XGA results for MelBSt.
Figure 3.

Long-term substrate or ligand binding activity for LeuT (a,b) and β2AR (c) solubilized in individual MGAs or DDM. Five MGAs (MGA-C11, MGA-C12, MGA-C13, MGA-C14 and MGA-C15) and three MGAs (MGA-C12, MGA-C13 and MGA-C14) were used for the evaluation with LeuT and β2AR, respectively. Two different detergent concentrations ((a) CMC +0.04 wt% and (b) CMC+0.2 wt%) were used for LeuT stability assessment while a single detergent concentration (CMC+0.2 wt%) was used for the β2AR stability experiment. LeuT activity was measured at regular intervals over the course of a 12 day incubation at room temperature via a scintillation proximity assay (SPA) using a radio-labelled substrate ([3H]-Leucine). β2AR ligand binding activity was assessed at regular intervals using the antagonist [3H]-dihydroalprenolol (DHA) over a three day incubation at room temperature. Error bars, SEM, n=3.
We next moved to the human β2 adrenergic receptor (β2AR), a G protein-coupled receptor (GPCR), for further detergent evaluation.[18] For this analysis, three MGAs, MGA-C12, MGA-C13 and MGA-C14, were selected as MGA-C10/C11 and MGA-C15 showed the worst behaviour for UapA and LeuT, respectively. The receptor was first expressed in Sf9 insect cells and then extracted from the membranes using a conventional detergent (DDM). The receptor was further purified in 0.1 wt% DDM. The DDM-purified receptor was diluted with individual MGAs- or DDM-containing buffers to give a final detergent concentration of the individual CMC +0.2 wt%. After dilution, the residual DDM concentration in the MGA samples was 0.0007 wt%. The receptor stability was addressed by measuring the ligand binding activity using a radio-labelled antagonist ([3H]-dihydroalprenolol, DHA).[19] As a preliminary study, the activity of the receptor solubilized in the individual MGAs or DDM was measured after a 30 min dilution to allow detergent reconstitution. The MGAs (MGA-C12, MGA-C13 and MGA-C14) showed an initial receptor activity comparable to both each other and DDM (Figure S5, Supporting Information). However, a clear difference in detergent efficacy was observed between the MGAs and DDM when the receptor stability was monitored at regular intervals over a three-day incubation at room temperature (Figure 3c). The DDM-solubilized receptor rapidly lost activity over time, resulting in only ≈15% retention of receptor activity at the end of the three-day incubation. A similar activity loss was observed for MGA-C12-solubilized receptor. In contrast, the MGA-C13 or MGA-C14-solubilized receptor lost ligand binding capacity much slower and additionally there was greater activity at the end of the incubation period (50–60%), with better performance for MGA-C14 relative to MGA-C13. In order to provide greater insights into the properties of the MGAs, DDM- or MGA-C13-solubilized β2AR was analyzed by size exclusion chromatography (SEC) following detergent exchange. MGA-C13 was chosen for this assessment as it showed effective stabilization of all four targeted membrane proteins. As can be seen in Figure S6a, Supporting Information, MGA-C13 formed smaller PDCs with the receptor than DDM, which may be favourable for membrane protein crystallization. Application of the DDM- or MGA-C13-solubilized β2AR to the SEC column in a detergent-free buffer revealed a significant difference between DDM and MGA-C13; a large decrease in intensity of the monodisperse peak (≈13 mL) and a concomitant appearance of the new broad aggregation peak (≈10 mL) were observed for the DDM-solubilized receptor in detergent-free buffer (Figure S6b, Supporting Information). In contrast, only a slight change in the chromatogram was observed for the MGA-C13-solubilized receptor under the same conditions. Thus, this MGA agent maintained receptor integrity even in a detergent-free buffer, presumably due to this detergent forming stronger interactions with the receptor than DDM.
In this study, we introduced mesitylene-cored glucosides (MGAs) with three alkyl chains and found that these agents were consistently more effective than DDM and di-alkylated versions (XGAs) for membrane protein stability. These agents were also clearly superior to the previously reported di-alkylated maltosides (XMAs) at stabilizing membrane proteins, particularly for LeuT and β2AR. The previous study had shown that the XMAs were inferior to DDM for these membrane proteins.[11] The well-behaved MGAs (MGA-C13 and MGA-C14) proved to be superior even to MNG-3, one of the most successful new amphiphiles,[5c,20] in terms of stabilizing UapA (Figure S7, Supporting Information). This finding appeared to be true for LeuT as well.[9h,j] It is hard to narrow down which structural features are responsible for the enhanced efficacy of the MGAs. However, due to the presence of three alkyl chains around a central ring structure, the MGA molecules have increased alkyl chain density (i.e., hydrophobic density) relative to the XGAs/XMAs with only two alkyl chains. Accordingly, the MGAs would generate a detergent micelle interior with increased hydrophobic density compared to the XGAs/XMAs. This may result in a stronger and thus more favourable detergent interaction with the protein hydrophobic surface, thus providing an optimal platform for effective protein encapsulation. The increased alkyl chain density also seemed to endow the MGAs with additional advantages. First, the MGAs were more water-soluble with increasing alkyl chain length than the equivalent XGAs and XMAs. Second, the MGAs formed relatively small micelles with a very low CMC value (1.5–8.0 μm), in stark contrast with the XGAs, which formed large liposomes with a high critical aggregation value (≈800 μm). Collectively, the current study revealed that the diverse detergent properties such as water solubility, self-assembly morphology and, more importantly, membrane protein stabilization efficacy could be significantly improved by modulating the alkyl chain density (i.e., hydrophobic density) of the detergent micelles. This detergent structure–property–efficacy relationship has not been studied in detail so far.
It is notable that the MGAs bear glucosides instead of maltosides as headgroups. In general, maltoside detergents (e.g., DDM) are more effective at stabilizing membrane proteins than glucoside agents (e.g., OG). However, glucoside agents are also widely used for membrane protein study, presumably as a result of the small headgroup (i.e., glucoside), which results in the formation of small PDCs. Small PDCs are often favourable for the generation of high quality protein crystals and for NMR-based protein structural study. Thus, glucoside detergents have distinctive advantages over maltoside agents even if those agents have relatively limited ability to stabilize membrane proteins. A similar conclusion was reached from the comparison of the GNGs with MNGs. For example, GNG-3 was generally inferior to the maltoside analogue (MNG-3) in terms of maintaining membrane protein stability,[9f,h,j] but this agent has successfully facilitated the high-resolution structure determinations of several membrane proteins.[10o,p,21] This analysis (glucosides vs. maltosides) implies that it is challenging to develop novel glucoside detergents with enhanced efficacy toward membrane protein stabilization relative to DDM, reflected by the fact that most novel detergents effective in this regard contain maltoside headgroups (e.g., MNGs, FAs and GDN). Very recently, a class of amphiphiles called NDTs have attracted substantial attention as these glucosides were shown to be superior to DDM for multiple membrane proteins.[9k] As the NDTs contain a hydrophilic neopentyl glycol (NG) linker to connect the glucose group with the hydrophobic alkyl chain, however, the headgroup of this agent is an NG-glucose conjugate rather than a glucoside group alone. The MGAs described here utilize a hydrophobic linker derived from dimethylmalonate to bridge the head and tail groups. Thus, these agents are the first examples of true glucoside detergents conferring enhanced protein stability compared to DDM, to multiple membrane proteins.
Conclusion
Detergent efficacy tends to vary depending on the characteristics of the target membrane proteins. Consequently, it is often the case that a detergent effective for one membrane protein is not favourable for another. Thus, it is extremely important to evaluate a newly developed agent with multiple membrane proteins to show general utility for membrane protein study. Furthermore, a detergent should be easily prepared in order for it to be accessible to the wider membrane protein community. Finding a detergent or detergent class with such general utility and ready accessibility is extremely challenging because multiple favourable properties including good water solubility, micelle formation, optimum hydrophile–lipophile balance (HLB) and small PDC formation need to be incorporated within a single molecular architecture along with convenient synthesis. In the present study, MGA-C13 and MGA-C14, showed general utility as these agents proved effective at stabilizing multiple membrane proteins. The results strongly indicate that these MGAs incorporate multiple favourable properties and thus have significant potential for membrane protein research. In addition, the design principles described here have significant potential for the development and optimization of other new detergents.
Supplementary Material
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (grant number 2008–0061891 and 2016R1A2B2011257 to P.S.C. and K.H.C.). This work was also supported by the National Science Foundation (grant number: MCB-1158085 to L.G.) and by the National Institutes of Health (grant number: R01 GM095538 to L.G.).
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under http://dx.doi.org/10.1002/chem.201603338. It contains experimental details, including the synthesis and characterization of the MGAs, and membrane protein stability assay.
References
- 1.a) Overington JP, Al-Lazikani B, Hopkins AL. Nat Rev Drug Discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]; b) Sanders CR, Myers JK. Annu Rev Biophys Biomol Struct. 2004;33:25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- 2.Lacapère JJ, Pebay-Peyroula E, Neumann JM, Etchebest C. Trends Biochem Sci. 2007;32:259–270. doi: 10.1016/j.tibs.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 3.http://blanco.biomol.uci.edu/mpstruc
- 4.a) Privé GG. Methods. 2007;41:388–397. doi: 10.1016/j.ymeth.2007.01.007. [DOI] [PubMed] [Google Scholar]; b) Chae PS, Laible PD, Gellman SH. Mol BioSyst. 2010;6:89–94. doi: 10.1039/b915162c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Proc Natl Acad Sci USA. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Newstead S, Ferrandon S, Iwata S. Protein Sci. 2008;17:466–472. doi: 10.1110/ps.073263108. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) He Y, Wang K, Yan N. Protein Cell. 2014;5:658–672. doi: 10.1007/s13238-014-0086-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Zhang QH, Tao HC, Hong WX. Methods. 2011;55:318–323. doi: 10.1016/j.ymeth.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kang HJ, Lee C, Drew D. Int J Biochem Cell Biol. 2013;45:636–644. doi: 10.1016/j.biocel.2012.12.018. [DOI] [PubMed] [Google Scholar]; c) Moraes I, Evans G, Sanchez-Weatherby J, Newstead S, Stewart PDS. Biochem Biophys Acta. 2014;1838:78–87. doi: 10.1016/j.bbamem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Tribet C, Audebert R, Popot JL. Proc Natl Acad Sci USA. 1996;93:15047–15050. doi: 10.1073/pnas.93.26.15047. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Champeil P, Menguy T, Tribet C, Popot JL, le Maire M. J Biol Chem. 2000;275:18623–18637. doi: 10.1074/jbc.M000470200. [DOI] [PubMed] [Google Scholar]; c) Nath A, Atkins WM, Sligar SG. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]; d) Orwick-Rydmark M, Lovett JE, Graziadei A, Lindholm L, Hicks MR, Watts A. Nano Lett. 2012;12:4687–4692. doi: 10.1021/nl3020395. [DOI] [PubMed] [Google Scholar]
- 8.a) McGregor CL, Chen L, Pomroy NC, Hwang P, Go S, Chakrabartty A, Prive GG. Nat Biotechnol. 2003;21:171–176. doi: 10.1038/nbt776. [DOI] [PubMed] [Google Scholar]; b) Tao HC, Lee SC, Moeller A, Roy RS, Siu FY, Zimmermann J, Stevens RC, Potter CS, Carragher B, Zhang QH. Nat Methods. 2013;10:759–761. doi: 10.1038/nmeth.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Chae PS, Wander MJ, Bowling AP, Laible PD, Gellman SH. ChemBioChem. 2008;9:1706–1709. doi: 10.1002/cbic.200800169. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chae PS, Cho KH, Wander MJ, Bae HE, Gellman SH, Laible PD. Biochem Biophys Acta. 2014;1838:278–286. doi: 10.1016/j.bbamem.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chae PS, Bae HE, Ehsan M, Hussain H, Kim JW. Org Biomol Chem. 2014;12:8480–8487. doi: 10.1039/c4ob01375a. [DOI] [PubMed] [Google Scholar]; d) Lee SC, Bennett BC, Hong WX, Fu Y, Baker KA, Marcoux J, Robinson CV, Ward AB, Halpert JR, Stevens RC, Stout CD, Yeager MJ, Zhang QH. Proc Natl Acad Sci USA. 2013;110:E1203–E1211. doi: 10.1073/pnas.1221442110. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Kruse AC, Manglik A, Cho KH, Nurva S, Gether U, Guan L, Loland CJ, Byrne B, Kobilka BK, Gellman SH. Chem Eur J. 2012;18:9485–9490. doi: 10.1002/chem.201200069. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Chae PS, Rana RR, Gotfryd K, Rasmussen SGF, Kruse AC, Cho KH, Capaldi S, Carlsson E, Kobilka B, Loland CJ, Gether U, Banerjee S, Byrne B, Lee JK, Gellman SH. Chem Commun. 2013;49:2287–2289. doi: 10.1039/c2cc36844g. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Cho KH, Bae HE, Das M, Gellman SH, Chae PS. Chem Asian J. 2014;9:632–638. doi: 10.1002/asia.201301303. [DOI] [PubMed] [Google Scholar]; h) Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot JL, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka B, Gellman SH. Nat Methods. 2010;7:1003–1090. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Cho KH, Byrne B, Chae PS. ChemBioChem. 2013;14:452–455. doi: 10.1002/cbic.201200759. [DOI] [PubMed] [Google Scholar]; j) Cho KH, Husri M, Amin A, Gotfryd K, Lee HJ, Go J, Kim JW, Loland CJ, Guan L, Byrne B, Chae PS. Analyst. 2015;140:3157–3163. doi: 10.1039/c5an00240k. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Sadaf A, Mortensen JS, Capaldi S, Tikhonova E, Hariharan P, Ribeiro O, Loland CJ, Guan L, Byrne B, Chae PS. Chem Sci. 2016;7:1933–1939. doi: 10.1039/c5sc02900g. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Ehsan M, Du Y, Scull NJ, Tikhonova E, Tarrasch J, Mortensen JS, Loland CJ, Skiniotis G, Guan L, Byrne B, Kobilka BK, Chae PS. J Am Chem Soc. 2016;138:3789–3796. doi: 10.1021/jacs.5b13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Rasmussen SGF, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, DeVree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi HJ, DeVree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kruse AC, Hu JX, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic-Jeremic J, Shah P, Shiloach J, Tate CG, Grisshammer R. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Kruse AC, Ring AM, Manglik A, Hu JX, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. Nature. 2013;502:575–579. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Rollauer SE, Tarry MJ, Graham JE, Jaaskelainen M, Jager F, Johnson S, Krehenbrink M, Liu SM, Lukey MJ, Marcoux J, McDowell MA, Rodriguez F, Roversi P, Stansfeld PJ, Robinson CV, Sansom MSP, Palmer T, Hogbom M, Berks BC, Lea SM. Nature. 2012;492:210–214. doi: 10.1038/nature11683. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Karakas E, Furukawa H. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Dickson V Kane, Pedi L, Long SB. Nature. 2014;516:213–218. doi: 10.1038/nature13913. [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Rasmussen SGF, DeVree BT, Zou YZ, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Shukla AK, Westfield GH, Xiao KH, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, Dosey AM, Su M, Liang CR, Gu LL, Shan JM, Chen X, Hanna R, Choi MJ, Yao XJ, Klink BU, Kahsai AW, Sidhu SS, Koide S, Penczek PA, Kossiakoff AA, Woods VL, Kobilka BK, Skiniotis G, Lefkowitz RJ. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Kellosalo J, Kajander T, Kogan K, Pokharel K, Goldman A. Science. 2012;337:473–476. doi: 10.1126/science.1222505. [DOI] [PubMed] [Google Scholar]; p) Frick A, Eriksson UK, Mattia F de, Ouml F, Berg K, Hedfalk R, Neutze W, de Grip J, Deen PMT, Tornroth-Horsefield S. Proc Natl Acad Sci USA. 2014;111:6305–6310. doi: 10.1073/pnas.1321406111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho KH, Du Y, Scull NJ, Hariharan P, Gotfryd K, Loland CJ, Guan L, Byrne B, Kobilka BK, Chae PS. Chem Eur J. 2015;21:10008–10013. doi: 10.1002/chem.201501083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chattopadhyay A, London E. Anal Biochem. 1984;139:408–412. doi: 10.1016/0003-2697(84)90026-5. [DOI] [PubMed] [Google Scholar]
- 13.a) Pantazopoulou A, Diallinas G. Mol Membr Biol. 2006;23:337–348. doi: 10.1080/09687860600738239. [DOI] [PubMed] [Google Scholar]; b) Alguel Y, Amillis S, Leung J, Lambrinidis G, Capaldi S, Scull NJ, Craven G, Iwata S, Armstrong A, Mikros E, Diallinas G, Cameron AD, Byrne B. Nat Commun. 2016;7:11336. doi: 10.1038/ncomms11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov AI, Mileni M, Chien EYT, Hanson MA, Stevens RC. Structure. 2008;16:351–359. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 15.a) Amin A, Hariharan P, Chae PS, Guan L. Biochemistry. 2015;54:5849–5855. doi: 10.1021/acs.biochem.5b00660. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ethayathulla AS, Yousef MS, Amin A, Leblanc G, Kaback HR, Guan L. Nat Commun. 2014;5:3009. doi: 10.1038/ncomms4009. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Amin A, Ethayathulla AS, Guan L. J Bacteriol. 2014;196:3134–3139. doi: 10.1128/JB.01868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, Overbeek R, Snead MA, Keller M, Aujay M, Huber R, Feldman RA, Short JM, Olsen GJ, Swanson RV. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 17.Quick M, Javitch JA. Proc Natl Acad Sci USA. 2007;104:3603–3608. doi: 10.1073/pnas.0609573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 19.Riva MA, Creese I. Mol Pharmacol. 1989;36:201–210. [PubMed] [Google Scholar]
- 20.Hauer F, Gerle C, Fischer N, Oshima A, Shinzawa-Itoh K, Shimada S, Yokoyama K, Fujiyoshi Y, Stark H. Structure. 2015;23:1769–1775. doi: 10.1016/j.str.2015.06.029. [DOI] [PubMed] [Google Scholar]
- 21.a) Dong YY, Pike ACW, Mackenzie A, McClenaghan C, Aryal P, Dong L, Quigley A, Grieben M, Goubin S, Mukhopadhyay S, Ruda GF, Clausen MV, Cao LS, Brennan PE, Burgess-Brown NA, Sansom MSP, Tucker SJ, Carpenter EP. Science. 2015;347:1256–1259. doi: 10.1126/science.1261512. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Quigley A, Dong YY, Pike ACW, Dong L, Shrestha L, Berridge G, Stansfeld PJ, Sansom MSP, Edwards AM, Bountra C, von Delft F, Bullock AN, Burgess-Brown NA, Carpenter EP. Science. 2013;339:1604–1607. doi: 10.1126/science.1231513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.